
Graph-Based Integration of Histone Modification Profiles


Abstract
:1. Introduction
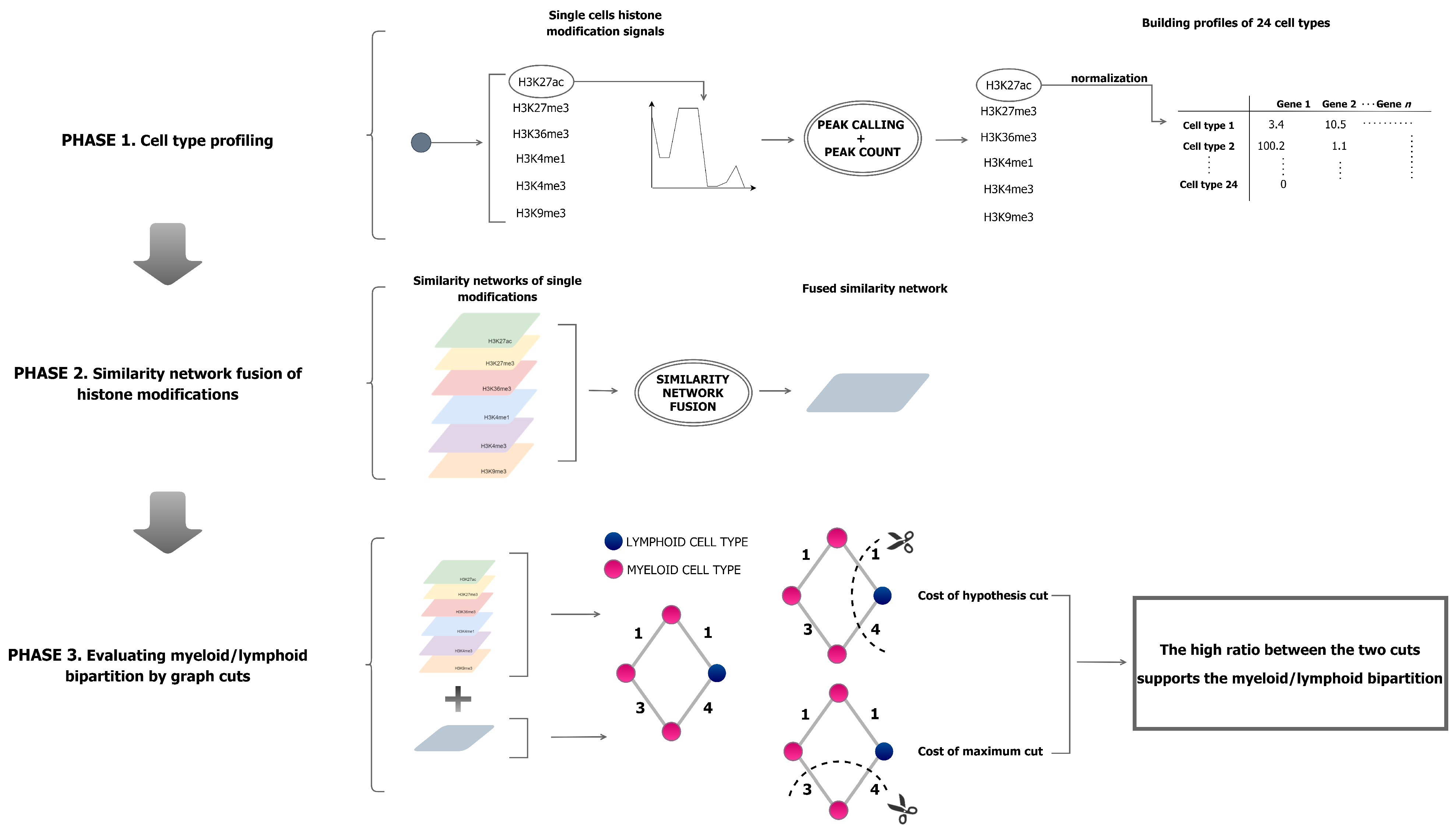
2. Materials and Methods
2.1. Peak Calling
2.2. Normalisation
2.3. Cell-Type Expression Profiles
2.4. Profile Integration
2.5. Hypothesis Testing
3. Results
3.1. Dataset
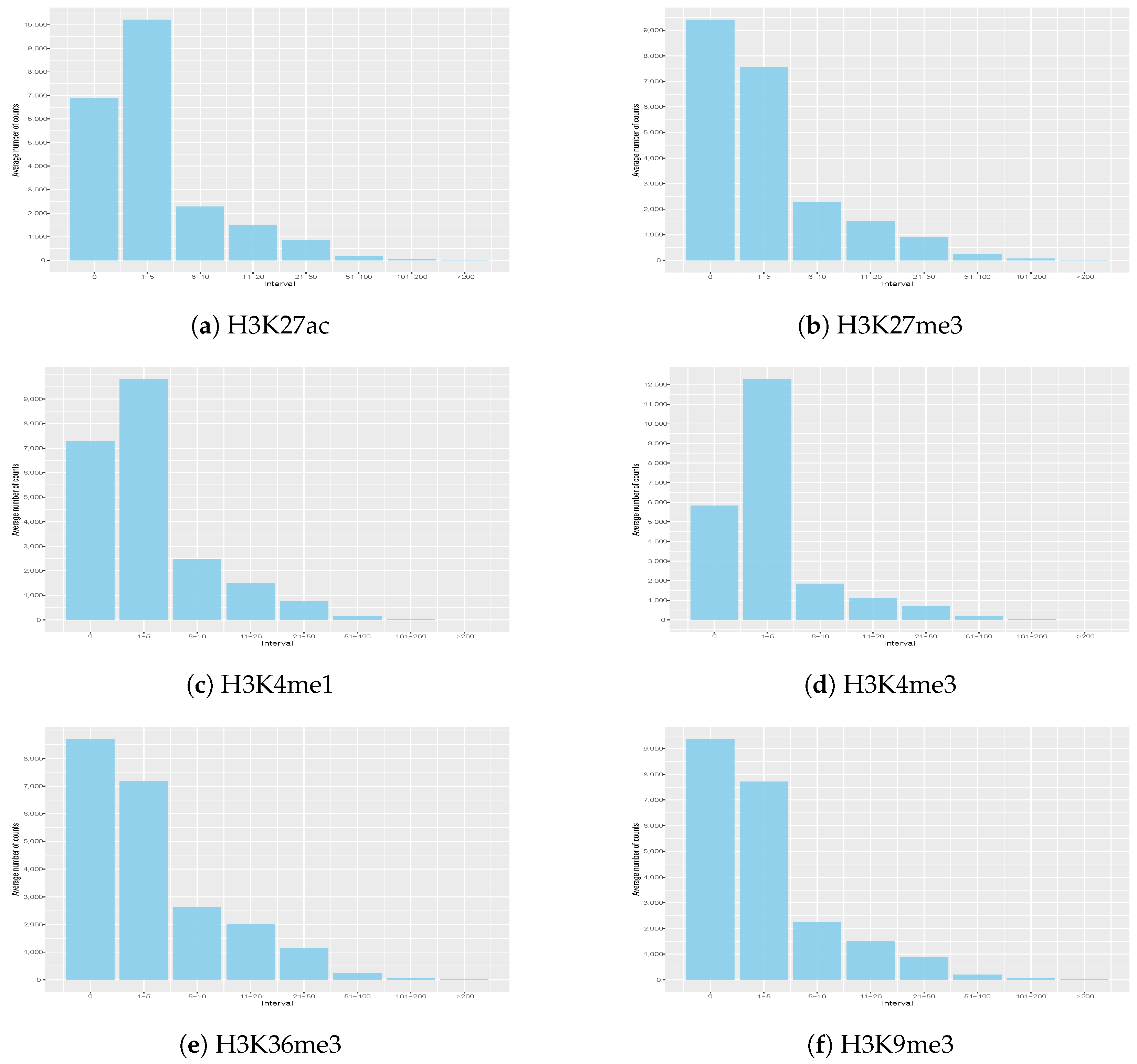
3.2. Histone Signal Distribution
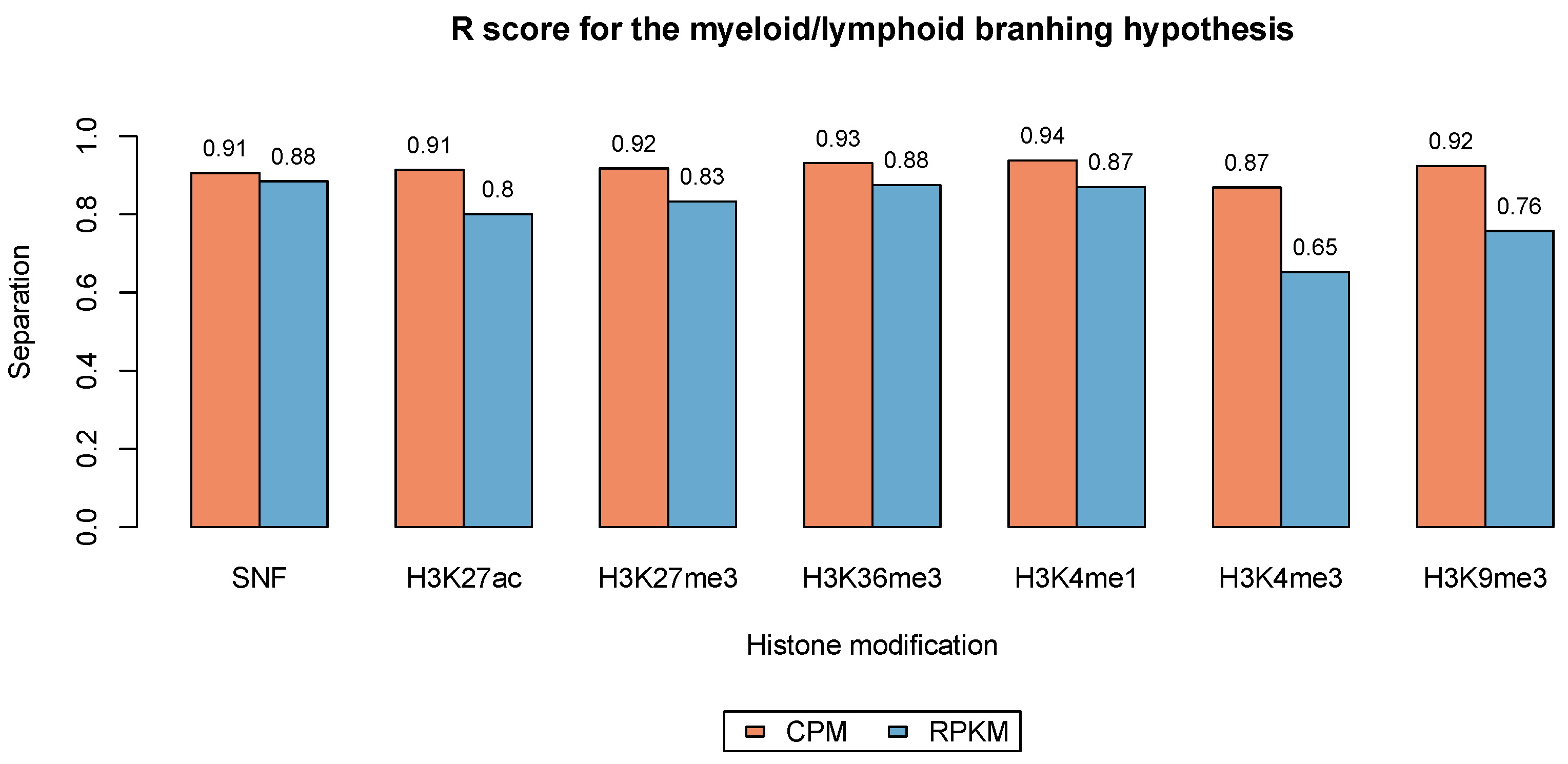
3.3. Phenotype Separation Evaluation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hizume, K.; Yoshimura, S.H.; Takeyasu, K. Linker histone H1 per se can induce three-dimensional folding of chromatin fiber. Biochemistry 2005, 44, 12978–12989. [Google Scholar] [CrossRef] [PubMed]
- Peterson, C.L.; Laniel, M.A. Histones and histone modifications. Curr. Biol. 2004, 14, R546–R551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, H. Histone modifications for human epigenome analysis. J. Hum. Genet. 2013, 58, 439–445. [Google Scholar] [CrossRef] [Green Version]
- Karlić, R.; Chung, H.R.; Lasserre, J.; Vlahoviček, K.; Vingron, M. Histone modification levels are predictive for gene expression. Proc. Natl. Acad. Sci. USA 2010, 107, 2926–2931. [Google Scholar] [CrossRef] [Green Version]
- Mardis, E.R. ChIP-seq: Welcome to the new frontier. Nat. Methods 2007, 4, 613–614. [Google Scholar] [CrossRef]
- O’Geen, H.; Echipare, L.; Farnham, P.J. Using ChIP-seq technology to generate high-resolution profiles of histone modifications. In Epigenetics Protocols; Springer: Berlin/Heidelberg, Germany, 2011; pp. 265–286. [Google Scholar]
- Bujold, D.; Morais, D.A.d.; Gauthier, C.; Côté, C.; Caron, M.; Kwan, T.; Chen, K.C.; Laperle, J.; Markovits, A.N.; Pastinen, T.; et al. The International Human Epigenome Consortium Data Portal. Cell Syst. 2016, 3, 496–499.E2. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, M.; Daujat, S.; Schneider, R. Lateral Thinking: How Histone Modifications Regulate Gene Expression. Trends Genet. 2016, 32, 42–56. [Google Scholar] [CrossRef] [Green Version]
- Bannister, A.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Pepke, S.; Wold, B.; Mortazavi, A. Computation for ChIP-seq and RNA-seq studies. Nat. Methods 2009, 6, S22–S32. [Google Scholar] [CrossRef]
- Vaissière, T.; Sawan, C.; Herceg, Z. Epigenetic interplay between histone modifications and DNA methylation in gene silencing. Mutat. Res. Mutat. Res. 2008, 659, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Jiang, J.; Wang, W.; Zhou, Z.H.; Tu, Z. Unsupervised metric fusion by cross diffusion. In Proceedings of the 2012 IEEE Conference on Computer Vision and Pattern Recognition, Providence, RI, USA, 16–21 June 2012; pp. 2997–3004. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Mezlini, A.; Demir, F.; Fiume, M.; Tu, Z.; Brudno, M.; Haibe-Kains, B.; Goldenberg, A. Similarity network fusion for aggregating data types on a genomic scale. Nat. Methods 2014, 11, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Prchal, J.T.; Throckmorton, D.W.; Carroll, A.J.; Fuson, E.W.; Gams, R.A.; Prchal, J.F. A common progenitor for human myeloid and lymphoid cells. Nature 1978, 274, 590–591. [Google Scholar] [CrossRef] [PubMed]
- Blahnik, K.R.; Dou, L.; O’Geen, H.; McPhillips, T.; Xu, X.; Cao, A.R.; Iyengar, S.; Nicolet, C.M.; Ludäscher, B.; Korf, I.; et al. Sole-Search: An integrated analysis program for peak detection and functional annotation using ChIP–seq data. Nucleic Acids Res. 2010, 38, e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621. [Google Scholar] [CrossRef]
- Lloyd, S. Least squares quantization in PCM. IEEE Trans. Inf. Theory 1982, 28, 129–137. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2017. [Google Scholar]
- Thorndike, R.L. Who belongs in the family? Psychometrika 1953, 18, 267–276. [Google Scholar] [CrossRef]
- Tritchler, D.; Parkhomenko, E.; Beyene, J. Filtering genes for cluster and network analysis. BMC Bioinform. 2009, 10, 193. [Google Scholar] [CrossRef] [Green Version]
- Karger, D.R. Global Min-cuts in RNC, and Other Ramifications of a Simple Min-Cut Algorithm. In Proceedings of the SODA, Austin, TX, USA, 25–27 January 1993; Volume 93, pp. 21–30. [Google Scholar]
- Ford, L.R.; Fulkerson, D.R. Maximal flow through a network. Can. J. Math. 1956, 8, 399–404. [Google Scholar] [CrossRef]
- Karp, R.M. Reducibility among combinatorial problems. In Complexity of Computer Computations; Springer: Berlin/Heidelberg, Germany, 1972; pp. 85–103. [Google Scholar]
- Bansal, V.; Bafna, V. HapCUT: An efficient and accurate algorithm for the haplotype assembly problem. Bioinformatics 2008, 24, i153–i159. [Google Scholar] [CrossRef] [Green Version]
- Stoer, M.; Wagner, F. A simple min-cut algorithm. J. ACM (JACM) 1997, 44, 585–591. [Google Scholar] [CrossRef]
- Church, B.V.; Williams, H.T.; Mar, J.C. Investigating skewness to understand gene expression heterogeneity in large patient cohorts. BMC Bioinform. 2019, 20, 1–14. [Google Scholar] [CrossRef] [PubMed]
- De Torrenté, L.; Zimmerman, S.; Suzuki, M.; Christopeit, M.; Greally, J.M.; Mar, J.C. The shape of gene expression distributions matter: How incorporating distribution shape improves the interpretation of cancer transcriptomic data. BMC Bioinform. 2020, 21, 562. [Google Scholar] [CrossRef] [PubMed]
- Alberti-Servera, L.; von Muenchow, L.; Tsapogas, P.; Capoferri, G.; Eschbach, K.; Beisel, C.; Ceredig, R.; Ivanek, R.; Rolink, A. Single-cell RNA sequencing reveals developmental heterogeneity among early lymphoid progenitors. EMBO J. 2017, 36, 3619–3633. [Google Scholar] [CrossRef]
- Perié, L.; Duffy, K.R.; Kok, L.; de Boer, R.J.; Schumacher, T.N. The branching point in erythro-myeloid differentiation. Cell 2015, 163, 1655–1662. [Google Scholar] [CrossRef] [Green Version]
- Mikkelsen, T.S.; Ku, M.; Jaffe, D.B.; Issac, B.; Lieberman, E.; Giannoukos, G.; Alvarez, P.; Brockman, W.; Kim, T.K.; Koche, R.P.; et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 2007, 448, 553–560. [Google Scholar] [CrossRef]
- Jacomy, M.; Venturini, T.; Heymann, S.; Bastian, M. ForceAtlas2, a continuous graph layout algorithm for handy network visualization designed for the Gephi software. PLoS ONE 2014, 9, e98679. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type | Origin | Lineage | H3K27ac | H3K27me3 | H3K36me3 | H3K4me1 | H3K4me3 | H4K9me3 |
|---|---|---|---|---|---|---|---|---|
| Alternatively activated macrophage | Blood | Myeloid | 7 | 7 | 7 | 7 | 7 | 7 |
| Band-form neutrophil | Bone marrow | Myeloid | 3 | 3 | 3 | 3 | 4 | 3 |
| CD14-positive, CD16-negative classical monocyte | Blood | Myeloid | 14 | 9 | 6 | 10 | 9 | 8 |
| CD34-negative, CD41-positive, CD42-positive megakaryocyte cell | Blood | Myeloid | 2 | 2 | 3 | 3 | 3 | 2 |
| CD38-negative naive B cell | Blood | Lymphoid | 4 | 5 | 6 | 5 | 7 | 7 |
| CD4-positive, alpha-beta T cell | Blood | Lymphoid | 9 | 9 | 9 | 9 | 9 | 9 |
| CD8-positive, alpha-beta T cell | Blood | Lymphoid | 6 | 5 | 5 | 5 | 5 | 5 |
| Central memory CD4-positive, alpha-beta T cell | Blood | Lymphoid | 1 | 1 | 1 | 1 | 2 | 1 |
| Class switched memory B cell | Blood | Lymphoid | 3 | 3 | 2 | 3 | 3 | 3 |
| Cytotoxic CD56-dim natural killer cell | Blood | Lymphoid | 4 | 4 | 4 | 5 | 6 | 5 |
| Effector memory CD8-positive, alpha-beta T cell | Blood | Lymphoid | 2 | 1 | 2 | 2 | 3 | 3 |
| Endothelial cell of umbilical vein (proliferating) | Blood | Lymphoid | 2 | 2 | 2 | 2 | 2 | 2 |
| Endothelial cell of umbilical vein (resting) | Blood | Lymphoid | 1 | 2 | 2 | 2 | 2 | 2 |
| Erythroblast | Blood | Myeloid | 2 | 2 | 2 | 2 | 2 | 2 |
| Inflammatory macrophage | Blood | Myeloid | 8 | 8 | 9 | 7 | 8 | 9 |
| Macrophage | Blood | Myeloid | 14 | 7 | 7 | 13 | 14 | 8 |
| Mature eosinophil | Blood | Myeloid | 2 | 2 | 2 | 2 | 2 | 2 |
| Mature neutrophil | Blood | Myeloid | 15 | 13 | 13 | 13 | 13 | 13 |
| Monocyte | Blood | Myeloid | 36 | 22 | 3 | 28 | 28 | 15 |
| Naive B cell | Blood | Lymphoid | 8 | 8 | 9 | 7 | 8 | 8 |
| Neutrophilic metamyelocyte | Bone marrow | Myeloid | 3 | 3 | 3 | 3 | 4 | 3 |
| Neutrophilic myelocyte | Bone marrow | Myeloid | 3 | 3 | 3 | 3 | 4 | 3 |
| Plasma cell | Bone marrow | Lymphoid | 3 | 3 | 3 | 3 | 3 | 3 |
| Segmented neutrophil of bone marrow | Bone marrow | Myeloid | 3 | 3 | 3 | 3 | 4 | 3 |
| Total | 155 | 127 | 109 | 141 | 152 | 126 |
| Modification | CPM | RPKM | INTERSECTION |
|---|---|---|---|
| H3K27ac | 5655 | 481 | 340 |
| H3K27me3 | 5294 | 235 | 184 |
| H3K36me3 | 6062 | 369 | 264 |
| H3K4me1 | 7309 | 248 | 206 |
| H3K4me3 | 5627 | 235 | 189 |
| H3K9me3 | 5295 | 383 | 280 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baccini, F.; Bianchini, M.; Geraci, F. Graph-Based Integration of Histone Modification Profiles. Mathematics 2022, 10, 1842. https://doi.org/10.3390/math10111842
Baccini F, Bianchini M, Geraci F. Graph-Based Integration of Histone Modification Profiles. Mathematics. 2022; 10(11):1842. https://doi.org/10.3390/math10111842
Chicago/Turabian StyleBaccini, Federica, Monica Bianchini, and Filippo Geraci. 2022. "Graph-Based Integration of Histone Modification Profiles" Mathematics 10, no. 11: 1842. https://doi.org/10.3390/math10111842