
Repositioning Drugs for Rare Diseases Based on Biological Features and Computational Approaches

 ,
,  ,
,  , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
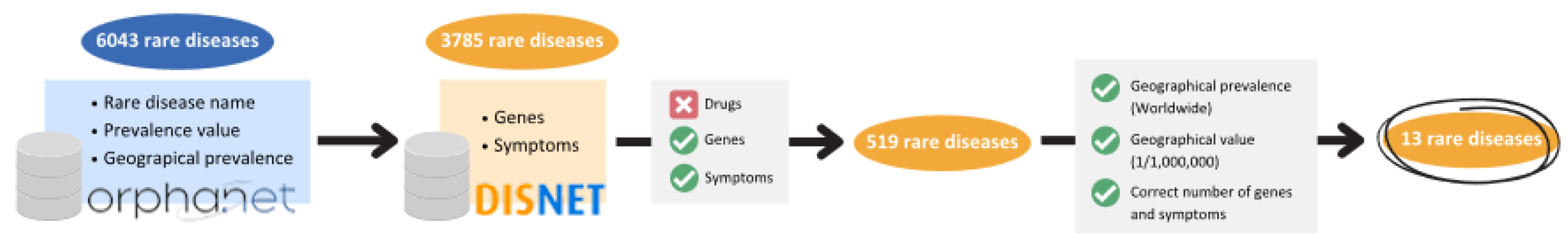
2.1.1. Data Acquisition and Integration
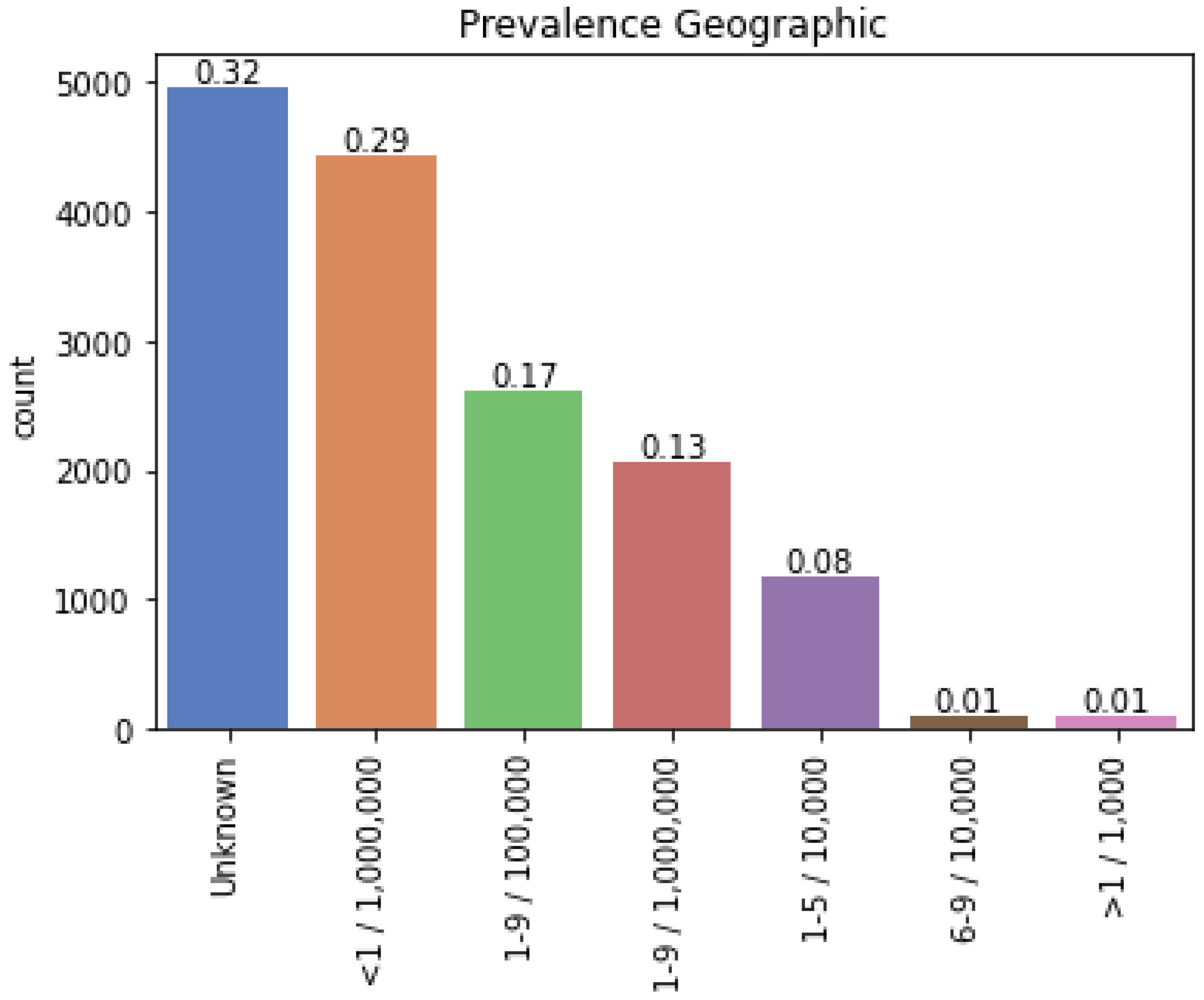
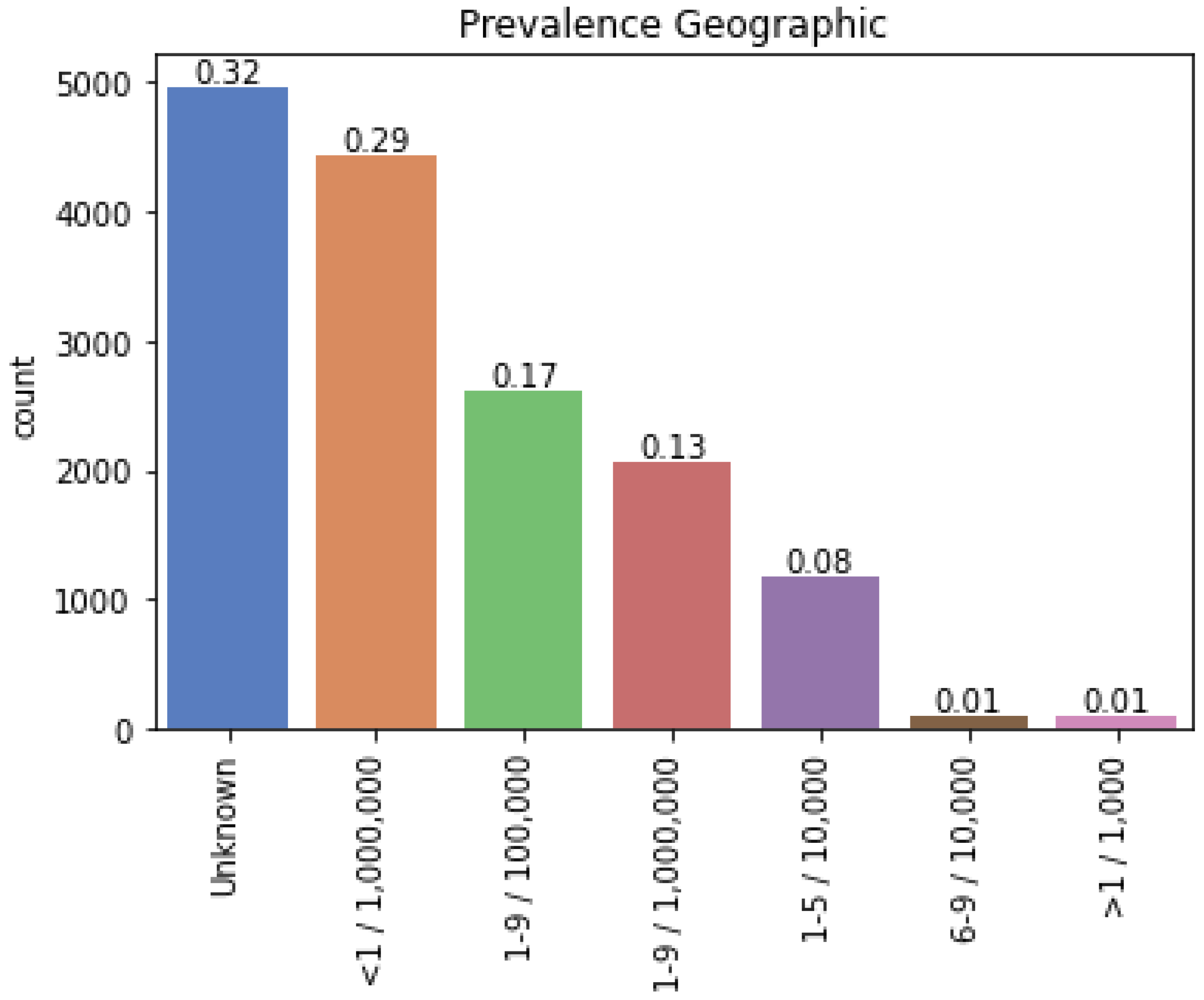
2.1.2. Selection of Rare Diseases
2.2. Methods
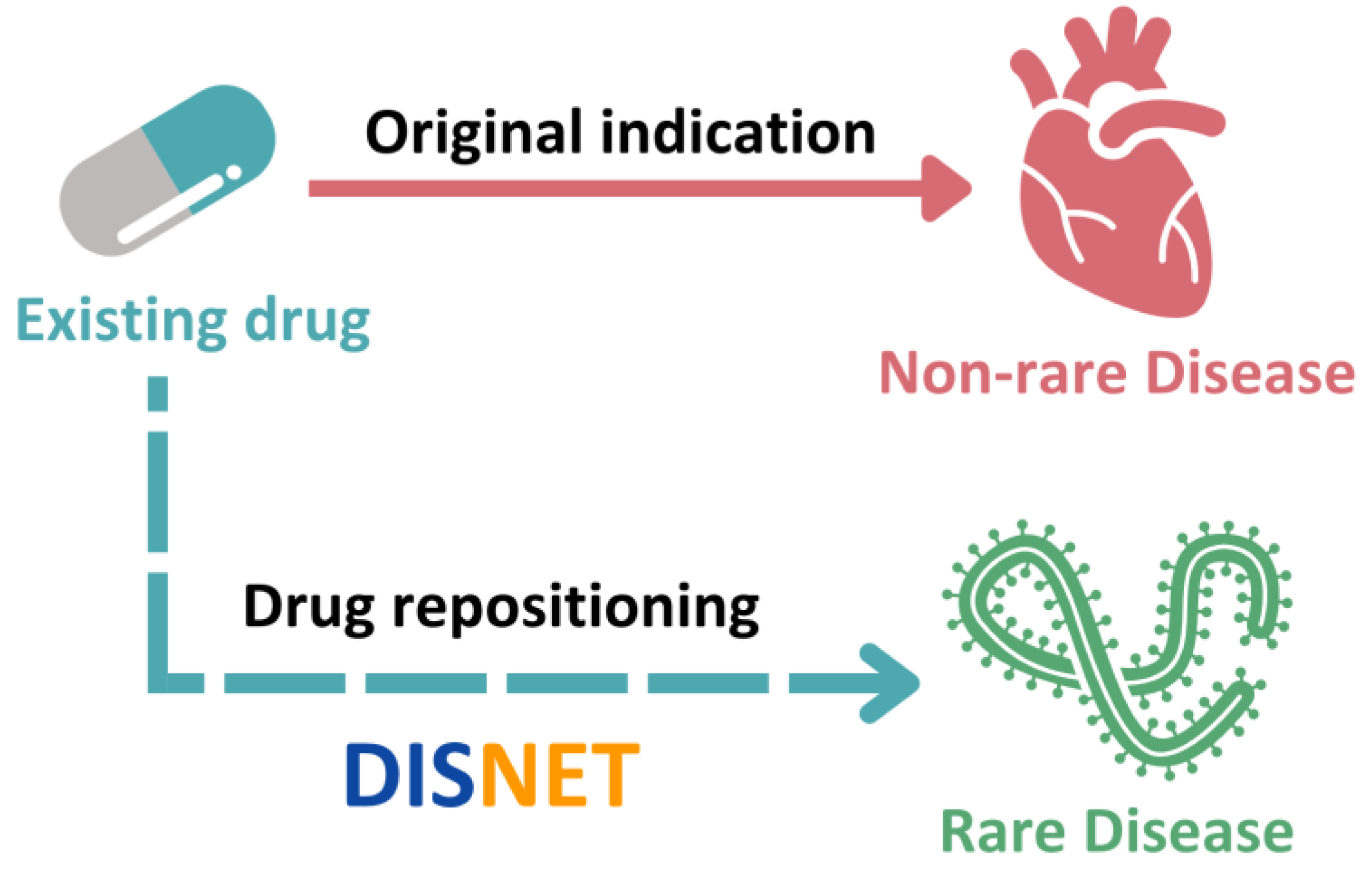
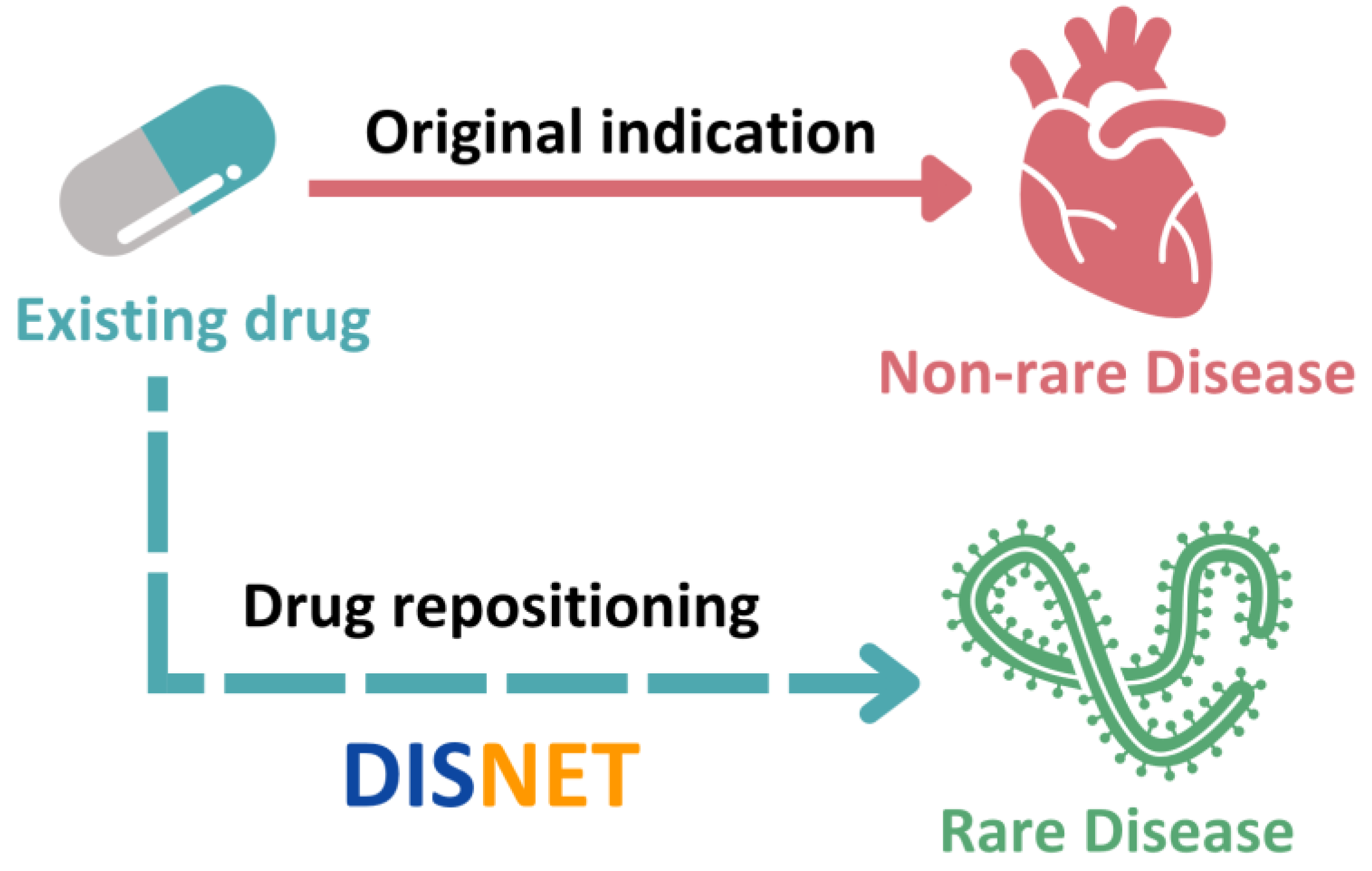
2.2.1. Drug Repositioning
- (1)
- Triples approach. The first strategy was to use triples for drug repositioning. A rare and a non-rare disease can be associated by a biological feature. The similarity between them through a specific feature is called triples. In this work, five kinds of triples were designed based upon five fundamental biological factors:
- Non-rare disease–Gene–Rare disease;
- Non-rare disease–Symptom–Rare disease;
- Non-rare disease–Protein interaction–Rare disease;
- Non-rare disease–Pathway–Rare disease;
- Non-rare disease–Variant–Rare disease.
- (2)
- Triples with Associated Target Approach. In this case, the triples are composed by the rare and non-rare diseases, and one biomedical feature. The difference with respect the previous method was that we forced the two diseases to share the gene that encoded the protein target of the non-rare-disease-associated drug. Then, the drug detection procedure was carried out as described in the above approach.
- (3)
- Direct Approach. An encoding gene for a drug target was associated with the rare disease. As a result, the drug can be used to treat rare diseases because there is a direct relation between the disease and the drug.
- (4)
- Paths Approach. This method is based on creating 6 strategies, named as paths, following the diseases’ biological characteristics and associated drugs. The drugs intersecting in these 6 paths (excluding those returning empty sets) are considered as the final list of this fourth computational approach.
- Rare disease–symptom–drug: we obtained the drugs which are indicated for the symptoms of the rare disease.
- Rare disease–symptom–disease–drug: we selected the drugs associated with the diseases that shared symptoms with the rare disease.
- Rare disease–symptom–gene–target–drug: given a rare disease and its symptoms, those non-rare diseases that present the same symptoms were searched. From these symptomatically similar diseases, we extracted their genes, targets and related drugs.
- Rare disease–gene–disease–drug: we obtained the drugs used for the diseases that shared genes with the rare disease.
- Rare disease–gene–protein–target–drug: in order to obtain the potential drugs, we obtained the genes, proteins and targets associated with these rare diseases.
- Rare disease–gene–protein–protein interaction–target–drug: from all of the rare diseases, we extracted the genes, proteins and the powerful protein–protein interactions data. After having obtained this information, we collected the associated target from which the corresponding drug.

2.2.2. Validation in Scientific Literature and Clinical Trials
- The names of the drugs and diseases were searched on Pubmed, Google, and clinicaltrials.gov.
- We checked the presence of results that related to both concepts.
- If positive results were obtained, all were selected. If the number was very high, the first 10 were selected.
- The chosen information was read.
- A conclusion was drawn for each relationship: whether the drug treats the studied disease or, on the contrary, causes it.
2.2.3. Drug Classification
- ATC 1st level: Anatomical or pharmacological groups.
- ATC 2nd level: Pharmacological or therapeutic subgroup.
- ATC 3rd and 4th levels: Chemical, pharmacological, or therapeutic subgroup.
- ATC 5th level: Chemical substance.
2.2.4. Phenotypical Similarity
2.2.5. Disease–Gene Associations (GDAs)
3. Results
3.1. Drug Repositioning
3.2. Validation in Clinical Trials
- Dexamethasone. It is used for the treatment of CMT disease [25].
- Amphetamine. Applying stimulant drugs such as this can help brain communication occur when the patient has this disease [29].
- Cisplatin together with doxorubicin produces the disease in combination with intrathecal cytosine arabinoside or methotrexate, which is another of the drugs that has been found for repositioning [32].
- Cytarabine. Another of the drugs obtained, cytarabine, is related to this line [33].
- Cocaine. A clinical case was collected where a woman developed this disease after the abusive consumption of cocaine [34].
- Deferoxamine. It is a risk factor for this disease when used for iron chelation [35].
- Furosemide. It produced locked-in syndrome in a case study when furosemide was being used to treat anasarca disease [36].
- Methamphetamine. The abuse of this substance can lead to neurological diseases that trigger locked-in syndrome [37].
- Methylprednisolone. It has been suggested to treat paraneoplastic symptoms but to a limited extent, because it is highly toxic [30].
- Alcohol. In accordance with the literature recommendation, patients with this diagnosis should avoid taking it [38].
- Silver sulfadiazine. A skin treatment to be applied on erosion areas affected by this pathology [39].
- Carbamazepine. It is stated that it should be avoided in conjunction with other sodium channel blockers because it can increase seizures despite being an anticonvulsant drug [43].
- Copper. The CNL6 gene participates in this disease. When the CNL6 gene is impaired, it produces the accumulation of biometals such as copper and this leads to the pathogenesis of the CNL6 disease. More research is needed on the function of the CNL6 gene [44]. Therefore, copper would not be an effective treatment against this disease.
- Dexamethasone. Its efficacy has not been proven 100%, more studies are needed to see if corticosteroids could influence the progression of the disease of the CNL3 gene by decreasing it [45]. What we hypothesize is that this anti-inflammatory treatment with corticosteroids may be beneficial in ameliorating some of the symptoms of juvenile CLN3 disease [45]. It also serves to alleviate the symptoms of another treatment that has been tried in this disease [46].
- Gentamicin sulfate. Literature evidence has been found for gentamicin. It can read premature termination codons (PTCs) and partially restore protein expression or function. PTC mutations are present in the CLN2 type of disease, and gentamicin can carry out this restoration [45].
- Methionine. It increases vacuolar acidification, which elevates the useful life of the vacuole. This mechanism affects a metabolic pathway in yeast proven for this disease [47].
- Valproic acid. It is effective in controlling seizures in this disease [43].
- Tamoxifen. It increases the production of cathepsin D, which helps to prevent this disease from occurring since it has been shown that many neurodegenerative diseases arise when there are low levels of cathepsin D or it is inactive, causing failures in one of the genes that produces this disease [48].
- Carvedilol. It is associated with this disease through congenital myotonia. It is used for heart problems, some of them produced by congenital myotonia [49].
- Albendazole and fluconazole. Both drugs cause liver failure and can, therefore, trigger liver failure [54].
- Ceftriaxone. It causes neonatal liver pathologies such as the one studied [57].
- Ciprofloxacin, sulfamethoxazole, and doxycycline. They are used to treat infections associated with haemochromatosis [58].
- Aspirin. It is used as a prophylactic treatment to prevent thromboembolism in patients with this disease, but its efficacy has not been fully demonstrated, and more studies are needed [62].
- Cyclosporine. It is used to perform immunosuppression that could be useful to perform heart transplants in patients with Emery–Dreifuss Muscular Dystrophy (EDMD) [59].
- Dantrolene. Anesthetic that was previously used for muscular dystrophies and may pose a risk to some patients [65].
- Enalapril, losartan, and enalaprilat. Both drugs are ACE inhibitors that are the first line of treatment for chronic heart failure although a substitute combination is being sought in this study [61]. In another article, this treatment is found to be used for heart failure [66]. They also help the blood vessels open wider and the heart can pump blood with less pressure [67].
- Isoproterenol. Atrial arrhythmias are a major problem in EDMD disease. Isoproterenol is used to prevent blockages in the heart or cardiac arrest, in a similar manner to epinephrine. In this study, it is administered to the patient at high levels but without causing arrhythmias [68].
- Metoprolol. It is indicated to control the heart rate and prevent further arrhythmias [68].
- Sirolimus. Rapamycin is a synonym, and it is used to prevent the progression of cardiomyopathies in mice. With it, a metabolic remodelling has been reached, which could be giving rise to a cardioprotective mechanism that slows the progression of EDMD and improves its prognosis [70].
- Valproic acid. It is used to treat the epileptic seizures that occur in this disease and other muscular dystrophies [63].
- Estradiol. It promotes mast cell release by causing increased histamine liberation resulting in hives/chronic urticaria (seen in women taking contraceptives or hormone replacement therapy) [74].
3.3. Drug Classification
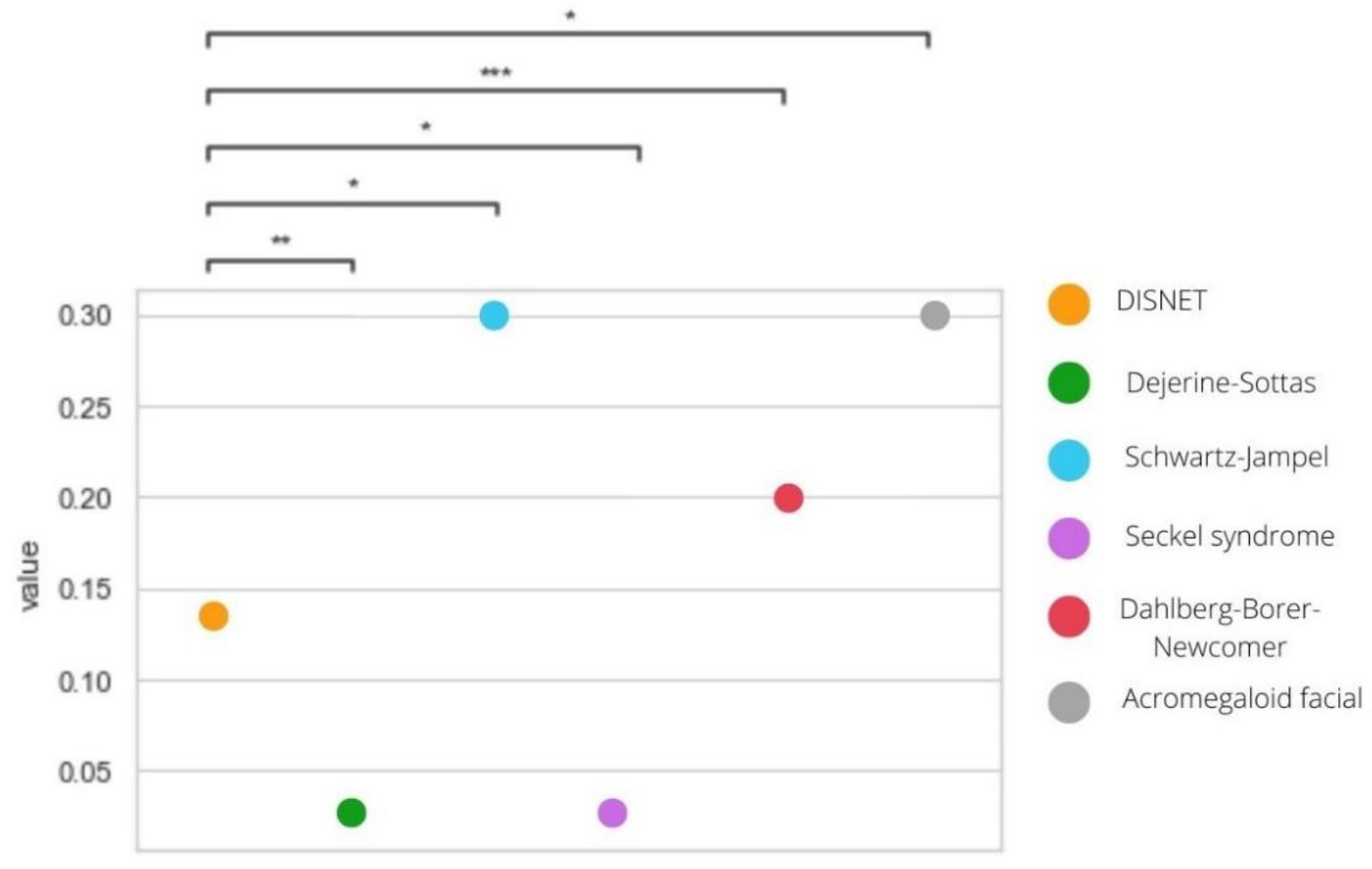
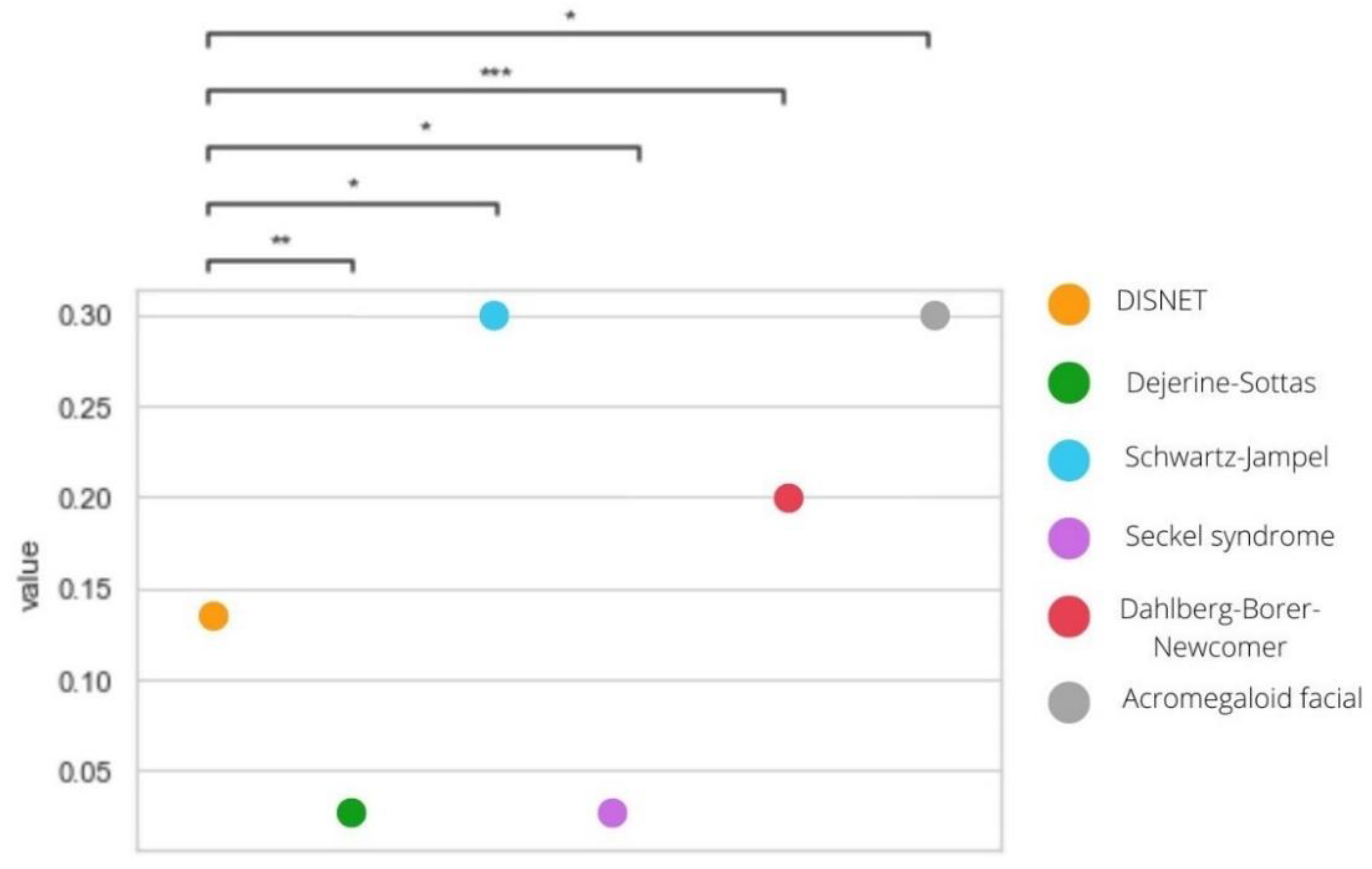
3.4. Phenotypical Similarity
3.5. Disease–Gene Associations (GDAs)
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Roessler, H.I.; Knoers, N.V.; van Haelst, M.M.; van Haaften, G. Drug Repurposing for Rare Diseases. Trends Pharmacol. Sci. 2021, 42, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Stolk, P.; Willemen, M.J.C.; Leufkens, H.G.M. Rare essentials drugs for rare diseases as essential medicines. Bull. World Health Organ. 2006, 84, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Tumiene, B.; Kristoffersson, U.; Hedley, V.; Kääriäinen, H. Rare diseases: Past achievements and future prospects. J. Commun. Genet. 2021, 12, 205–206. [Google Scholar] [CrossRef] [PubMed]
- Nguengang Wakap, S.; Lambert, D.M.; Olry, A.; Rodwell, C.; Gueydan, C.; Lanneau, V.; Murphy, D.; Le Cam, Y.; Rath, A. Estimating cumulative point prevalence of rare diseases: Analysis of the Orphanet database. Eur. J. Hum. Genet. 2020, 28, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Delavan, B.; Roberts, R.; Huang, R.; Bao, W.; Tong, W.; Liu, Z. Computational drug repositioning for rare diseases in the era of precision medicine. Drug Discov. Today 2018, 23, 382–394. [Google Scholar] [CrossRef] [PubMed]
- Sardana, D.; Zhu, C.; Zhang, M.; Gudivada, R.C.; Yang, L.; Jegga, A. Drug repositioning for orphan diseases. Briefings Bioinform. 2011, 12, 346–356. [Google Scholar] [CrossRef]
- Joppi, R.; Bertele’, V.; Garattini, S. Orphan drugs, orphan diseases. The first decade of orphan drug legislation in the EU. Eur. J. Clin. Pharmacol. 2013, 69, 1009–1024. [Google Scholar] [CrossRef]
- DiMasi, J.A.; Grabowski, H.G.; Hansen, R.W. Innovation in the pharmaceutical industry: New estimates of R&D costs. J. Health Econ. 2016, 47, 20–33. [Google Scholar] [CrossRef]
- Collins, F. An audience with...Francis Collins. Interviewed by Asher Mullard. Nat. Rev. Drug Discov. 2010, 10, 14. [Google Scholar] [CrossRef]
- Ko, Y. Computational Drug Repositioning: Current Progress and Challenges. Appl. Sci. 2020, 10, 5076. [Google Scholar] [CrossRef]
- Nosengo, N. Can you teach old drugs new tricks? Nature 2016, 534, 314–316. [Google Scholar] [CrossRef] [PubMed]
- Shahreza, M.L.; Ghadiri, N.; Mousavi, S.R.; Varshosaz, J.; Green, J. A review of network-based approaches to drug repositioning. Briefings Bioinform. 2018, 19, 878–892. [Google Scholar] [CrossRef]
- Santamaría, L.P.; Carro, E.U.; Uzquiano, M.D.; Ruiz, E.M.; Gallardo, Y.P.; Rodríguez-González, A. A data-driven methodology towards evaluating the potential of drug repurposing hypotheses. Comput. Struct. Biotechnol. J. 2021, 19, 4559–4573. [Google Scholar] [CrossRef] [PubMed]
- Turanli, B.; Altay, O.; Borén, J.; Turkez, H.; Nielsen, J.; Uhlen, M.; Arga, K.Y.; Mardinoglu, A. Systems biology based drug repositioning for development of cancer therapy. Semin. Cancer Biol. 2021, 68, 47–58. [Google Scholar] [CrossRef]
- Shim, J.S.; Liu, J.O. Recent Advances in Drug Repositioning for the Discovery of New Anticancer Drugs. Int. J. Biol. Sci. 2014, 10, 654–663. [Google Scholar] [CrossRef] [PubMed]
- Fiscon, G.; Conte, F.; Farina, L.; Paci, P. SAveRUNNER: A network-based algorithm for drug repurposing and its application to COVID-19. PLoS Comput. Biol. 2021, 17, e1008686. [Google Scholar] [CrossRef]
- Gysi, D.M.; Do Valle, Í.; Zitnik, M.; Ameli, A.; Gan, X.; Varol, O.; Ghiassian, S.D.; Patten, J.J.; Davey, R.A.; Loscalzo, J.; et al. Network medicine framework for identifying drug-repurposing opportunities for COVID-19. Proc. Natl. Acad. Sci. USA 2021, 118, e2025581118. [Google Scholar] [CrossRef]
- Rameshrad, M.; Ghafoori, M.; Mohammadpour, A.H.; Nayeri, M.J.D.; Hosseinzadeh, H. A comprehensive review on drug repositioning against coronavirus disease 2019 (COVID19). Naunyn-Schmiedebergs Arch. fur Exp. Pathol. und Pharmakol. 2020, 393, 1137–1152. [Google Scholar] [CrossRef]
- Santamaría, L.P.; Uzquiano, M.D.; Carro, E.U.; Ortiz-Roldán, N.; Gallardo, Y.P.; Rodríguez-González, A. Integrating heterogeneous data to facilitate COVID-19 drug repurposing. Drug Discov. Today 2021, 27, 558–566. [Google Scholar] [CrossRef]
- Shahreza, M.L.; Ghadiri, N.; Green, J.R. A computational drug repositioning method applied to rare diseases: Adrenocortical carcinoma. Sci. Rep. 2020, 10, 8846. [Google Scholar] [CrossRef]
- Corbett, A.; Pickett, J.; Burns, A.; Corcoran, J.; Dunnett, S.; Edison, P.; Hagan, J.J.; Holmes, C.; Jones, E.; Katona, C.; et al. Drug repositioning for Alzheimer’s disease. Nat. Rev. Drug Discov. 2012, 11, 833–846. [Google Scholar] [CrossRef] [PubMed]
- Ballard, C.; Aarsland, D.; Cummings, J.; O’Brien, J.; Mills, R.; Molinuevo, J.L.; Fladby, T.; Williams, G.; Doherty, P.; Corbett, A.; et al. Drug repositioning and repurposing for Alzheimer disease. Nat. Rev. Neurol. 2020, 16, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Fiscon, G.; Conte, F.; Amadio, S.; Volonté, C.; Paci, P. Drug Repurposing: A Network-based Approach to Amyotrophic Lateral Sclerosis. Neurotherapeutics 2021, 18, 1678–1691. [Google Scholar] [CrossRef] [PubMed]
- Lagunes-García, G.; Rodríguez-González, A.; Prieto-Santamaría, L.; Del Valle, E.P.G.; Zanin, M.; Menasalvas-Ruiz, E. DISNET: A framework for extracting phenotypic disease information from public sources. PeerJ 2020, 8, e8580. [Google Scholar] [CrossRef] [PubMed]
- Kikukawa, Y.; Hata, H.; Ueda, M.; Yamashita, T.; Nasu, S.; Ide, K.; Ueno, S.; Ando, Y.; Mitsuya, H.; Okuno, Y. Successful Treatment of Amyloid Light-chain Amyloidosis in a Charcot-Marie-Tooth Disease Patient with Lenalidomide, Cyclophosphamide, and Dexamethasone. Intern. Med. 2016, 55, 2707–2712. [Google Scholar] [CrossRef]
- Nakamura, T.; Hashiguchi, A.; Suzuki, S.; Uozumi, K.; Tokunaga, S.; Takashima, H. Vincristine exacerbates asymptomatic Charcot–Marie–Tooth disease with a novel EGR2 mutation. Neurogenetics 2012, 13, 77–82. [Google Scholar] [CrossRef]
- Zhao, L.-J.; Xue, H.-X.; Fu, W.-Y.; Cui, H.-D.; Yang, L.-L.; Zhang, N. High-dose thalidomide increases the risk of peripheral neuropathy in the treatment of ankylosing spondylitis. Neural Regen. Res. 2015, 10, 814–818. [Google Scholar] [CrossRef]
- Bastuji-Garin, S.; Ochonisky, S.; Bouche, P.; Gherardi, R.K.; Duguet, C.; Djerradine, Z.; Poli, F.; Revuz, J. Incidence and Risk Factors for Thalidomide Neuropathy: A Prospective Study of 135 Dermatologic Patients. J. Investig. Dermatol. 2002, 119, 1020–1026. [Google Scholar] [CrossRef]
- De Massari, D.; Ruf, C.A.; Furdea, A.; Matuz, T.; Van Der Heiden, L.; Halder, S.; Silvoni, S.; Birbaumer, N. Brain communication in the locked-in state. Brain 2013, 136, 1989–2000. [Google Scholar] [CrossRef]
- Pistoia, F.; Conson, M.; Sarà, M. Opsoclonus-Myoclonus Syndrome in Patients with Locked-in Syndrome: A Therapeutic Porthole with Gabapentin. Mayo Clin. Proc. 2010, 85, 527–531. [Google Scholar] [CrossRef] [Green Version]
- Pistoia, F.; Sacco, S.; Sarà, M.; Franceschini, M.; Carolei, A. Intrathecal Baclofen: Effects on Spasticity, Pain, and Consciousness in Disorders of Consciousness and Locked-in Syndrome. Curr. Pain Headache Rep. 2015, 19, 466. [Google Scholar] [CrossRef] [PubMed]
- Kleinschmidt-DeMasters, B.K.; Yeh, M. ‘Locked-in syndrome’ after intrathecal cytosine arabinoside therapy for malignant immunoblastic lymphoma. Cancer 1992, 70, 2504–2507. [Google Scholar] [CrossRef]
- Doan, T.; Lacayo, N.; Fisher, P.; Liao, Y.J. Dorsolateral Midbrain MRI Abnormalities and Ocular Motor Deficits Following Cytarabine-Based Chemotherapy for Acute Myelogenous Leukemia. J. Neuro-Ophthalmol. 2011, 31, 52–53. [Google Scholar] [CrossRef] [PubMed]
- Ali, O.; Bueno, M.G.; Duong-Pham, T.; Gunawardhana, N.; Tran, D.H.; Chow, R.D.; Verceles, A.C. Cocaine as a rare cause of locked-in syndrome: A case report. J. Med. Case Rep. 2019, 13, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Maffini, F.; Cocorocchio, E.; Pruneri, G.; Bonomo, G.; Peccatori, F.; Chiapparini, L.; Di Vincenzo, S.; Martinelli, G.; Viale, G. Locked-in syndrome after basilary artery thrombosis by mucormycosis masquerading as meningoencephalitis in a lymphoma patient. Ecancermedicalscience 2013, 7. [Google Scholar] [CrossRef]
- Ormonde, C.; Cabral, R.; Serpa, S. Osmotic Demyelination Syndrome in a Patient with Hypokalemia but No Hyponatremia. Case Rep. Nephrol. 2020, 2020, 3618763–3618764. [Google Scholar] [CrossRef]
- Baker, S.; Oster, J.; Liu, A. Central pontine myelinolysis in a patient with methamphetamine abuse. Brain Behav. Immun. Health 2020, 10, 100166. [Google Scholar] [CrossRef]
- Alto, A.W.; Clarcq, L. Cutaneous and systemic manifestations of mastocytosis. Am. Fam. Phys. 1999, 59, 3047. [Google Scholar]
- Rayinda, T.; Oktarina, D.A.M.; Danarti, R. Diffuse cutaneous mastocytosis masquerading as linear IgA bullous dermatosis of childhood. Dermatol. Rep. 2021, 13. [Google Scholar] [CrossRef]
- Carter, M.C.; Uzzaman, A.; Scott, L.M.; Metcalfe, D.D.; Quezado, Z. Pediatric Mastocytosis: Routine Anesthetic Management for a Complex Disease. Anesthesia Analg. 2008, 107, 422–427. [Google Scholar] [CrossRef]
- Czarny, J.; Lange, M.; Ługowska-Umer, H.; Nowicki, R.J. Cutaneous mastocytosis treatment: Strategies, limitations and perspectives. Adv. Dermatol. Allergol. 2018, 35, 541–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandru, F.; Petca, R.-C.; Costescu, M.; Dumitrașcu, M.; Popa, A.; Petca, A.; Miulescu, R.-G. Cutaneous Mastocytosis in Childhood—Update from the Literature. J. Clin. Med. 2021, 10, 1474. [Google Scholar] [CrossRef] [PubMed]
- Babacan-Yildiz, G.; Hanagasi, H.; Gurvit, H.; Sirin, G.; Solakoglu, S.; Kucuk, O.S. A Rare Dementing Disease: Adult Neuronal Ceroid Lipofuscinoses. J. Neuropsychiatr. Clin Neurosci. 2012, 24, 493–498. [Google Scholar] [CrossRef]
- Mukherjee, A.B.; Appu, A.P.; Sadhukhan, T.; Casey, S.; Mondal, A.; Zhang, Z.; Bagh, M.B. Emerging new roles of the lysosome and neuronal ceroid lipofuscinoses. Mol. Neurodegener. 2019, 14, 4. [Google Scholar] [CrossRef] [PubMed]
- Kohan, R.; Cismondi, I.A.; Oller-Ramirez, A.M.; Guelbert, N.; Anzolini, V.T.; Alonso, G.; Mole, S.; De Kremer, R.D.; De Halac, I.N. Therapeutic Approaches to the Challenge of Neuronal Ceroid Lipofuscinoses. Curr. Pharm. Biotechnol. 2011, 12, 867–883. [Google Scholar] [CrossRef]
- Schaefers, J.; van der Giessen, L.J.; Klees, C.; Jacobs, E.H.; Sieverdink, S.; Dremmen, M.H.G.; Spoor, J.K.H.; van der Ploeg, A.T.; Hout, J.M.P.V.D.; Huidekoper, H.H. Presymptomatic treatment of classic late-infantile neuronal ceroid lipofuscinosis with cerliponase alfa. Orphanet J. Rare Dis. 2021, 16, 221. [Google Scholar] [CrossRef]
- Faller, K.M.; Gutierrez-Quintana, R.; Mohammed, A.; Rahim, A.A.; Tuxworth, R.I.; Wager, K.; Bond, M. The neuronal ceroid lipofuscinoses: Opportunities from model systems. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2015, 1852, 2267–2278. [Google Scholar] [CrossRef]
- Mijanovic, O.; Petushkova, A.; Brankovic, A.; Turk, B.; Solovieva, A.; Nikitkina, A.; Bolevich, S.; Timashev, P.; Parodi, A.; Zamyatnin, A. Cathepsin D—Managing the Delicate Balance. Pharmaceutics 2021, 13, 837. [Google Scholar] [CrossRef]
- Muarrak, H.S.; Muarrak, D.P.; Rojas, M.B.; Condales, E.S.; Quintana, Y.R. Effects of the use of carvedilol in dilated cardiomyopathy in pediatric age. Mediciego 2015, 21, 106–116. [Google Scholar]
- Alderton, G.K.; Joenje, H.; Varon, R.; Børglum, A.D.; Jeggo, P.A.; O’Driscoll, M. Seckel syndrome exhibits cellular features demonstrating defects in the ATR-signalling pathway. Hum. Mol. Genet. 2004, 13, 3127–3138. [Google Scholar] [CrossRef]
- Nishijima, H.; Nishitani, H.; Saito, N.; Nishimoto, T. Caffeine mimics adenine and 2′-deoxyadenosine, both of which inhibit the guanine-nucleotide exchange activity of RCC1 and the kinase activity of ATR. Genes Cells Devoted Mol. Cell. Mech. 2003, 8, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Penera, T.; Gugig, R.; Davis, J.; McDiarmid, S.; Vargas, J.; Rosenthal, P.; Berquist, W.; Heyman, M.B.; Ament, M.E. Outcome of acetaminophen overdose in pediatric patients and factors contributing to hepatotoxicity. J. Pediatr. 1997, 130, 300–304. [Google Scholar] [CrossRef]
- Van Swelm, R.P.L.; Laarakkers, C.M.M.; Blous, L.; Peters, J.G.P.; Davidson, E.N.B.; Van Der Kraan, P.M.; Swinkels, D.W.; Masereeuw, R.; Russel, F. Acute Acetaminophen Intoxication Leads to Hepatic Iron Loading by Decreased Hepcidin Synthesis. Toxicol. Sci. 2012, 129, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Grama, A.; Aldea, C.O.; Burac, L.; Delean, D.; Bulata, B.; Sirbe, C.; Duca, E.; Boghitoiu, D.; Coroleuca, A.; Pop, T.L. Etiology and Outcome of Acute Liver Failure in Children—The Experience of a Single Tertiary Care Hospital from Romania. Children 2020, 7, 282. [Google Scholar] [CrossRef]
- Scotet, V.; Mérour, M.-C.; Mercier, A.-Y.; Chanu, B.; Le Faou, T.; Raguénes, O.; Le Gac, G.; Mura, C.; Nousbaum, J.-B.; Férec, C. Hereditary Hemochromatosis: Effect of Excessive Alcohol Consumption on Disease Expression in Patients Homozygous for the C282Y Mutation. Am. J. Epidemiol. 2003, 158, 129–134. [Google Scholar] [CrossRef]
- Adams, P.C.; Agnew, S. Alcoholism in hereditary hemochromatosis revisited: Prevalence and clinical consequences among homozygous siblings. Hepatology 1996, 23, 724–727. [Google Scholar] [CrossRef]
- Sociedad Argentina de Pediatría. Consensus on hyperbilirubinemia of the first trimester of life. Arch. Argent. Pediatr. 2020, 118, S12–S49. [Google Scholar] [CrossRef]
- Khan, F.A.; Fisher, M.A.; Khakoo, R.A. Association of hemochromatosis with infectious diseases: Expanding spectrum. Int. J. Infect. Dis. 2007, 11, 482–487. [Google Scholar] [CrossRef]
- Blagova, O.; Nedostup, A.; Shumakov, D.; Poptsov, V.; Shestak, A.; Zaklyasminskaya, E. Dilated cardiomyopathy with severe arrhythmias in Emery-Dreifuss muscular dystrophy: From ablation to heart transplantation. J. Atr. Fibrillation 2016, 9, 1468. [Google Scholar] [CrossRef]
- Faiella, W.; Bessoudo, R. Cardiac manifestations in Emery–Dreifuss muscular dystrophy. Can. Med. Assoc. J. 2018, 190, E1414–E1417. [Google Scholar] [CrossRef]
- Weintraub, R.G.; Semsarian, C.; Macdonald, P. Dilated cardiomyopathy. Lancet 2017, 390, 400–414. [Google Scholar] [CrossRef]
- Boriani, G.; Gallina, M.; Merlini, L.; Bonne, G.; Toniolo, D.; Amati, S.; Biffi, M.; Martignani, C.; Frabetti, L.; Bonvicini, M.; et al. Clinical Relevance of Atrial Fibrillation/Flutter, Stroke, Pacemaker Implant, and Heart Failure in Emery-Dreifuss Muscular Dystrophy. Stroke 2003, 34, 901–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manta, P.; Terzis, G.; Papadimitriou, C.; Kontou, C.; Vassilopoulos, D. Emerin expression in tubular aggregates. Acta Neuropathol. 2004, 107, 546–552. [Google Scholar] [CrossRef] [PubMed]
- Nelson, I.; Stojkovic, T.; Allamand, V.; Leturcq, F.; Bécane, H.-M.; Babuty, D.; Toutain, A.; Béroud, C.; Richard, P.; Romero, N.B.; et al. Laminin α2 Deficiency-Related Muscular Dystrophy Mimicking Emery-Dreifuss and Collagen VI related Diseases. J. Neuromuscul. Dis. 2015, 2, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Lerman, J. Perioperative management of the paediatric patient with coexisting neuromuscular disease. Br. J. Anaesth. 2011, 107 (Suppl. 1), i79–i89. [Google Scholar] [CrossRef]
- Sanna, T.; Russo, A.D.; Toniolo, D.; Vytopil, M.; Pelargonio, G.; De Martino, G.; Ricci, E.; Silvestri, G.; Giglio, V.; Messano, L.; et al. Cardiac features of Emery-Dreifuss muscular dystrophy caused by lamin A/C gene mutations. Eur. Heart J. 2003, 24, 2227–2236. [Google Scholar] [CrossRef]
- Muscular Dystrophy News Home—Muscular Dystrophy News. Available online: https://musculardystrophynews.com/ (accessed on 7 July 2022).
- Butt, K.; Ambati, S. Atrial arrhythmias in Emery-Dreifuss muscular dystrophy: Approach to successful ablation. Heart Case Rep. 2020, 6, 318–321. [Google Scholar] [CrossRef]
- Bădilă, E.; Lungu, I.; Grumezescu, A.; Udriște, A.S. Diagnosis of Cardiac Abnormalities in Muscular Dystrophies. Medicina 2021, 57, 488. [Google Scholar] [CrossRef]
- Vignier, N.; Muchir, A. An Omics View of Emery–Dreifuss Muscular Dystrophy. J. Pers. Med. 2020, 10, 50. [Google Scholar] [CrossRef]
- Bielski, L.Y.; Orlandi, A.M.; Boquete, H.R. Inhibidores de tirosina cinasa y disfunción tiroidea. J. Pers. Med. 2016, 53, 96–105. [Google Scholar] [CrossRef]
- Paez, H.A.A.; Siabato, M.R. Reacciones cutáneas severas secundarias al uso de Lamotrigina. Acta Neurol. Colomb. 2014, 30, 128–133. [Google Scholar]
- Bota, R.G.; Ligasan, A.P.; Najdowski, T.G.; Novac, A. Acute Hypersensitivity Syndrome Caused by Valproic Acid: A Review of the Literature and a Case Report. Perm. J. 2011, 15, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Kasperska-Zajac, A.; Brzoza, Z.; Rogala, B. Sex hormones and urticaria. J. Dermatol. Sci. 2008, 52, 79–86. [Google Scholar] [CrossRef]
- Sam, A.; Tan, T.; Meeran, K. Insulin-mediated “pseudoacromegaly”. Hormones 2011, 10, 156–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, O.L.; Despaigne, M.S.; Palay, D.; Cascaret, A.R. Acromegalia: Diagnóstico y tratamiento. Medisan 2015, 19, 403–416. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CUIs | Disease Name | N. Genes | N. Symptoms |
|---|---|---|---|
| C0011195 | Dejerine–Sottas syndrome | 31 | 10 |
| C0023944 | Locked-In Syndrome | 1 | 17 |
| C0024054 | Lown–Ganong–Levine syndrome | 1 | 6 |
| C0024901 | Diffuse cutaneous mastocytosis | 1 | 237 |
| C0027877 | Congenital neuronal ceroid lipofuscinosis | 38 | 52 |
| C0036391 | Schwartz–Jampel syndrome | 23 | 5 |
| C0265202 | Seckel syndrome | 15 | 4 |
| C0268059 | Neonatal hemochromatosis | 1 | 43 |
| C0549463 | X-Linked Lymphoproliferative Disorder | 11 | 1 |
| C0751337 | X-Linked Emery–Dreifuss Muscular Dystrophy | 44 | 32 |
| C0869083 | Dahlberg–Borer–Newcomer syndrome | 12 | 2 |
| C1852146 | Vibratory urticaria | 1 | 11 |
| C0796280 | Acromegaloid facial appearance syndrome | 1 | 90 |
| Diseases | Approaches | |||||
|---|---|---|---|---|---|---|
| Triples | T. Target | Direct DR | Paths | All | TT.DDR.P * | |
| Dejerine–Sottas syndrome | 6 | 17 | 17 | 2 | 0 | 2 |
| Locked-In Syndrome | 46 | 0 | 0 | 80 | 44 | 80 |
| Lown–Ganong–Levine syndrome | 0 | 0 | 0 | 965 | 965 | 965 |
| Diffuse cutaneous mastocytosis | 7 | 0 | 0 | 4 | 2 | 4 |
| Congenital neuronal ceroid lipofuscinosis | 2 | 0 | 0 | 48 | 2 | 48 |
| Schwartz–Jampel syndrome | 533 | 0 | 30 | 15 | 10 | 14 |
| Seckel syndrome | 6 | 2 | 2 | 0 | 0 | 2 |
| Neonatal hemochromatosis | 2 | 0 | 0 | 91 | 0 | 91 |
| X-Linked Lymphoproliferative Disorder | 0 | 0 | 7 | 1 | 1 | 1 |
| X-Linked Emery–Dreifuss Muscular Dystrophy | 10 | 0 | 0 | 126 | 10 | 126 |
| Dahlberg–Borer–Newcomer syndrome | 1 | 8 | 0 | 0 | 0 | 8 |
| Vibratory urticaria | 2 | 0 | 0 | 0 | 2 | 0 |
| Acromegaloid facial appearance syndrome | 35 | 2 | 2 | 0 | 2 | 2 |
| Diseases | Computational Drugs | Drugs Clinical Trials | Drugs Effects | Drugs Toxic |
|---|---|---|---|---|
| Dejerine–Sottas syndrome | 2 | 2 | 1 | 1 |
| Locked-In Syndrome | 80 | 11 | 2 | 9 |
| Diffuse cutaneous mastocytosis | 4 | 4 | 3 | 1 |
| Congenital neuronal ceroid lipofuscinosis | 48 | 7 | 5 | 2 |
| Schwartz–Jampel syndrome | 14 | 1 | 1 | 0 |
| Seckel syndrome | 2 | 1 | 0 | 1 |
| Neonatal hemochromatosis | 91 | 8 | 3 | 5 |
| X-Linked Lymphoproliferative Disorder | 1 | 0 | 0 | 0 |
| X-Linked Emery–Dreifuss Muscular Dystrophy | 126 | 15 | 11 | 4 |
| Dahlberg–Borer–Newcomer syndrome | 8 | 8 | 0 | 8 |
| Vibratory urticaria | 2 | 2 | 0 | 2 |
| Acromegaloid facial appearance syndrome | 2 | 1 | 1 | 0 |
| Disease | PS. DISNET Diseases (Mean Jaccard) | PS. Rare—Non-Rare Diseases (Mean Jaccard) | p-Value |
|---|---|---|---|
| Dejerine–Sottas syndrome | 0.0466 | 0.0904 | 0.0112 |
| Schwartz–Jampel syndrome | 0.0500 | 0.8425 | 0.0000 |
| Seckel syndrome | 0.0607 | 0.0312 | 0.0173 |
| Dahlberg–Borer–Newcomer syndrome | 0.0396 | 0.1996 | 0.0000 |
| Acromegaloid facial appearance syndrome | 0.0471 | 0.0319 | 0.0000 |
| Disease | Drug | Gen Symbol | DSI | DPI | GDA |
|---|---|---|---|---|---|
| Dejerine–Sottas syndrome | Dexamethasone | NR0B1 | 0.512 | 0.621 | 0.02 |
| Thalidomide | PTGS2 | 0.338 | 0.897 | 0.02 | |
| Thalidomide | TNF | 0.263 | 0.966 | 0.02 | |
| Schwartz–Jampel syndrome | Carvedilol | VEGFA | 0.298 | 0.897 | 0.3 |
| Seckel syndrome | Caffeine | ATM | 0.401 | 0.862 | 0.02 |
| Dahlberg–Borer–Newcomer syndrome | Sorafenib | BRAF | 0.352 | 0.793 | 0.2 |
| Fostamatinib | |||||
| Vemurafenib | |||||
| Encorafenib | |||||
| Regorafenib | |||||
| Dabrafenib mesylate | |||||
| Sorafenib tosylate | |||||
| Dabrafenib | |||||
| Fostamatinib | ICK | 0.602 | 0.621 | 0.2 | |
| Acromegaloid facial appearance syndrome | Glyburide | ABCC9 | 0.59 | 0.517 | 0.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Otero-Carrasco, B.; Prieto Santamaría, L.; Ugarte Carro, E.; Caraça-Valente Hernández, J.P.; Rodríguez-González, A. Repositioning Drugs for Rare Diseases Based on Biological Features and Computational Approaches. Healthcare 2022, 10, 1784. https://doi.org/10.3390/healthcare10091784
Otero-Carrasco B, Prieto Santamaría L, Ugarte Carro E, Caraça-Valente Hernández JP, Rodríguez-González A. Repositioning Drugs for Rare Diseases Based on Biological Features and Computational Approaches. Healthcare. 2022; 10(9):1784. https://doi.org/10.3390/healthcare10091784
Chicago/Turabian StyleOtero-Carrasco, Belén, Lucía Prieto Santamaría, Esther Ugarte Carro, Juan Pedro Caraça-Valente Hernández, and Alejandro Rodríguez-González. 2022. "Repositioning Drugs for Rare Diseases Based on Biological Features and Computational Approaches" Healthcare 10, no. 9: 1784. https://doi.org/10.3390/healthcare10091784
APA StyleOtero-Carrasco, B., Prieto Santamaría, L., Ugarte Carro, E., Caraça-Valente Hernández, J. P., & Rodríguez-González, A. (2022). Repositioning Drugs for Rare Diseases Based on Biological Features and Computational Approaches. Healthcare, 10(9), 1784. https://doi.org/10.3390/healthcare10091784