
Transition to Multicellularity and Peto Paradox

{kind=link}
Abstract
:1. Introduction
1.1. Innovations
1.2. Organization of the Paper
2. Model
2.1. Chemical Kinetics with Random Stress Parameters
2.2. Extended Model with Anti-Stress Response Encoded by Genes
2.3. Assumptions
2.4. Stochastic Equations for Perturbations Induced by Stress Fluctuations
2.5. Model with Compartments
2.6. Associated Optimization Problems
2.7. Stochastic Equation for Feedback Evolution
3. Materials and Methods
4. Stochastic Stability
5. Main Results
6. Proof of Theorems 1 and 2
7. Transitions to Complex GRN
8. Morphogenesis Models and the Number of Coding Genes
8.1. Brief Overview of Models
8.2. Number of Genes
9. Multicellularity and Peto’s Paradox
9.1. How Multicellularity Supports Homeostasis
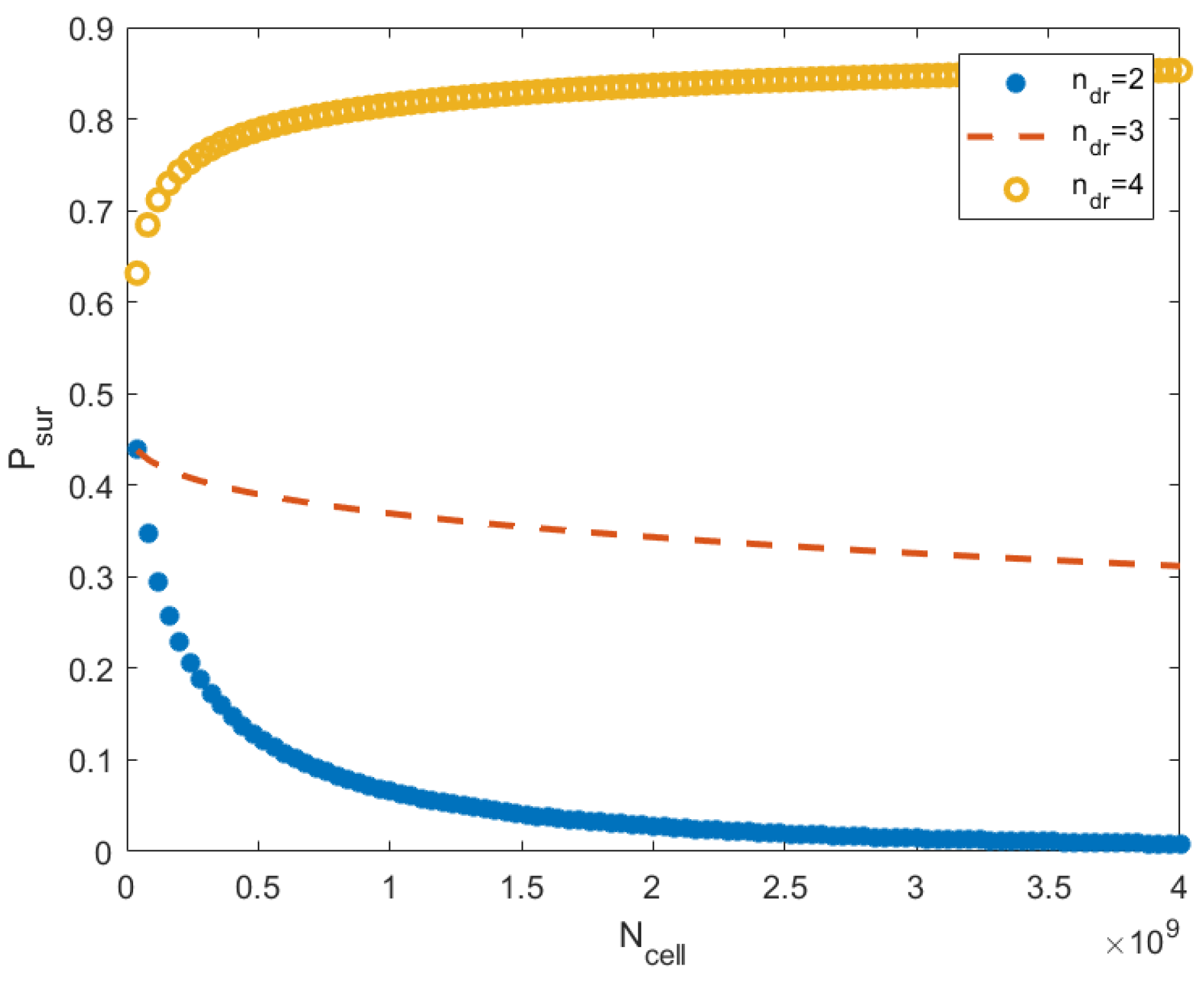
9.2. Peto’s Paradox: When a Large Mass Can Help
10. Concluding Remarks
Funding
Data Availability Statement
Conflicts of Interest
Appendix A
Appendix A.1. Estimates for Approximations by RBF
Appendix A.2. Estimates for RBFN
Appendix A.3. Encoding GRN
Appendix A.4. Estimate of Ventsel–Freidlin Distance
References
- Koonin, E.V. The Logic of Chance: The Nature and Origin of Biological Evolution; FT Press: NewYork, NY, USA, 2011. [Google Scholar]
- Iranzo, J.; Lobkovsky, A.E.; Wolf, Y.I.; Koonin, E.V. Virus-host arms race at the joint origin of multicellularity and programmed cell death. Cell Cycle 2014, 13, 3083–3088. [Google Scholar] [CrossRef]
- Koonin, E.V. Viruses and mobile elements as drivers of evolutionary transitions. Phil. Trans. R. Soc. B 2016, 371, 20150442. [Google Scholar] [CrossRef] [PubMed]
- Maynard Smith, J.; Schatzmary, E. The Major Transitions in Evolution; Oxford University Press: Oxford, UK, 1995. [Google Scholar]
- Fisher, R.A. The Genetical Theory of Natural Selection; Clarendon Press: Oxford, UK, 1930. [Google Scholar]
- Orr, H.A. Adaptation and cost of complexity. Evolution 2000, 54, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Orr, H.A. The genetic theory of adaptation: A brief history. Nat. Rev. Genet. 2005, 6, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Peto, R.; Roe, F.J.C.; Lee, P.N.; Levy, L.; Clack, J. Cancer and ageing in mice and men. Br. J. Cancer 1975, 32, 411–426. [Google Scholar] [CrossRef] [PubMed]
- Caulin, A.F.; Maley, C.C. Peto’s Paradox: Evolution’s prescription for cancer prevention. Trends Ecol. Evol. 2011, 26, 175–182. [Google Scholar] [CrossRef]
- Kobayashi, H.; Man, S. cquired multicellular-mediated resistance to alkylating agents in cancer. Proc. Natl. Acad. Sci. USA 1993, 90, 3294–3298. [Google Scholar] [CrossRef]
- Domazet-Loso, T.; Tautz, D. Phylostratigraphic tracking of cancer genes suggests a link to the emergence of multicellularity in metazoa. BMC Biol. 2010, 8, 66. [Google Scholar] [CrossRef]
- Dang, C. Links between metabolism and cancer. Genes Dev. 2012, 26, 877–890. [Google Scholar] [CrossRef]
- Vakulenko, S.; Grigoriev, D. Deep gene networks and response to stress. Mathematics 2021, 9, 3028. [Google Scholar] [CrossRef]
- Hansberg, W.; Aguirre, J.; Rís-Momberg, M.; Rangel, P.; Peraza, L.; Montes de Oca, Y.; Cano-Domínguez, N. Cell differentiation as a response to oxidative stress. Br. Mycol. Soc. Symp. Ser. 2008, 27, 235–257. [Google Scholar]
- Tower, J. Stress and stem cells. Wiley Interdiscip. Rev. Dev. Biol. 2012, 1, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Bornstein, S.; Steenblock, C.; Chrousos, G.P.; Schally, A.V.; Beuschlein, F.; Kline, G.; Krone, N.P.; Licinio, J.; Wong, M.L.; Ullmann, E.; et al. Stress-inducible-stem cells: A new view on endocrine, metabolic and mental disease? Mol. Psychiatry 2019, 24, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Greaves, R.B.; Dietmann, S.; Smith, A.; Stepney, S.; Halley, J.D. A conceptual and computational framework for modelling and understanding the non-equilibrium gene regulatory networks of mouse embryonic stem cells. PLoS Comput. Biol. 2017, 13, e1005713. [Google Scholar] [CrossRef]
- Jiang, P.; Ludwig, M.L.; Kreitman, M.; Reinitz, J. Natural variation of the expression pattern of the segmentation gene even-skipped in Drosophila melanogaster. Dev. Biol. 2015, 405, 173–181. [Google Scholar] [CrossRef]
- Mjolsness, E.; Sharp, D.H.; Reinitz, J. A connectionist model of development. J. Theor. Biol. 1991, 152, 429–453. [Google Scholar] [CrossRef]
- Valiant, L.G. Evolvability. J. ACM 2006, 120, 1–19. [Google Scholar]
- Vakulenko, S.; Grigoriev, D. Instability, complexity, and evolutionz. Zap. Nauchn. Sem. POMI 2008, 360, 31–69. [Google Scholar]
- Gromov, M.; Carbone, A. Mathematical slices of molecular biology. Gaz. MathéMaticiens 2001, 88A. [Google Scholar]
- Vakulenko, S. Complexity and Evolution of Dissipative Systems; de Gruyter: Berlin, Germany, 2014. [Google Scholar]
- Ventsel, D.A.; Freidlin, M.I. Random Perturbations of Dynamic Systems; Springer: New York, NY, USA, 1984. [Google Scholar]
- Reinitz, J.; Sharp, D.H. Mechanism of formation of eve stripes. Mech. Dev. 1995, 49, 133–158. [Google Scholar] [CrossRef]
- Valiant, L.G. Evolvability. J. ACM 2009, 56, 1–21. [Google Scholar] [CrossRef]
- Horsthemke, W.; Lefever, R. Noise-induced Transitions; Springer: Berlin/Heidelberg, Germany, 1984. [Google Scholar]
- Chen, B.; Abuassba, A.O. Compartmental Models with Application to Pharmacokinetics. Procedia Comput. Sci. 2021, 187, 60–70. [Google Scholar] [CrossRef]
- Gillespie, D.T. Exact Stochastic Simulation of Coupled Chemical Reactions. J. Phys. Chem. 1977, 81, 2340–2361. [Google Scholar] [CrossRef]
- Henry, D. Geometric Theory of Semilinear Parabolic Equations; Springer: Berlin/Heidelberg, Germany, 1981. [Google Scholar]
- Szendro, I.G.; Schenk, M.F.; Franke, J.; Krug, J.; de Visser, J.A.G. Quantitative analyses of empirical fitness landscapes. J. Stat. Mech. Theory Exp. 2013, 2013, P01005. [Google Scholar] [CrossRef]
- Jiang, P.; Reinitz, J.; Kreitman, M. The effect of mutational robustness on the evolvability of multicellular organisms and eukaryotic cells. J. Evol. Biol. 2023, 36, 906–924. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Liu, X.; Rong, Y.; Xu, Z. Almost optimal estimates for approximation and learning by radial basis function networks. Mach. Learn. 2014, 95, 147–164. [Google Scholar] [CrossRef]
- Turing, A. The chemical basis of morphogenesis. Phil. Trans. Roy. Soc. B 1952, 237, 37–72. [Google Scholar]
- Cooke, J.; Zeeman, E. A Clock and Wavefront model for control of the number of repeated structures during animal morphogenesis. J. Theor. Biol. 1976, 58, 455–476. [Google Scholar] [CrossRef]
- Meinhardt, H. Models of Biological Pattern Formation; Academic Press: London, UK, 1982; pp. 1–215. [Google Scholar]
- Baker, R.; Schnell, S.; Maini, P. A clock and wavefront mechanism for somite formation. Dev. Biol. 2006, 293, 116–126. [Google Scholar] [CrossRef]
- Kishan, K.; Rui, L.; Cui, F.; Yu, Q.; Haake, A.R. GNE: A deep learning framework for gene network inference by aggregating biological information. BMC Syst. Biol. 2019, 13, 38. [Google Scholar]
- Shen, Z.; Yang, H.; Zhang, S. Nonlinear Approximation via Compositions. Neural Netw. 2019, 119, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Surkova, S.; Spirov, A.V.; Gursky, V.V.; Janssens, H.; Kim, A.R.; Radulescu, O.; Vanario-Alonso, C.E.; Sharp, D.H.; Samsonova, M.; Reinitz, J.; et al. Canalization of gene expression in the Drosophila blastoderm by gap gene cross regulation. PLoS Biol. 2009, 7, e1000049. [Google Scholar]
- Murray, J.D. Mathematical Biology, 3rd ed.; Springer: Berlin/Heidelberg, Germany, 2002. [Google Scholar] [CrossRef]
- Crampin, E.J.; Gaffney, E.A.; Maini, P.K. Reaction and diffusion on growing domains: Scenarios for robust pattern formation. Bull. Math. Biol. 1999, 61, 1093–1120. [Google Scholar] [CrossRef] [PubMed]
- Krause, A.L.; Gaffney, E.A.; Maini, P.K.; Kika, V. Modern Perspectives on Near-Equilibrium Analysis of Turing Systems. Philos. Trans. A 2019, 379, 20200268. [Google Scholar] [CrossRef]
- Nicolis, G.; Prigogine, I. Self-Organization in Nonequilibrium Systems: From Dissipative Structures to Order through Fluctuations; Wiley and Sons: New York, NY, USA; London, UK; Sydney, Australia; Toronto, ON, Canada, 1977. [Google Scholar]
- Haken, H. Synergetics: An Introduction: Nonequilibrium Phase Transitions and Self-Organization in Physics, Chemistry, and Biology; Springer: Berlin/Heidelberg, Germany; New York, NY, USA, 1978. [Google Scholar]
- Vakulenko, S.; Volpert, V. Generalized travelling waves for perturbed monotone reaction-diffusion systems. Nonlinear Anal. 2001, 46, 757–776. [Google Scholar] [CrossRef]
- Brenner, S. Life’s code script. Nature 2012, 482, 461. [Google Scholar] [CrossRef]
- Maroto, M.; Bone, R.A.; Dale, J. Somitogenesis. Development 2012, 139, 2453–2456. [Google Scholar] [CrossRef]
- Resende, T.; Ferreira, M.; Teillet, M.A.; Tavares, A.T.; Andrade, R.P.; Palmeirim, I. Sonic hedgehog in temporal control of somite formation. Proc. Natl. Acad. Sci. USA 2010, 107, 12907–12912. [Google Scholar] [CrossRef]
- Pourquié, O. The chick embryo: A leading model for model in somitogenesis studies. Mech. Dev 2004, 121, 1069–1079. [Google Scholar] [CrossRef]
- Heltberg, M.L.; Krishna, S.; Jensen, M. On chaotic dynamics in transcription factors and the associated effects in differential gene regulation. Nat. Commun. 2019, 10, 71. [Google Scholar] [CrossRef]
- Wolpert, L.; Tickle, C.; Jessell, T. Principles of development; Oxford University Press: Cambridge, UK, 2002. [Google Scholar]
- Reinitz, J.; Vakulenko, S.; Sudakow, I.; Grigoriev, D. Robust morphogenesis by chaotic dynamics. Sci. Rep. 2023, 13, 7482. [Google Scholar] [CrossRef] [PubMed]
- Maroudas-Sacks, Y.; Keren, K. Mechanical Patterning in Animal Morphogenesis. Annu. Rev. Cell Dev. Biol. 2021, 37, 469–493. [Google Scholar] [CrossRef] [PubMed]
- Sudakow, I.; Vakulenko, S.; Grigoriev, D. Excitable media store and transfer complicated information via topological defect motion. Commun. Nonlinear Sci. Numer. Simul. 2023, 116, 106844. [Google Scholar] [CrossRef]
- Vakulenko, S.; Grigoriev, D. Complexity of gene circuits, Pfaffian functions and the morphogenesis problem. Compte Rendu Math. 2003, 337, 721–724. [Google Scholar] [CrossRef]
- Vakulenko, S.; Radulescu, O.; Reinitz, J. Size Regulation in the Segmentation of Drosophila: Interacting Interfaces between Localized Domains of Gene Expression Ensure Robust Spatial Patterning. Phys. Rev. Lett. 2009, 103, 168102–168106. [Google Scholar] [CrossRef]
- Iranzo, J.; Martincorena, I.; Koonin, E. Cancer-mutation network and the number and specificity of driver mutations. Proc. Natl. Acad. Sci. USA 2018, 115, E6010–E6019. [Google Scholar] [CrossRef]
- Longa, H.; Miller, S.F.; Strauss, C.; Zhao, C.; Cheng, L.; Ye, Z.; Griffin, K.; Te, R.; Lee, H.; Chen, C.; et al. Antibiotic treatment enhances the genome-wide mutation rate of target cells. Proc. Natl. Acad. Sci. USA 2016, 113. [Google Scholar] [CrossRef]
- Milholland, B.; Dong, X.; Zhang, L.; Hao, X.; Suh, Y.; Vijg, J. Differences between germline and somatic mutation rates in humans and mice. Nat Commun. 2017, 8, 15183. [Google Scholar] [CrossRef]
- Kimura, M. The Neutral Theory of Molecular Evolution; Cambridge University Press: Cambridge, UK, 1983. [Google Scholar]
- Keizer, J. Statistical Thermodynamics of Nonequilibrium Processes; Springer: New York, NY, USA, 1987. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vakulenko, S. Transition to Multicellularity and Peto Paradox. Mathematics 2023, 11, 5003. https://doi.org/10.3390/math11245003
Vakulenko S. Transition to Multicellularity and Peto Paradox. Mathematics. 2023; 11(24):5003. https://doi.org/10.3390/math11245003
Chicago/Turabian StyleVakulenko, Sergey. 2023. "Transition to Multicellularity and Peto Paradox" Mathematics 11, no. 24: 5003. https://doi.org/10.3390/math11245003
APA StyleVakulenko, S. (2023). Transition to Multicellularity and Peto Paradox. Mathematics, 11(24), 5003. https://doi.org/10.3390/math11245003