
Methamphetamine Dysregulates Macrophage Functions and Autophagy to Mediate HIV Neuropathogenesis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. General Culture Methods, HIV Infection, and Key Reagents
2.2. Measurement of HIV Infection
2.3. Mass Spectrometry Analysis
2.4. Phagocytosis Experiments
2.5. Reactive Oxygen Species Measurement
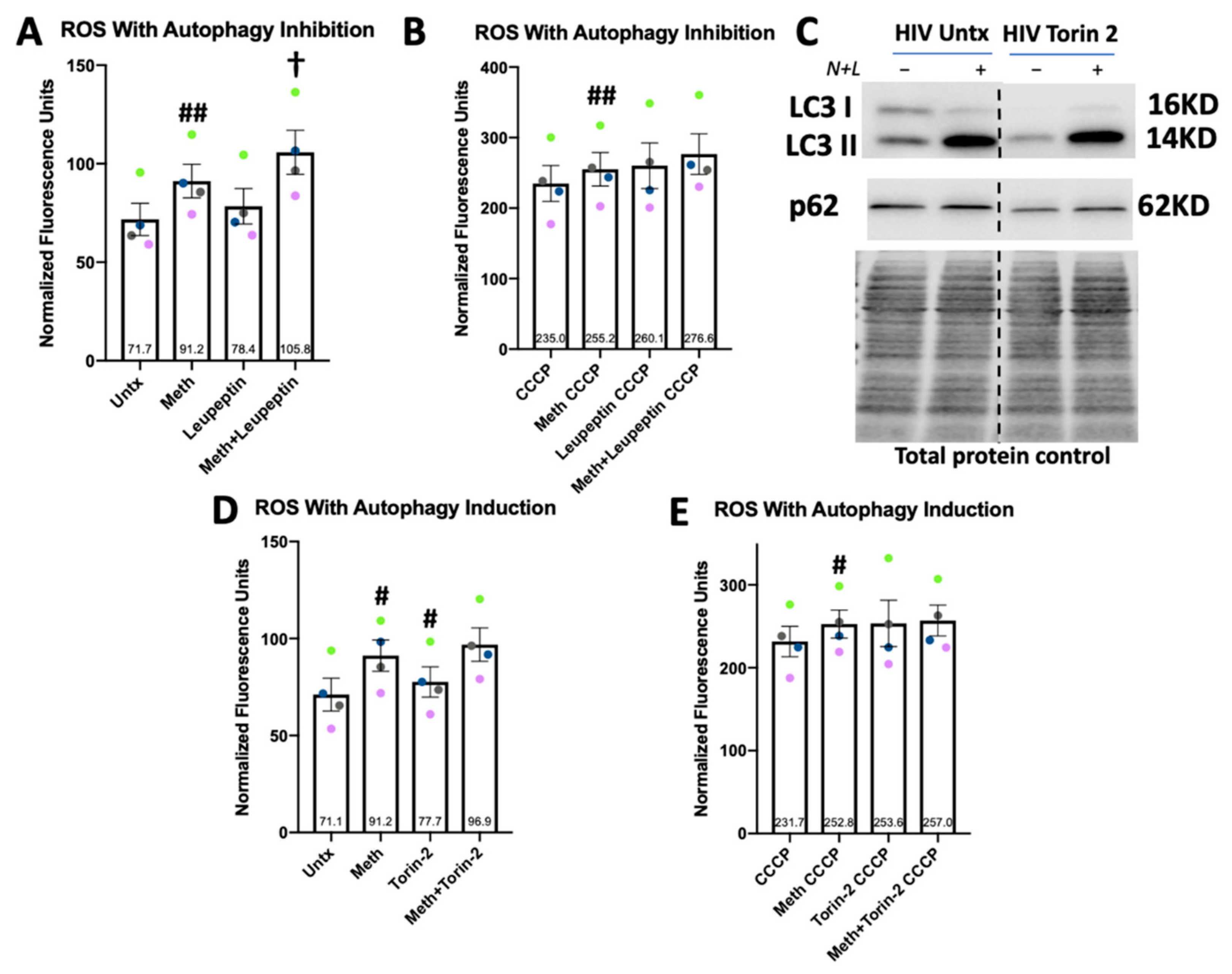
2.6. Autophagic Flux and p62 Experiments
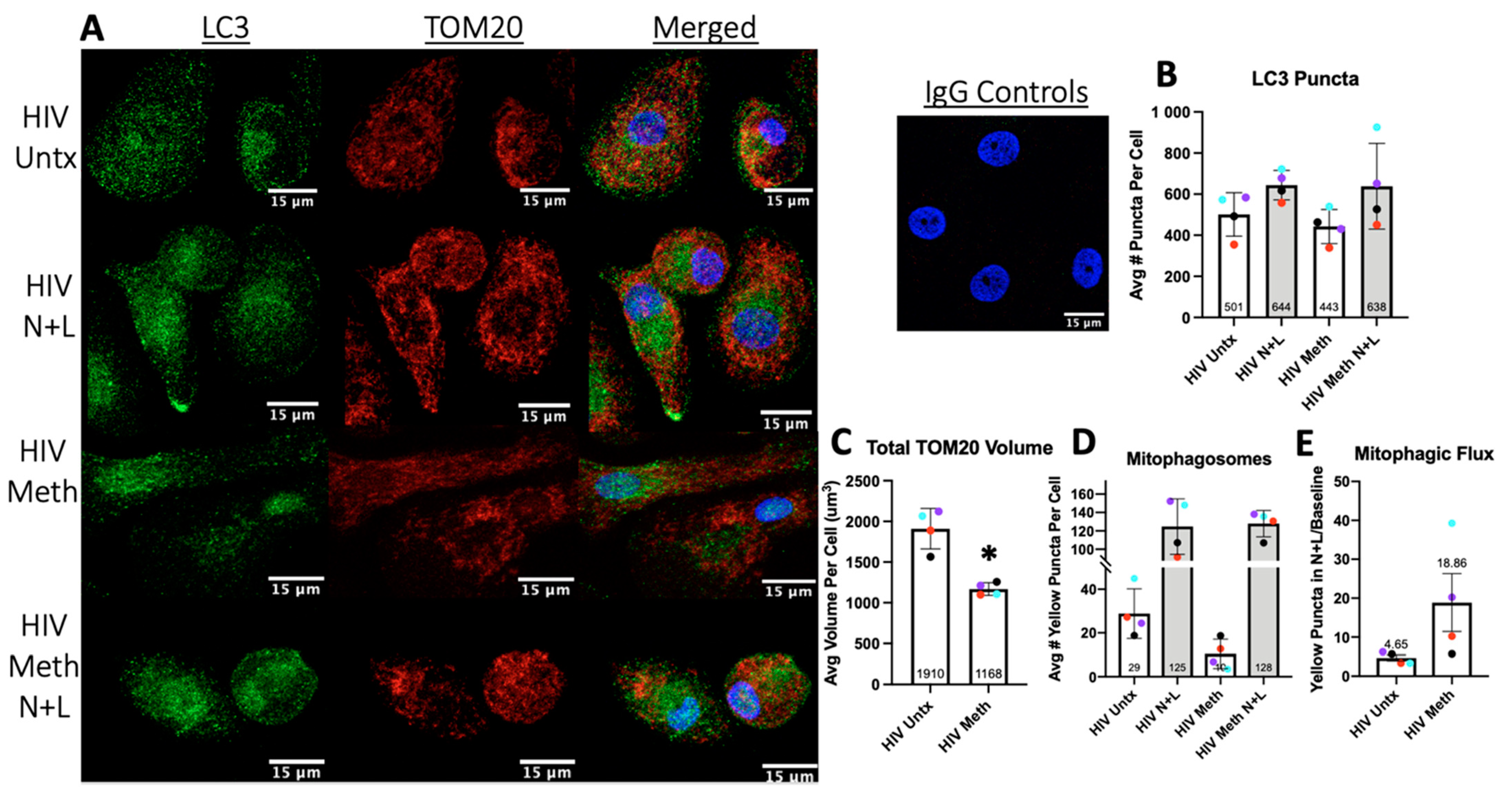
2.7. LC3 and TOM20 Immunofluorescence Studies of Mitophagy
2.8. Statistical Analyses for Functional Assays
3. Results
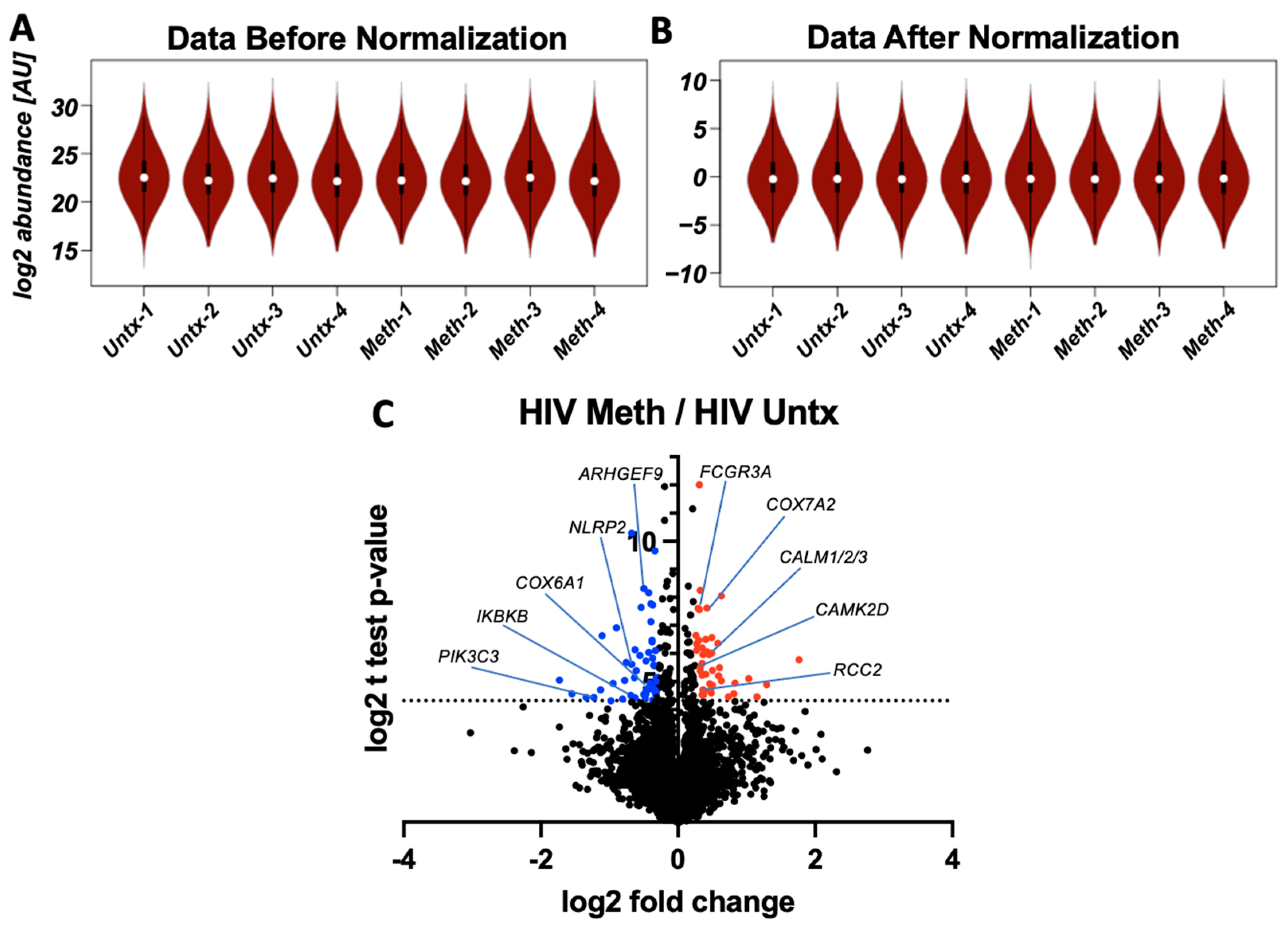
3.1. Methamphetamine Alters Protein Expression in HIV-Infected Macrophages
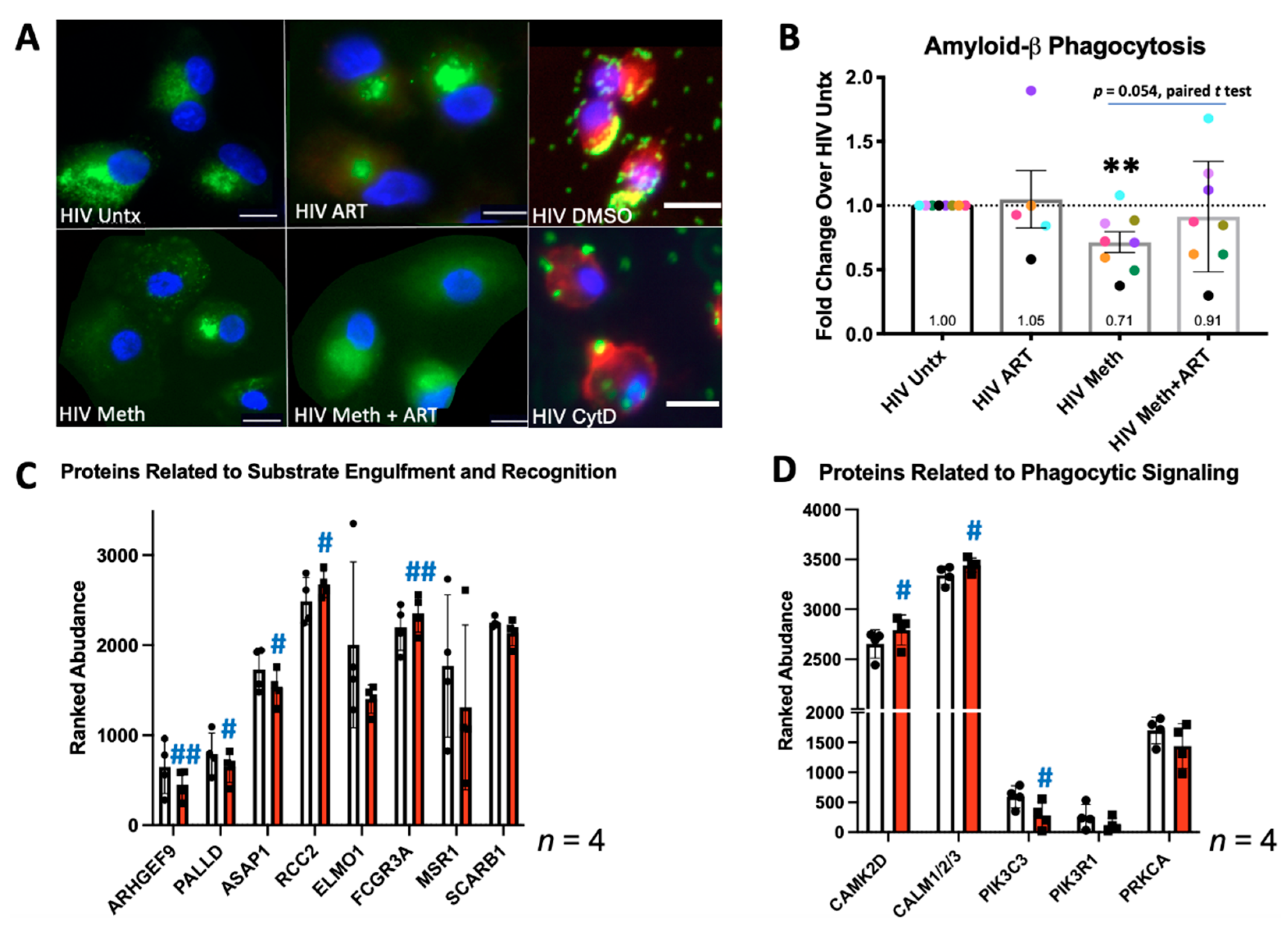
3.2. Meth Inhibits Phagocytosis and Alters Expression of Phagocytic Proteins
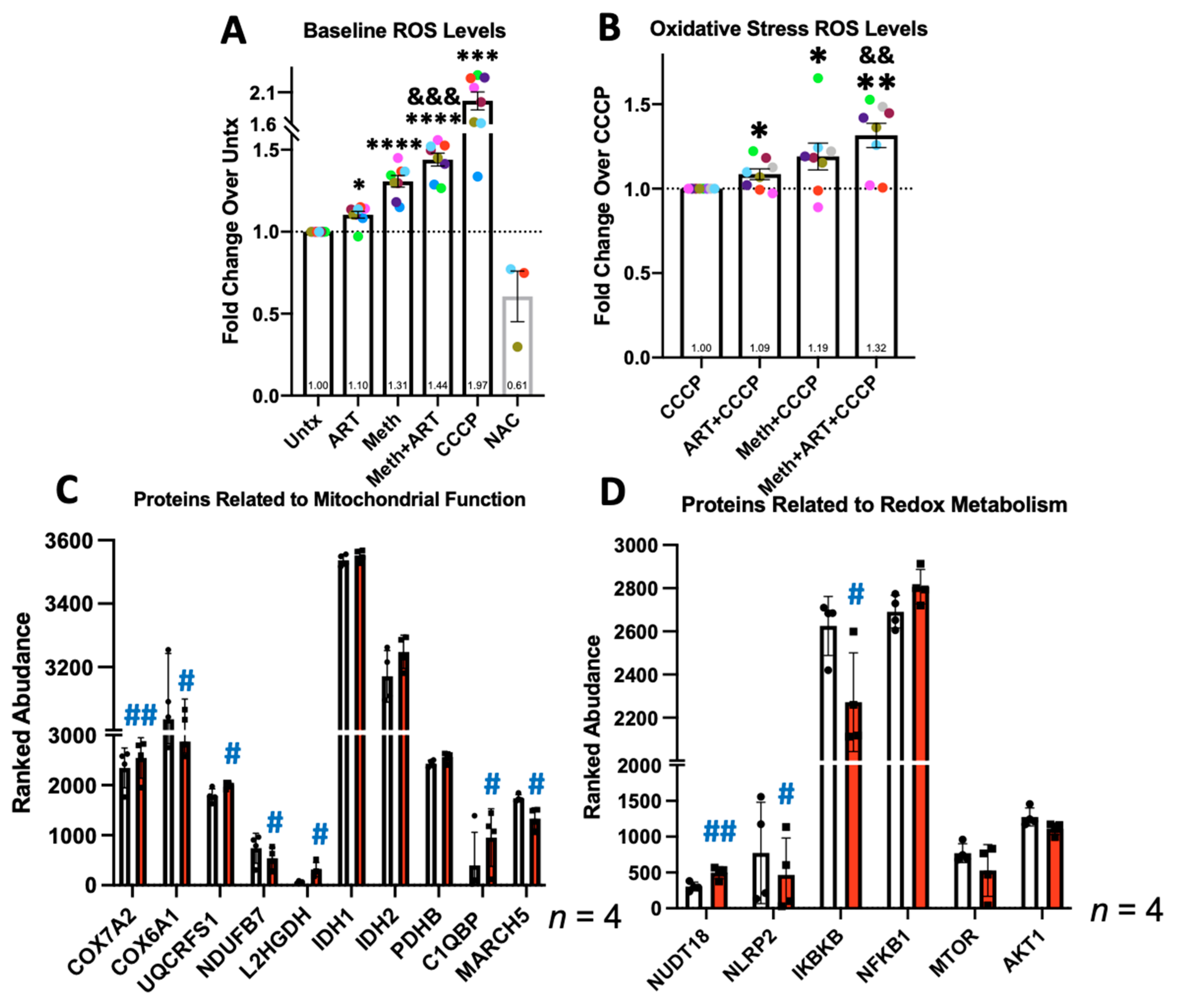
3.3. Meth Increases ROS and Changes Expression of Proteins Involved in Related Metabolic Processes
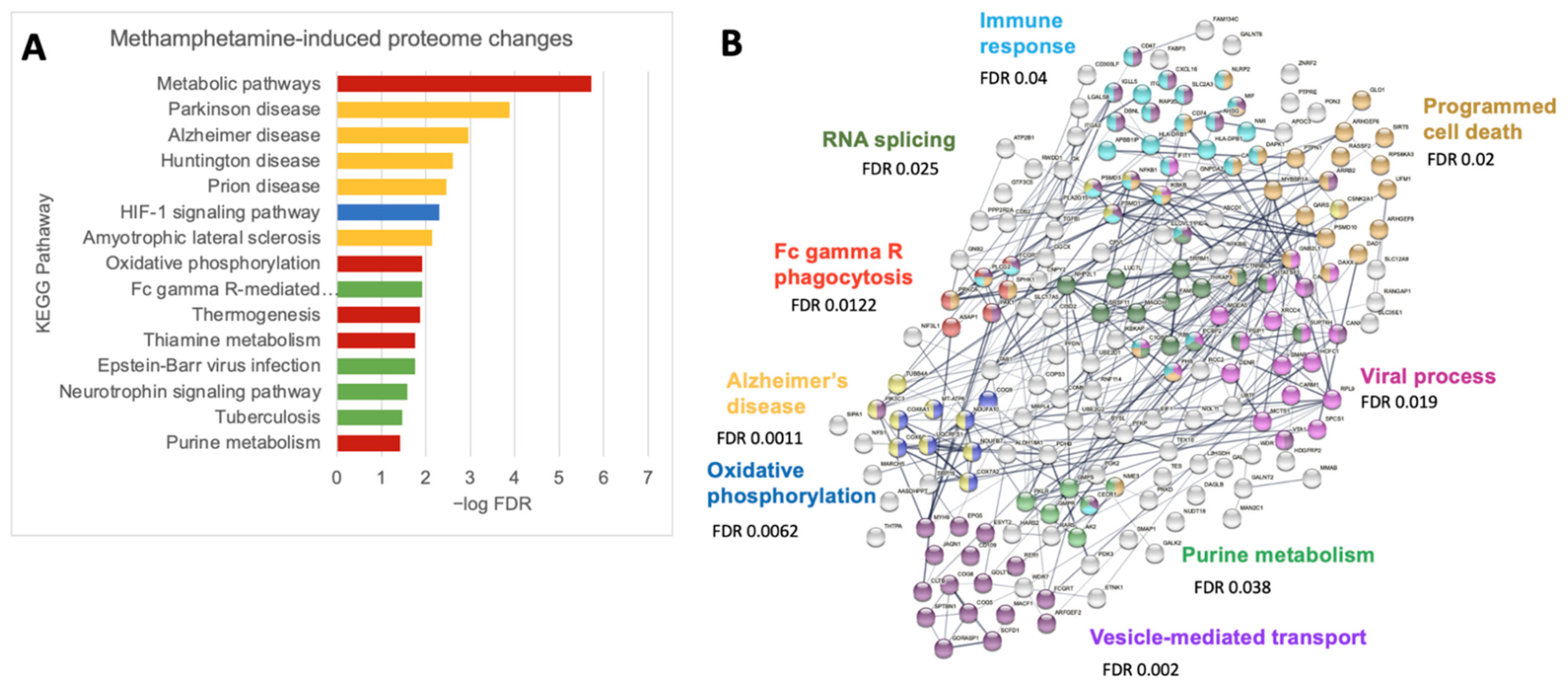
3.4. Meth Increases Protein Pathways Involved in Macrophage Functions and Neurodegenerative Diseases
3.5. Meth Induces Autophagy, Inhibits Its Completion, and May Reduce p62 Degradation
3.6. Meth May Induce Mitophagy to Decrease Total Mitochondrial Content
3.7. Drug Inhibited Lysosomal Degradation Increases ROS in Meth-Treated Macrophages
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- UNAIDS Reference. 2021. Available online: https://www.unaids.org/sites/default/files/media_asset/JC3032_AIDS_Data_book_2021_En.pdf (accessed on 20 March 2022).
- Cheney, L.; Barbaro, J.M.; Berman, J.W. Antiretroviral Drugs Impact Autophagy with Toxic Outcomes. Cells 2021, 10, 909. [Google Scholar] [CrossRef] [PubMed]
- Nasi, M.; De Biasi, S.; Gibellini, L.; Bianchini, E.; Pecorini, S.; Bacca, V.; Guaraldi, G.; Mussini, C.; Pinti, M.; Cossarizza, A. Ageing and inflammation in patients with HIV infection. Clin. Exp. Immunol. 2017, 187, 44–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spooner, R.; Ranasinghe, S.; Urasa, S.; Yoseph, M.; Koipapi, S.; Mukaetova-Ladinska, E.B.; Lewis, T.; Howlett, W.; Dekker, M.; Kisoli, A.; et al. HIV—Associated Neurocognitive Disorders (HAND): The first longitudinal follow up of a cART-treated cohort of older people in sub-Saharan Africa. J. Acquir. Immune Defic. Syndr. 2022, 90, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-San-Juan, J.; Arrieta-Aldea, I.; Arnau-Barres, I.; Garcia-Escobar, G.; Lerma-Chipirraz, E.; Perez-Garcia, P.; Marcos, A.; Blasco-Hernando, F.; Gonzalez-Mena, A.; Canas, E.; et al. Factors associated to neurocognitive impairment in older adults living with HIV. Eur. J. Med. Res. 2022, 27, 15. [Google Scholar] [CrossRef]
- Saylor, D.; Dickens, A.M.; Sacktor, N.; Haughey, N.; Slusher, B.; Pletnikov, M.; Mankowski, J.L.; Brown, A.; Volsky, D.J.; McArthur, J.C. HIV-associated neurocognitive disorder—Pathogenesis and prospects for treatment. Nat. Rev. Neurol. 2016, 12, 234–248. [Google Scholar] [CrossRef]
- Caruana, G.; Vidili, G.; Serra, P.A.; Bagella, P.; Spanu, A.; Fiore, V.; Calvisi, D.F.; Manetti, R.; Rocchitta, G.; Nuvoli, S.; et al. The burden of HIV-associated neurocognitive disorder (HAND) in post-HAART era: A multidisciplinary review of the literature. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 2290–2301. [Google Scholar]
- Byrd, D.A.; Fellows, R.P.; Morgello, S.; Franklin, D.; Heaton, R.K.; Deutsch, R.; Atkinson, J.H.; Clifford, D.B.; Collier, A.C.; Marra, C.M.; et al. Neurocognitive impact of substance use in HIV infection. J. Acquir. Immune Defic. Syndr. 2011, 58, 154–162. [Google Scholar] [CrossRef] [Green Version]
- Applebaum, A.J.; Otto, M.W.; Richardson, M.A.; Safren, S.A. Contributors to neuropsychological impairment in HIV-infected and HIV-uninfected opiate-dependent patients. J. Clin. Exp. Neuropsychol. 2010, 32, 579–589. [Google Scholar] [CrossRef] [Green Version]
- Meyer, V.J.; Rubin, L.H.; Martin, E.; Weber, K.M.; Cohen, M.H.; Golub, E.T.; Valcour, V.; Young, M.A.; Crystal, H.; Anastos, K.; et al. HIV and recent illicit drug use interact to affect verbal memory in women. J. Acquir. Immune Defic. Syndr. 2013, 63, 67–76. [Google Scholar] [CrossRef] [Green Version]
- Grov, C.; Westmoreland, D.; Morrison, C.; Carrico, A.W.; Nash, D. The Crisis We Are Not Talking About: One-in-Three Annual HIV Seroconversions Among Sexual and Gender Minorities Were Persistent Methamphetamine Users. J. Acquir. Immune Defic. Syndr. 2020, 85, 272–279. [Google Scholar] [CrossRef]
- Fulcher, J.A.; Javanbakht, M.; Shover, C.L.; Ragsdale, A.; Brookmeyer, R.; Shoptaw, S.; Gorbach, P.M. Comparative impact of methamphetamine and other drug use on viral suppression among sexual minority men on antiretroviral therapy. Drug Alcohol Depend. 2021, 221, 108622. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.; Lee, J.Y.; Fulcher, J.A.; Roach, M.E.; Dilworth, S.E.; Chahine, A.; Pallikkuth, S.; Fuchs, D.; Pahwa, S.; Carrico, A.W. Getting to the point: Methamphetamine injection is associated with biomarkers relevant to HIV pathogenesis. Drug Alcohol Depend. 2020, 213, 108133. [Google Scholar] [CrossRef] [PubMed]
- Rappaport, J.; Volsky, D.J. Role of the macrophage in HIV-associated neurocognitive disorders and other comorbidities in patients on effective antiretroviral treatment. J. Neurovirol. 2015, 21, 235–241. [Google Scholar] [CrossRef] [Green Version]
- Ko, A.; Kang, G.; Hattler, J.B.; Galadima, H.I.; Zhang, J.; Li, Q.; Kim, W.K. Macrophages but not Astrocytes Harbor HIV DNA in the Brains of HIV-1-Infected Aviremic Individuals on Suppressive Antiretroviral Therapy. J. Neuroimmune Pharmacol. 2018, 14, 110–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debaisieux, S.; Lachambre, S.; Gross, A.; Mettling, C.; Besteiro, S.; Yezid, H.; Henaff, D.; Chopard, C.; Mesnard, J.M.; Beaumelle, B. HIV-1 Tat inhibits phagocytosis by preventing the recruitment of Cdc42 to the phagocytic cup. Nat. Commun. 2015, 6, 6211. [Google Scholar] [CrossRef] [Green Version]
- Le-Bury, G.; Niedergang, F. Defective Phagocytic Properties of HIV-Infected Macrophages: How Might They Be Implicated in the Development of Invasive Salmonella Typhimurium? Front. Immunol. 2018, 9, 531. [Google Scholar] [CrossRef] [Green Version]
- Hauser, K.F.; Knapp, P.E. Interactions of HIV and drugs of abuse: The importance of glia, neural progenitors, and host genetic factors. Int. Rev. Neurobiol. 2014, 118, 231–313. [Google Scholar]
- Herb, M.; Schramm, M. Functions of ROS in Macrophages and Antimicrobial Immunity. Antioxidants 2021, 10, 313. [Google Scholar] [CrossRef]
- Ko, J.H.; Yoon, S.O.; Lee, H.J.; Oh, J.Y. Rapamycin regulates macrophage activation by inhibiting NLRP3 inflammasome-p38 MAPK-NFkappaB pathways in autophagy- and p62-dependent manners. Oncotarget 2017, 8, 40817–40831. [Google Scholar] [CrossRef] [Green Version]
- Takikita, S.; Takano, T.; Narita, T.; Takikita, M.; Ohno, M.; Shimada, M. Neuronal apoptosis mediated by IL-1 beta expression in viral encephalitis caused by a neuroadapted strain of the mumps virus (Kilham Strain) in hamsters. Exp. Neurol. 2001, 172, 47–59. [Google Scholar] [CrossRef]
- Leymarie, O.; Lepont, L.; Berlioz-Torrent, C. Canonical and Non-Canonical Autophagy in HIV-1 Replication Cycle. Viruses 2017, 9, 270. [Google Scholar] [CrossRef] [PubMed]
- Nardacci, R.; Amendola, A.; Ciccosanti, F.; Corazzari, M.; Esposito, V.; Vlassi, C.; Taibi, C.; Fimia, G.M.; Del Nonno, F.; Ippolito, G.; et al. Autophagy plays an important role in the containment of HIV-1 in nonprogressor-infected patients. Autophagy 2014, 10, 1167–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dever, S.M.; Rodriguez, M.; Lapierre, J.; Costin, B.N.; El-Hage, N. Differing roles of autophagy in HIV-associated neurocognitive impairment and encephalitis with implications for morphine co-exposure. Front. Microbiol. 2015, 6, 653. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Masliah, E.; Spector, S.A. Autophagy is increased in postmortem brains of persons with HIV-1-associated encephalitis. J. Infect. Dis. 2011, 203, 1647–1657. [Google Scholar] [CrossRef] [Green Version]
- Kyei, G.B.; Dinkins, C.; Davis, A.S.; Roberts, E.; Singh, S.B.; Dong, C.; Wu, L.; Kominami, E.; Ueno, T.; Yamamoto, A.; et al. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J. Cell Biol. 2009, 186, 255–268. [Google Scholar] [CrossRef]
- Campbell, G.R.; Rawat, P.; Bruckman, R.S.; Spector, S.A. Human Immunodeficiency Virus Type 1 Nef Inhibits Autophagy through Transcription Factor EB Sequestration. PLoS Pathog. 2015, 11, e1005018. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.Y.; Lu, J.H. Autophagy and Macrophage Functions: Inflammatory Response and Phagocytosis. Cells 2019, 9, 70. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Cuervo, A.M.; Ravikumar, B.; Sarkar, S.; Korolchuk, V.; Kaushik, S.; Klionsky, D.J. In search of an “autophagomometer”. Autophagy 2009, 5, 585–589. [Google Scholar] [CrossRef]
- Johansen, T.; Lamark, T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2014, 7, 279–296. [Google Scholar] [CrossRef]
- Yoo, S.M.; Jung, Y.K. A Molecular Approach to Mitophagy and Mitochondrial Dynamics. Mol. Cells 2018, 41, 18–26. [Google Scholar] [PubMed]
- Barbaro, J.M.; Cuervo, A.M.; Berman, J.W. HIV Increases the Inhibitory Impact of Morphine and Antiretrovirals on Autophagy in Primary Human Macrophages: Contributions to Neuropathogenesis. Cells 2021, 10, 2183. [Google Scholar] [CrossRef] [PubMed]
- Heideveld, E.; Horcas-Lopez, M.; Lopez-Yrigoyen, M.; Forrester, L.M.; Cassetta, L.; Pollard, J.W. Methods for macrophage differentiation and in vitro generation of human tumor associated-like macrophages. Methods Enzymol. 2020, 632, 113–131. [Google Scholar] [PubMed]
- Talloczy, Z.; Martinez, J.; Joset, D.; Ray, Y.; Gacser, A.; Toussi, S.; Mizushima, N.; Nosanchuk, J.D.; Goldstein, H.; Loike, J.; et al. Methamphetamine inhibits antigen processing, presentation, and phagocytosis. PLoS Pathog. 2008, 4, e28. [Google Scholar] [CrossRef]
- Schepers, R.J.; Oyler, J.M.; Joseph, R.E., Jr.; Cone, E.J.; Moolchan, E.T.; Huestis, M.A. Methamphetamine and amphetamine pharmacokinetics in oral fluid and plasma after controlled oral methamphetamine administration to human volunteers. Clin. Chem. 2003, 49, 121–132. [Google Scholar] [CrossRef] [Green Version]
- Best, B.M.; Letendre, S.L.; Koopmans, P.; Rossi, S.S.; Clifford, D.B.; Collier, A.C.; Gelman, B.B.; Marra, C.M.; McArthur, J.C.; McCutchan, J.A.; et al. Low cerebrospinal fluid concentrations of the nucleotide HIV reverse transcriptase inhibitor, tenofovir. J. Acquir. Immune Defic. Syndr. 2012, 59, 376–381. [Google Scholar] [CrossRef] [Green Version]
- Van Den Hof, M.; Blokhuis, C.; Cohen, S.; Scherpbier, H.J.; Wit, F.; Pistorius, M.C.M.; Kootstra, N.A.; Teunissen, C.E.; Mathot, R.A.A.; Pajkrt, D. CNS penetration of ART in HIV-infected children. J. Antimicrob. Chemother. 2018, 73, 484–489. [Google Scholar] [CrossRef]
- Letendre, S.L.; Mills, A.M.; Tashima, K.T.; Thomas, D.A.; Min, S.S.; Chen, S.; Song, I.H.; Piscitelli, S.C. ING116070: A study of the pharmacokinetics and antiviral activity of dolutegravir in cerebrospinal fluid in HIV-1-infected, antiretroviral therapy-naive subjects. Clin. Infect. Dis. 2014, 59, 1032–1037. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Stransky, S.; Aguilan, J.; Brenowitz, M.; Sidoli, S. DNA methylation and hydroxymethylation analysis using a high throughput and low bias direct injection mass spectrometry platform. MethodsX 2021, 8, 101585. [Google Scholar] [CrossRef]
- Weiner, A.K.; Sidoli, S.; Diskin, S.J.; Garcia, B.A. Graphical Interpretation and Analysis of Proteins and their Ontologies (GiaPronto): A One-Click Graph Visualization Software for Proteomics Data Sets. Mol. Cell Proteom. 2018, 17, 1426–1431. [Google Scholar] [CrossRef] [Green Version]
- Ninkovic, J.; Roy, S. Morphine decreases bacterial phagocytosis by inhibiting actin polymerization through cAMP-, Rac-1-, and p38 MAPK-dependent mechanisms. Am. J. Pathol. 2012, 180, 1068–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, Y.S.; Choi, S.E.; Koh, H.C. PGAM5 regulates PINK1/Parkin-mediated mitophagy via DRP1 in CCCP-induced mitochondrial dysfunction. Toxicol. Lett. 2018, 284, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.P.; Hu, L.F.; Zheng, H.F.; Mao, C.J.; Hu, W.D.; Xiong, K.P.; Wang, F.; Liu, C.F. Application and interpretation of current autophagy inhibitors and activators. Acta Pharmacol. Sin. 2013, 34, 625–635. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Barres, B.A. Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 2018, 18, 225–242. [Google Scholar] [CrossRef] [PubMed]
- Andras, I.E.; Toborek, M. Amyloid beta accumulation in HIV-1-infected brain: The role of the blood brain barrier. IUBMB Life 2013, 65, 43–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Achim, C.L.; Adame, A.; Dumaop, W.; Everall, I.P.; Masliah, E. Increased accumulation of intraneuronal amyloid beta in HIV-infected patients. J. Neuroimmune Pharmacol. 2009, 4, 190–199. [Google Scholar] [CrossRef] [Green Version]
- Liang, H.; Wang, X.; Chen, H.; Song, L.; Ye, L.; Wang, S.H.; Wang, Y.J.; Zhou, L.; Ho, W.Z. Methamphetamine enhances HIV infection of macrophages. Am. J. Pathol. 2008, 172, 1617–1624. [Google Scholar] [CrossRef]
- Tanna, C.E.; Goss, L.B.; Ludwig, C.G.; Chen, P.W. Arf GAPs as Regulators of the Actin Cytoskeleton-An Update. Int. J. Mol. Sci. 2019, 20, 442. [Google Scholar] [CrossRef] [Green Version]
- Uribe-Querol, E.; Rosales, C. Phagocytosis: Our Current Understanding of a Universal Biological Process. Front. Immunol. 2020, 11, 1066. [Google Scholar] [CrossRef]
- Wang, P.; Zhang, W.; Wang, L.; Liang, W.; Cai, A.; Gao, Y.; Chen, L. RCC2 Interacts with Small GTPase RalA and Regulates Cell Proliferation and Motility in Gastric Cancer. Onco. Targets Ther. 2020, 13, 3093–3103. [Google Scholar] [CrossRef] [Green Version]
- Gumienny, T.L.; Brugnera, E.; Tosello-Trampont, A.C.; Kinchen, J.M.; Haney, L.B.; Nishiwaki, K.; Walk, S.F.; Nemergut, M.E.; Macara, I.G.; Francis, R.; et al. CED-12/ELMO, a novel member of the CrkII/Dock180/Rac pathway, is required for phagocytosis and cell migration. Cell 2001, 107, 27–41. [Google Scholar] [CrossRef] [Green Version]
- Bonilla, D.L.; Bhattacharya, A.; Sha, Y.; Xu, Y.; Xiang, Q.; Kan, A.; Jagannath, C.; Komatsu, M.; Eissa, N.T. Autophagy regulates phagocytosis by modulating the expression of scavenger receptors. Immunity 2013, 39, 537–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tebar, F.; Chavero, A.; Agell, N.; Lu, A.; Rentero, C.; Enrich, C.; Grewal, T. Pleiotropic Roles of Calmodulin in the Regulation of KRas and Rac1 GTPases: Functional Diversity in Health and Disease. Int. J. Mol. Sci. 2020, 21, 3680. [Google Scholar] [CrossRef] [PubMed]
- Smith, F.D.; Samelson, B.K.; Scott, J.D. Discovery of cellular substrates for protein kinase A using a peptide array screening protocol. Biochem. J. 2011, 438, 103–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gozzelino, L.; De Santis, M.C.; Gulluni, F.; Hirsch, E.; Martini, M. PI(3,4)P2 Signaling in Cancer and Metabolism. Front. Oncol. 2020, 10, 360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, R.; Gu, J.; Zong, S.; Wu, M.; Yang, M. Structure and mechanism of mitochondrial electron transport chain. Biomed. J. 2018, 41, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Roosterman, D.; Cottrell, G.S. Rethinking the Citric Acid Cycle: Connecting Pyruvate Carboxylase and Citrate Synthase to the Flow of Energy and Material. Int. J. Mol. Sci. 2021, 22, 604. [Google Scholar] [CrossRef]
- Ma, S.; Sun, R.; Jiang, B.; Gao, J.; Deng, W.; Liu, P.; He, R.; Cui, J.; Ji, M.; Yi, W.; et al. L2hgdh Deficiency Accumulates l-2-Hydroxyglutarate with Progressive Leukoencephalopathy and Neurodegeneration. Mol. Cell Biol. 2017, 37, e00492-16. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Huang, J.; Gao, M.; Guo, G.; Zeng, S.; Chen, X.; Wang, X.; Gong, Z.; Yan, Y. Current perspectives on the clinical implications of oxidative RNA damage in aging research: Challenges and opportunities. Geroscience 2021, 43, 487–505. [Google Scholar] [CrossRef]
- Yoo, Y.S.; Park, Y.Y.; Kim, J.H.; Cho, H.; Kim, S.H.; Lee, H.S.; Kim, T.H.; Kim, Y.S.; Lee, Y.; Kim, C.J.; et al. The mitochondrial ubiquitin ligase MARCH5 resolves MAVS aggregates during antiviral signalling. Nat. Commun. 2015, 6, 7910. [Google Scholar] [CrossRef] [Green Version]
- Gwinn, M.R.; Vallyathan, V. Respiratory burst: Role in signal transduction in alveolar macrophages. J. Toxicol. Environ. Health B Crit. Rev. 2006, 9, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Lamark, T.; Svenning, S.; Johansen, T. Regulation of selective autophagy: The p62/SQSTM1 paradigm. Essays Biochem. 2017, 61, 609–624. [Google Scholar] [PubMed]
- Wu, Y.H.; Chou, T.F.; Young, L.; Hsieh, F.Y.; Pan, H.Y.; Mo, S.T.; Brown, S.B.; Chen, R.H.; Kimchi, A.; Lai, M.Z. Tumor suppressor death-associated protein kinase 1 inhibits necroptosis by p38 MAPK activation. Cell Death Dis. 2020, 11, 305. [Google Scholar] [CrossRef]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhu, Y.; Wang, D.; Chen, M.; Gao, P.; Xiao, W.; Rao, G.; Wang, X.; Jin, H.; Xu, L.; et al. Morphine induces Beclin 1- and ATG5-dependent autophagy in human neuroblastoma SH-SY5Y cells and in the rat hippocampus. Autophagy 2014, 6, 386–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, T.; Kondo, S.; Le, W.; Jankovic, J. The role of autophagy-lysosome pathway in neurodegeneration associated with Parkinson’s disease. Brain 2008, 131, 1969–1978. [Google Scholar] [CrossRef]
- Schofield, J.H.; Schafer, Z.T. Mitochondrial Reactive Oxygen Species and Mitophagy: A Complex and Nuanced Relationship. Antioxid. Redox. Signal. 2021, 34, 517–530. [Google Scholar] [CrossRef]
- Esteban-Martinez, L.; Sierra-Filardi, E.; McGreal, R.S.; Salazar-Roa, M.; Marino, G.; Seco, E.; Durand, S.; Enot, D.; Grana, O.; Malumbres, M.; et al. Programmed mitophagy is essential for the glycolytic switch during cell differentiation. EMBO J. 2017, 36, 1688–1706. [Google Scholar] [CrossRef]
- Sun, X.; Chang, R.; Tang, Y.; Luo, S.; Jiang, C.; Jia, H.; Xu, Q.; Dong, Z.; Liang, Y.; Loor, J.J.; et al. Transcription factor EB (TFEB)-mediated autophagy protects bovine mammary epithelial cells against H2O2-induced oxidative damage in vitro. J. Anim. Sci. Biotechnol. 2021, 12, 35. [Google Scholar] [CrossRef]
- Park, S.; Sim, H.; Lee, K. Rapamycin-resistant and torin-sensitive mTOR signaling promotes the survival and proliferation of leukemic cells. BMB Rep. 2016, 49, 63–68. [Google Scholar] [CrossRef] [Green Version]
- Wainwright, J.J.; Mikre, M.; Whitley, P.; Dawson, E.; Huskey, A.; Lukowiak, A.; Giroir, B.P. Analysis of Drug Test Results Before and After the US Declaration of a National Emergency Concerning the COVID-19 Outbreak. JAMA 2020, 324, 1674–1677. [Google Scholar] [CrossRef] [PubMed]
- Leon-Rivera, R.; Morsey, B.; Niu, M.; Fox, H.S.; Berman, J.W. Interactions of Monocytes, HIV, and ART Identified by an Innovative scRNAseq Pipeline: Pathways to Reservoirs and HIV-Associated Comorbidities. mBio 2020, 11, e01037-20. [Google Scholar] [CrossRef] [PubMed]
- Leon-Rivera, R.; Veenstra, M.; Donoso, M.; Tell, E.; Eugenin, E.A.; Morgello, S.; Berman, J.W. Central Nervous System (CNS) Viral Seeding by Mature Monocytes and Potential Therapies to Reduce CNS Viral Reservoirs in the cART Era. mBio 2021, 12, e03633-20. [Google Scholar] [CrossRef] [PubMed]
- Kadiu, I.; Ricardo-Dukelow, M.; Ciborowski, P.; Gendelman, H.E. Cytoskeletal protein transformation in HIV-1-infected macrophage giant cells. J. Immunol. 2007, 178, 6404–6415. [Google Scholar] [CrossRef] [Green Version]
- Ciborowski, P.; Kadiu, I.; Rozek, W.; Smith, L.; Bernhardt, K.; Fladseth, M.; Ricardo-Dukelow, M.; Gendelman, H.E. Investigating the human immunodeficiency virus type 1-infected monocyte-derived macrophage secretome. Virology 2007, 363, 198–209. [Google Scholar] [CrossRef] [Green Version]
- Vural, A.; Kehrl, J.H. Autophagy in macrophages: Impacting inflammation and bacterial infection. Scientifica 2014, 2014, 825463. [Google Scholar] [CrossRef] [Green Version]
- Tao, H.; Yancey, P.G.; Blakemore, J.L.; Zhang, Y.; Ding, L.; Jerome, W.G.; Brown, J.D.; Vickers, K.C.; Linton, M.F. Macrophage SR-BI modulates autophagy via VPS34 complex and PPARalpha transcription of Tfeb in atherosclerosis. J. Clin. Investig. 2021, 131, e94229. [Google Scholar] [CrossRef]
- Akay, C.; Cooper, M.; Odeleye, A.; Jensen, B.K.; White, M.G.; Vassoler, F.; Gannon, P.J.; Mankowski, J.; Dorsey, J.L.; Buch, A.M.; et al. Antiretroviral drugs induce oxidative stress and neuronal damage in the central nervous system. J. Neurovirol. 2014, 20, 39–53. [Google Scholar] [CrossRef] [Green Version]
- Shi, C.S.; Shenderov, K.; Huang, N.N.; Kabat, J.; Abu-Asab, M.; Fitzgerald, K.A.; Sher, A.; Kehrl, J.H. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 2012, 13, 255–263. [Google Scholar] [CrossRef]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [Green Version]
- Korbecki, J.; Bajdak-Rusinek, K.; Kupnicka, P.; Kapczuk, P.; Siminska, D.; Chlubek, D.; Baranowska-Bosiacka, I. The Role of CXCL16 in the Pathogenesis of Cancer and Other Diseases. Int. J. Mol. Sci. 2021, 22, 3490. [Google Scholar] [CrossRef] [PubMed]
- Wetzel, K.S.; Yi, Y.; Elliott, S.T.C.; Romero, D.; Jacquelin, B.; Hahn, B.H.; Muller-Trutwin, M.; Apetrei, C.; Pandrea, I.; Collman, R.G. CXCR6-Mediated Simian Immunodeficiency Virus SIVagmSab Entry into Sabaeus African Green Monkey Lymphocytes Implicates Widespread Use of Non-CCR5 Pathways in Natural Host Infections. J. Virol. 2017, 91, e01626-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burns, A.; Ciborowski, P. Acute exposure to methamphetamine alters TLR9-mediated cytokine expression in human macrophage. Immunobiology 2016, 221, 199–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilija Pun, N.; Park, P.H. Role of p62 in the suppression of inflammatory cytokine production by adiponectin in macrophages: Involvement of autophagy and p21/Nrf2 axis. Sci. Rep. 2017, 7, 393. [Google Scholar] [CrossRef]
- Foroughi, K.; Khaksari, M.; Rahmati, M.; Bitaraf, F.S.; Shayannia, A. Apelin-13 Protects PC12 Cells Against Methamphetamine-Induced Oxidative Stress, Autophagy and Apoptosis. Neurochem. Res. 2019, 44, 2103–2112. [Google Scholar] [CrossRef]
- Foroughi, K.; Jahanbani, S.; Khaksari, M.; Shayannia, A. Obestatin attenuated methamphetamine-induced PC12 cells neurotoxicity via inhibiting autophagy and apoptosis. Hum. Exp. Toxicol. 2020, 39, 301–310. [Google Scholar] [CrossRef]
- Tan, X.H.; Zhang, K.K.; Xu, J.T.; Qu, D.; Chen, L.J.; Li, J.H.; Wang, Q.; Wang, H.J.; Xie, X.L. Luteolin alleviates methamphetamine-induced neurotoxicity by suppressing PI3K/Akt pathway-modulated apoptosis and autophagy in rats. Food Chem. Toxicol. 2020, 137, 111179. [Google Scholar] [CrossRef]
- Nixon, R.A.; Yang, D.S. Autophagy failure in Alzheimer’s disease—Locating the primary defect. Neurobiol. Dis. 2011, 43, 38–45. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef]
- Chilunda, V.; Calderon, T.M.; Martinez-Aguado, P.; Berman, J.W. The impact of substance abuse on HIV-mediated neuropathogenesis in the current ART era. Brain Res. 2019, 1724, 146426. [Google Scholar] [CrossRef]
- Liu, Y.; Meng, F.Z.; Wang, X.; Wang, P.; Liu, J.B.; Hu, W.H.; Young, W.B.; Ho, W.Z. Methamphetamine facilitates HIV infection of primary human monocytes through inhibiting cellular viral restriction factors. Cell Biosci. 2021, 11, 194. [Google Scholar] [CrossRef] [PubMed]
- Niu, M.; Morsey, B.; Lamberty, B.G.; Emanuel, K.; Yu, F.; Leon-Rivera, R.; Berman, J.W.; Gaskill, P.J.; Matt, S.M.; Ciborowski, P.S.; et al. Methamphetamine Increases the Proportion of SIV-Infected Microglia/Macrophages, Alters Metabolic Pathways, and Elevates Cell Death Pathways: A Single-Cell Analysis. Viruses 2020, 12, 1297. [Google Scholar] [CrossRef] [PubMed]
- Viola, A.; Munari, F.; Sanchez-Rodriguez, R.; Scolaro, T.; Castegna, A. The Metabolic Signature of Macrophage Responses. Front. Immunol. 2019, 10, 1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Corrigendum: Macrophage Polarization: Different Gene Signatures in M1(LPS+) vs. Classically and M2(LPS-) vs. Alternatively Activated Macrophages. Front. Immunol. 2020, 11, 234. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wu, F.; Xue, L.; Wang, B.; Li, J.; Chen, Y.; Chen, T. Methamphetamine causes neurotoxicity by promoting polarization of macrophages and inflammatory response. Hum. Exp. Toxicol. 2018, 37, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Arranz, A.; Doxaki, C.; Vergadi, E.; De La Torre, Y.M.; Vaporidi, K.; Lagoudaki, E.D.; Ieronymaki, E.; Androulidaki, A.; Venihaki, M.; Margioris, A.N.; et al. Akt1 and Akt2 protein kinases differentially contribute to macrophage polarization. Proc. Natl. Acad. Sci. USA 2012, 109, 9517–9522. [Google Scholar] [CrossRef] [Green Version]
- Krutetskaya, Z.I.; Milenina, L.S.; Antonov, V.G.; Nozdrachev, A.D. Sigma-1 Receptor Agonist Amitriptyline Inhibits Store-Dependent Ca(2+) Entry in Macrophages. Dokl. Biochem. Biophys. 2019, 488, 307–310. [Google Scholar] [CrossRef]
- Robson, M.J.; Turner, R.C.; Naser, Z.J.; McCurdy, C.R.; Huber, J.D.; Matsumoto, R.R. SN79, a sigma receptor ligand, blocks methamphetamine-induced microglial activation and cytokine upregulation. Exp. Neurol. 2013, 247, 134–142. [Google Scholar] [CrossRef] [Green Version]
- Shen, K.; Zhang, Y.; Lv, X.; Chen, X.; Zhou, R.; Nguyen, L.K.; Wu, X.; Yao, H. Molecular Mechanisms Involving Sigma-1 Receptor in Cell Apoptosis of BV-2 Microglial Cells Induced by Methamphetamine. CNS Neurol. Disord. Drug Targets 2016, 15, 857–865. [Google Scholar] [CrossRef]
- Chao, J.; Zhang, Y.; Du, L.; Zhou, R.; Wu, X.; Shen, K.; Yao, H. Author Correction: Molecular mechanisms underlying the involvement of the sigma-1 receptor in methamphetamine-mediated microglial polarization. Sci. Rep. 2021, 11, 6142. [Google Scholar] [CrossRef]
- Rosen, D.A.; Seki, S.M.; Fernandez-Castaneda, A.; Beiter, R.M.; Eccles, J.D.; Woodfolk, J.A.; Gaultier, A. Modulation of the sigma-1 receptor-IRE1 pathway is beneficial in preclinical models of inflammation and sepsis. Sci. Transl. Med. 2019, 11, eaau5266. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barbaro, J.M.; Sidoli, S.; Cuervo, A.M.; Berman, J.W. Methamphetamine Dysregulates Macrophage Functions and Autophagy to Mediate HIV Neuropathogenesis. Biomedicines 2022, 10, 1257. https://doi.org/10.3390/biomedicines10061257
Barbaro JM, Sidoli S, Cuervo AM, Berman JW. Methamphetamine Dysregulates Macrophage Functions and Autophagy to Mediate HIV Neuropathogenesis. Biomedicines. 2022; 10(6):1257. https://doi.org/10.3390/biomedicines10061257
Chicago/Turabian StyleBarbaro, John M., Simone Sidoli, Ana Maria Cuervo, and Joan W. Berman. 2022. "Methamphetamine Dysregulates Macrophage Functions and Autophagy to Mediate HIV Neuropathogenesis" Biomedicines 10, no. 6: 1257. https://doi.org/10.3390/biomedicines10061257
APA StyleBarbaro, J. M., Sidoli, S., Cuervo, A. M., & Berman, J. W. (2022). Methamphetamine Dysregulates Macrophage Functions and Autophagy to Mediate HIV Neuropathogenesis. Biomedicines, 10(6), 1257. https://doi.org/10.3390/biomedicines10061257