
The Role of Inflammatory Cytokines in the Pathogenesis of Colorectal Carcinoma—Recent Findings and Review

 , , , ,
, , , ,
Abstract
:1. Introduction
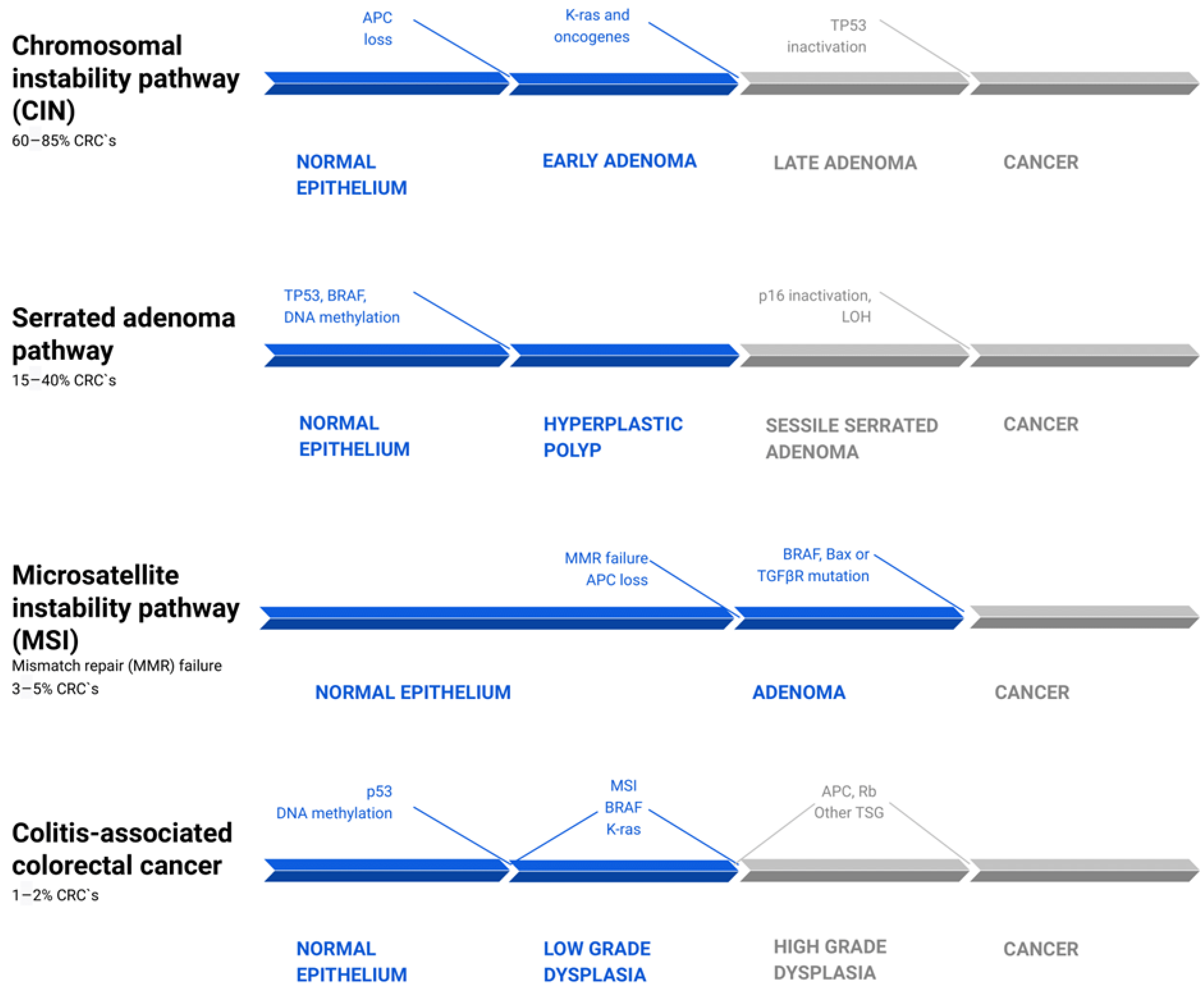
2. Inflammation in the Pathways of Sporadic and Colitis-Associated Colorectal Carcinogenesis
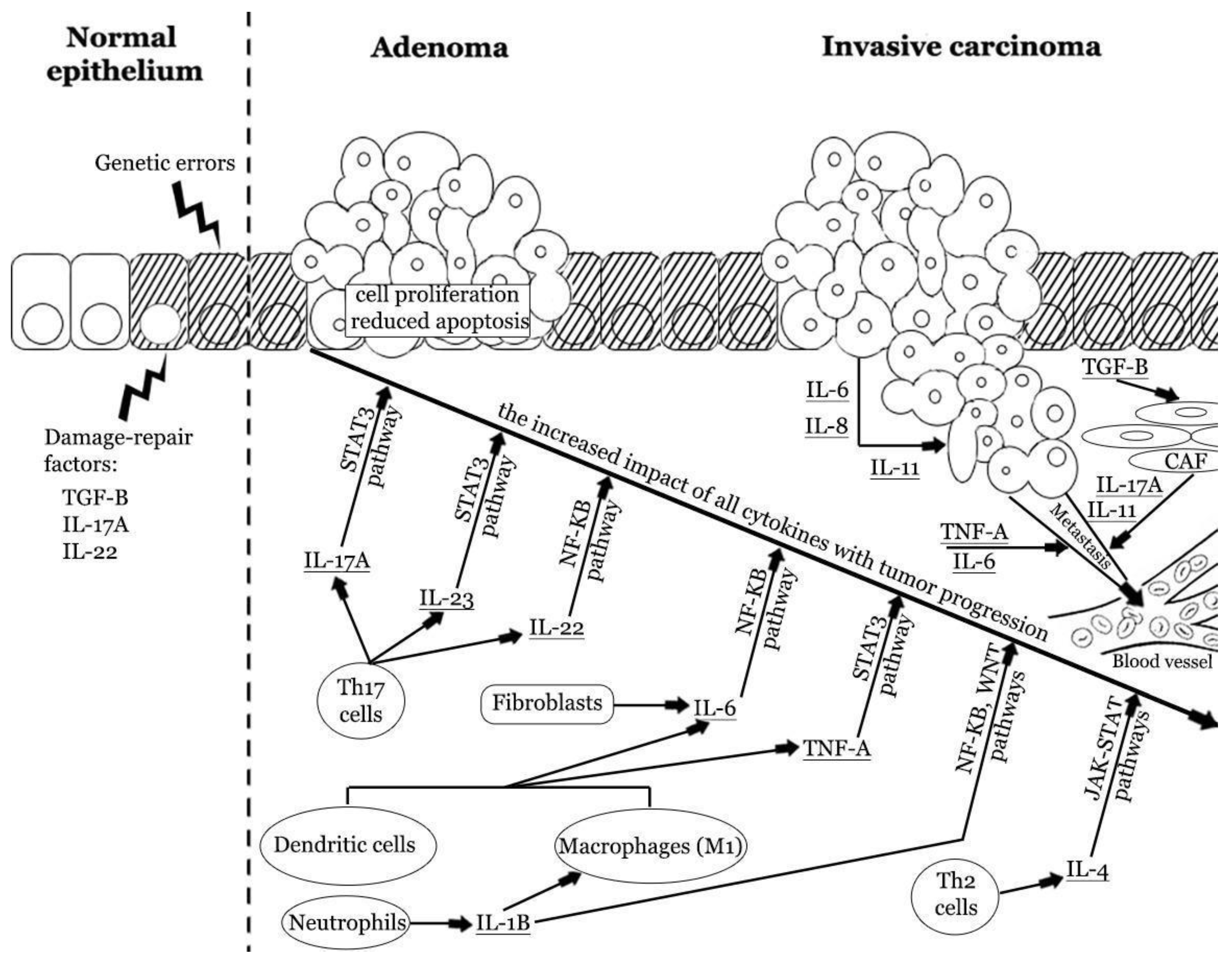
3. Cytokines, Tumor Microenvironment, and Epithelial–Mesenchymal Transition
4. The Mediatory Role of Interleukins in Colorectal Carcinogenesis
4.1. IL-1β
4.2. IL-4
4.3. IL-6
4.4. IL-7
4.5. IL-8
4.6. IL-10
4.7. IL-11
4.8. IL-17
4.9. IL-21
4.10. IL-22
4.11. IL-23
4.12. IL-33
4.13. IFN-γ
4.14. TGF-β
4.15. TNF-α
4.16. GM-CSF
5. Diagnostic and Therapeutic Implications
6. Final Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data availability
Conflicts of Interest
References
- Rawla, P.; Sunkara, T.; Barsouk, A. Epidemiology of Colorectal Cancer: Incidence, Mortality, Survival, and Risk Factors. Przegląd Gastroenterol 2019, 14, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briukhovetska, D.; Dörr, J.; Endres, S.; Libby, P.; Dinarello, C.A.; Kobold, S. Interleukins in Cancer: From Biology to Therapy. Nat. Rev. Cancer 2021, 21, 481–499. [Google Scholar] [CrossRef]
- Carethers, J.M.; Jung, B.H. Genetics and Genetic Biomarkers in Sporadic Colorectal Cancer. Gastroenterology 2015, 149, 1177–1190. [Google Scholar] [CrossRef] [Green Version]
- Ullman, T.A.; Itzkowitz, S.H. Intestinal Inflammation and Cancer. Gastroenterology 2011, 140, 1807–1816. [Google Scholar] [CrossRef]
- Grivennikov, S.I. Inflammation and Colorectal Cancer: Colitis-Associated Neoplasia. Semin. Immunopathol. 2013, 35, 229–244. [Google Scholar] [CrossRef]
- Dulai, P.S.; Sandborn, W.J.; Gupta, S. Colorectal Cancer and Dysplasia in Inflammatory Bowel Disease: A Review of Disease Epidemiology, Pathophysiology, and Management. Cancer Prev. Res. 2016, 9, 887–894. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Linstra, R.; van Vugt, M.A.T.M. Genomic Instability, Inflammatory Signaling and Response to Cancer Immunotherapy. Biochim. Biophys. Acta Rev. Cancer 2022, 1877, 188661. [Google Scholar] [CrossRef]
- Pino, M.S.; Chung, D.C. The Chromosomal Instability Pathway in Colon Cancer. Gastroenterology 2010, 138, 2059–2072. [Google Scholar] [CrossRef] [Green Version]
- Robles, A.I.; Traverso, G.; Zhang, M.; Roberts, N.J.; Khan, M.A.; Joseph, C.; Lauwers, G.Y.; Selaru, F.M.; Popoli, M.; Pittman, M.E.; et al. Whole-Exome Sequencing Analyses of Inflammatory Bowel Disease-Associated Colorectal Cancers. Gastroenterology 2016, 150, 931–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grivennikov, S.I.; Cominelli, F. Colitis-Associated and Sporadic Colon Cancers: Different Diseases, Different Mutations? Gastroenterology 2016, 150, 808–810. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.R.; Chang, D.K. Colorectal Cancer in Inflammatory Bowel Disease: The Risk, Pathogenesis, Prevention and Diagnosis. World J. Gastroenterol. 2014, 20, 9872–9881. [Google Scholar] [CrossRef] [PubMed]
- Műzes, G.; Molnár, B.; Tulassay, Z.; Sipos, F. Changes of the Cytokine Profile in Inflammatory Bowel Diseases. World J. Gastroenterol. 2012, 18, 5848–5861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stolfi, C.; Rizzo, A.; Franzè, E.; Rotondi, A.; Fantini, M.C.; Sarra, M.; Caruso, R.; Monteleone, I.; Sileri, P.; Franceschilli, L.; et al. Involvement of Interleukin-21 in the Regulation of Colitis-Associated Colon Cancer. J. Exp. Med. 2011, 208, 2279–2290. [Google Scholar] [CrossRef]
- Krzystek-Korpacka, M.; Zawadzki, M.; Neubauer, K.; Bednarz-Misa, I.; Górska, S.; Wiśniewski, J.; Witkiewicz, W.; Gamian, A. Elevated Systemic Interleukin-7 in Patients with Colorectal Cancer and Individuals at High Risk of Cancer: Association with Lymph Node Involvement and Tumor Location in the Right Colon. Cancer Immunol. Immunother. 2017, 66, 171–179. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; de Haar, C.; Chen, M.; Deuring, J.; Gerrits, M.M.; Smits, R.; Xia, B.; Kuipers, E.J.; van der Woude, C.J. Disease-Related Expression of the IL6/STAT3/SOCS3 Signalling Pathway in Ulcerative Colitis and Ulcerative Colitis-Related Carcinogenesis. Gut 2010, 59, 227–235. [Google Scholar] [CrossRef]
- Franzè, E.; Monteleone, I.; Cupi, M.L.; Mancia, P.; Caprioli, F.; Marafini, I.; Colantoni, A.; Ortenzi, A.; Laudisi, F.; Sica, G.; et al. Interleukin-34 Sustains Inflammatory Pathways in the Gut. Clin. Sci. 2015, 129, 271–280. [Google Scholar] [CrossRef]
- Nemeth, Z.H.; Bogdanovski, D.A.; Barratt-Stopper, P.; Paglinco, S.R.; Antonioli, L.; Rolandelli, R.H. Crohn’s Disease and Ulcerative Colitis Show Unique Cytokine Profiles. Cureus 2017, 9, e1177. [Google Scholar] [CrossRef] [Green Version]
- El-Bassat, H.; AboAli, L.; El Yamany, S.; Al Shenawy, H.; Al Din, R.A.; Taha, A. Interleukin-23p19 Expression in Patients with Ulcerative Colitis and Its Relation to Disease Severity. Adv. Dig. Med. 2016, 3, 88–94. [Google Scholar] [CrossRef] [Green Version]
- Khalyfa, A.A.; Punatar, S.; Aslam, R.; Yarbrough, A. Exploring the Inflammatory Pathogenesis of Colorectal Cancer. Diseases 2021, 9, 79. [Google Scholar] [CrossRef] [PubMed]
- Yaeger, R.; Paroder, V.; Bates, D.D.B.; Capanu, M.; Chou, J.; Tang, L.; Chatila, W.; Schultz, N.; Hersch, J.; Kelsen, D. Systemic Chemotherapy for Metastatic Colitis-Associated Cancer Has a Worse Outcome Than Sporadic Colorectal Cancer: Matched Case Cohort Analysis. Clin. Colorectal Cancer 2020, 19, e151–e156. [Google Scholar] [CrossRef] [PubMed]
- Malki, A.; ElRuz, R.A.; Gupta, I.; Allouch, A.; Vranic, S.; Al Moustafa, A.-E. Molecular Mechanisms of Colon Cancer Progression and Metastasis: Recent Insights and Advancements. Int. J. Mol. Sci. 2020, 22, 130. [Google Scholar] [CrossRef] [PubMed]
- Leggett, B.; Whitehall, V. Role of the Serrated Pathway in Colorectal Cancer Pathogenesis. Gastroenterology 2010, 138, 2088–2100. [Google Scholar] [CrossRef]
- Mundade, R.; Imperiale, T.F.; Prabhu, L.; Loehrer, P.J.; Lu, T. Genetic Pathways, Prevention, and Treatment of Sporadic Colorectal Cancer. Oncoscience 2014, 1, 400–406. [Google Scholar] [CrossRef] [Green Version]
- Munkholm, P. Review Article: The Incidence and Prevalence of Colorectal Cancer in Inflammatory Bowel Disease. Aliment. Pharmacol. Ther. 2003, 18, 1–5. [Google Scholar] [CrossRef]
- Lakatos, P.-L.; Lakatos, L. Risk for Colorectal Cancer in Ulcerative Colitis: Changes, Causes and Management Strategies. World J. Gastroenterol. 2008, 14, 3937–3947. [Google Scholar] [CrossRef] [Green Version]
- Snover, D.C. Update on the Serrated Pathway to Colorectal Carcinoma. Hum. Pathol. 2011, 42, 1–10. [Google Scholar] [CrossRef]
- Lin, Y.; He, Z.; Ye, J.; Liu, Z.; She, X.; Gao, X.; Liang, R. Progress in Understanding the IL-6/STAT3 Pathway in Colorectal Cancer. Onco Targets Ther. 2020, 13, 13023–13032. [Google Scholar] [CrossRef]
- Zhang, L.; Li, J.; Li, L.; Zhang, J.; Wang, X.; Yang, C.; Li, Y.; Lan, F.; Lin, P. IL-23 Selectively Promotes the Metastasis of Colorectal Carcinoma Cells with Impaired Socs3 Expression via the STAT5 Pathway. Carcinogenesis 2014, 35, 1330–1340. [Google Scholar] [CrossRef]
- Jana, A.; Krett, N.L.; Guzman, G.; Khalid, A.; Ozden, O.; Staudacher, J.J.; Bauer, J.; Baik, S.H.; Carroll, T.; Yazici, C.; et al. NFkB Is Essential for Activin-Induced Colorectal Cancer Migration via Upregulation of PI3K-MDM2 Pathway. Oncotarget 2017, 8, 37377–37393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selaru, F.M.; Xu, Y.; Yin, J.; Zou, T.; Liu, T.C.; Mori, Y.; Abraham, J.M.; Sato, F.; Wang, S.; Twigg, C.; et al. Artificial Neural Networks Distinguish among Subtypes of Neoplastic Colorectal Lesions. Gastroenterology 2002, 122, 606–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.; Scurrah, C.R.; McKinley, E.T.; Simmons, A.J.; Ramirez-Solano, M.A.; Zhu, X.; Markham, N.O.; Heiser, C.N.; Vega, P.N.; Rolong, A.; et al. Differential Pre-Malignant Programs and Microenvironment Chart Distinct Paths to Malignancy in Human Colorectal Polyps. Cell 2021, 184, 6262–6280. [Google Scholar] [CrossRef] [PubMed]
- Sakai, E.; Fukuyo, M.; Ohata, K.; Matsusaka, K.; Doi, N.; Mano, Y.; Takane, K.; Abe, H.; Yagi, K.; Matsuhashi, N.; et al. Genetic and Epigenetic Aberrations Occurring in Colorectal Tumors Associated with Serrated Pathway. Int. J. Cancer 2016, 138, 1634–1644. [Google Scholar] [CrossRef] [Green Version]
- Cooks, T.; Pateras, I.S.; Tarcic, O.; Solomon, H.; Schetter, A.J.; Wilder, S.; Lozano, G.; Pikarsky, E.; Forshew, T.; Rosenfeld, N.; et al. Mutant p53 Prolongs NF-κB Activation and Promotes Chronic Inflammation and Inflammation-Associated Colorectal Cancer. Cancer Cell 2013, 23, 634–646. [Google Scholar] [CrossRef] [Green Version]
- Vuletić, A.; Mirjačić Martinović, K.; Tišma Miletić, N.; Zoidakis, J.; Castellvi-Bel, S.; Čavić, M. Cross-Talk Between Tumor Cells Undergoing Epithelial to Mesenchymal Transition and Natural Killer Cells in Tumor Microenvironment in Colorectal Cancer. Front. Cell Dev. Biol. 2021, 9, 750022. [Google Scholar] [CrossRef]
- Szylberg, Ł.; Janiczek, M.; Popiel, A.; Marszałek, A. Expression of COX-2, IL-1β, TNF-α and IL-4 in Epithelium of Serrated Adenoma, Adenoma and Hyperplastic Polyp. Arch. Med. Sci. 2016, 12, 172–178. [Google Scholar] [CrossRef]
- Marszałek, A.; Szylberg, L.; Wiśniewska, E.; Janiczek, M. Impact of COX-2, IL-1β, TNF-α, IL-4 and IL-10 on the Process of Carcinogenesis in the Large Bowel. Pol. J. Pathol. 2012, 63, 221–227. [Google Scholar] [CrossRef]
- Li, J.; Chen, D.; Shen, M. Tumor Microenvironment Shapes Colorectal Cancer Progression, Metastasis, and Treatment Responses. Front. Med. 2022, 9, 869010. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, J.; Zhang, N.; Chen, M.; Wang, H.; Zhu, D. Targeting the Tumour Immune Microenvironment for Cancer Therapy in Human Gastrointestinal Malignancies. Cancer Lett. 2019, 458, 123–135. [Google Scholar] [CrossRef]
- De Simone, V.; Franzè, E.; Ronchetti, G.; Colantoni, A.; Fantini, M.C.; Di Fusco, D.; Sica, G.S.; Sileri, P.; MacDonald, T.T.; Pallone, F.; et al. Th17-Type Cytokines, IL-6 and TNF-α Synergistically Activate STAT3 and NF-kB to Promote Colorectal Cancer Cell Growth. Oncogene 2015, 34, 3493–3503. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Tan, W.; Wang, C. Tumor-Associated Macrophage-Derived Cytokines Enhance Cancer Stem-like Characteristics through Epithelial-Mesenchymal Transition. Onco Targets Ther. 2018, 11, 3817–3826. [Google Scholar] [CrossRef] [Green Version]
- Ribatti, D.; Tamma, R.; Annese, T. Epithelial-Mesenchymal Transition in Cancer: A Historical Overview. Transl. Oncol. 2020, 13, 100773. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Chen, B.; Zhu, Z.; Ye, W.; Zeng, J.; Liu, G.; Wang, S.; Gao, J.; Xu, G.; Huang, Z. Prognostic Value of ZEB-1 in Solid Tumors: A Meta-Analysis. BMC Cancer 2019, 19, 635. [Google Scholar] [CrossRef] [PubMed]
- Pavlič, A.; Urh, K.; Štajer, K.; Boštjančič, E.; Zidar, N. Epithelial-Mesenchymal Transition in Colorectal Carcinoma: Comparison Between Primary Tumor, Lymph Node and Liver Metastases. Front. Oncol. 2021, 11, 662806. [Google Scholar] [CrossRef]
- Haist, M.; Stege, H.; Grabbe, S.; Bros, M. The Functional Crosstalk between Myeloid-Derived Suppressor Cells and Regulatory T Cells within the Immunosuppressive Tumor Microenvironment. Cancers 2021, 13, 210. [Google Scholar] [CrossRef]
- Ayob, A.Z.; Ramasamy, T.S. Cancer Stem Cells as Key Drivers of Tumour Progression. J. Biomed. Sci. 2018, 25, 20. [Google Scholar] [CrossRef]
- Li, J.; Huang, L.; Zhao, H.; Yan, Y.; Lu, J. The Role of Interleukins in Colorectal Cancer. Int. J. Biol. Sci. 2020, 16, 2323–2339. [Google Scholar] [CrossRef]
- Wei, X.; Zhang, Y.; Yang, Z.; Sha, Y.; Pan, Y.; Chen, Y.; Cai, L. Analysis of the Role of the Interleukins in Colon Cancer. Biol. Res. 2020, 53, 20. [Google Scholar] [CrossRef]
- Chen, J.; Gong, C.; Mao, H.; Li, Z.; Fang, Z.; Chen, Q.; Lin, M.; Jiang, X.; Hu, Y.; Wang, W.; et al. E2F1/SP3/STAT6 Axis Is Required for IL-4-Induced Epithelial-Mesenchymal Transition of Colorectal Cancer Cells. Int. J. Oncol. 2018, 53, 567–578. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Anderson, D.E.; Baecher-Allan, C.; Hastings, W.D.; Bettelli, E.; Oukka, M.; Kuchroo, V.K.; Hafler, D.A. IL-21 and TGF-Beta Are Required for Differentiation of Human T(H)17 Cells. Nature 2008, 454, 350–352. [Google Scholar] [CrossRef] [PubMed]
- Rébé, C.; Ghiringhelli, F. Interleukin-1β and Cancer. Cancers 2020, 12, 1791. [Google Scholar] [CrossRef] [PubMed]
- Kakkar, R.; Lee, R.T. The IL-33/ST2 Pathway: Therapeutic Target and Novel Biomarker. Nat. Rev. Drug Discov. 2008, 7, 827–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelfo, V.; Romaniello, D.; Mazzeschi, M.; Sgarzi, M.; Grilli, G.; Morselli, A.; Manzan, B.; Rihawi, K.; Lauriola, M. Roles of IL-1 in Cancer: From Tumor Progression to Resistance to Targeted Therapies. Int. J. Mol. Sci. 2020, 21, 6009. [Google Scholar] [CrossRef]
- Crucitti, A.; Corbi, M.; Tomaiuolo, P.M.; Fanali, C.; Mazzari, A.; Lucchetti, D.; Migaldi, M.; Sgambato, A. Laparoscopic Surgery for Colorectal Cancer Is Not Associated with an Increase in the Circulating Levels of Several Inflammation-Related Factors. Cancer Biol. Ther. 2015, 16, 671–677. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wang, L.; Pappan, L.; Galliher-Beckley, A.; Shi, J. IL-1β Promotes Stemness and Invasiveness of Colon Cancer Cells through Zeb1 Activation. Mol. Cancer 2012, 11, 87. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Zhang, H.; Xia, J.; Hou, J.; Wang, Y.; Yang, T.; Wang, S.; Zhang, X.; Chen, X.; Wu, X. Interleukin-1β Induces Intercellular Adhesion Molecule-1 Expression, Thus Enhancing the Adhesion between Mesenchymal Stem Cells and Endothelial Progenitor Cells via the p38 MAPK Signaling Pathway. Int. J. Mol. Med. 2018, 41, 1976–1982. [Google Scholar] [CrossRef] [Green Version]
- Ishimoto, T.; Izumi, D.; Sakamoto, Y.; Miyamoto, Y.; Baba, H. Molecular Insights into Colorectal Cancer Stem Cell Regulation by Environmental Factors. J. Cancer Metastasis Treat. 2015, 1, 156–162. [Google Scholar] [CrossRef] [Green Version]
- Duchartre, Y.; Kim, Y.-M.; Kahn, M. The Wnt Signaling Pathway in Cancer. Crit. Rev. Oncol. Hematol. 2016, 99, 141–149. [Google Scholar] [CrossRef] [Green Version]
- Hai Ping, P.; Feng Bo, T.; Li, L.; Nan Hui, Y.; Hong, Z. IL-1β/NF-Kb Signaling Promotes Colorectal Cancer Cell Growth through miR-181a/PTEN Axis. Arch. Biochem. Biophys. 2016, 604, 20–26. [Google Scholar] [CrossRef] [Green Version]
- He, C.; Yu, T.; Shi, Y.; Ma, C.; Yang, W.; Fang, L.; Sun, M.; Wu, W.; Xiao, F.; Guo, F.; et al. MicroRNA 301A Promotes Intestinal Inflammation and Colitis-Associated Cancer Development by Inhibiting BTG1. Gastroenterology 2017, 152, 1434–1448. [Google Scholar] [CrossRef] [PubMed]
- Kaler, P.; Galea, V.; Augenlicht, L.; Klampfer, L. Tumor Associated Macrophages Protect Colon Cancer Cells from TRAIL-Induced Apoptosis through IL-1beta-Dependent Stabilization of Snail in Tumor Cells. PLoS ONE 2010, 5, e11700. [Google Scholar] [CrossRef] [PubMed]
- Mukund, K.; Syulyukina, N.; Ramamoorthy, S.; Subramaniam, S. Right and Left-Sided Colon Cancers—Specificity of Molecular Mechanisms in Tumorigenesis and Progression. BMC Cancer 2020, 20, 317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, W.; Zhang, C.; Wang, X.; Dou, D.; Chen, D.; Li, J. Resolving the Difference between Left-Sided and Right-Sided Colorectal Cancer by Single-Cell Sequencing. JCI Insight 2022, 7, e152616. [Google Scholar] [CrossRef]
- Haabeth, O.A.W.; Lorvik, K.B.; Hammarström, C.; Donaldson, I.M.; Haraldsen, G.; Bogen, B.; Corthay, A. Inflammation Driven by Tumour-Specific Th1 Cells Protects against B-Cell Cancer. Nat. Commun. 2011, 2, 240. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Yang, Z.; Deng, B.; Wu, D.; Quan, Y.; Min, Z. Interleukin 1β/1RA Axis in Colorectal Cancer Regulates Tumor Invasion, Proliferation and Apoptosis via Autophagy. Oncol. Rep. 2020, 43, 908–918. [Google Scholar] [CrossRef] [Green Version]
- Dmitrieva-Posocco, O.; Dzutsev, A.; Posocco, D.F.; Hou, V.; Yuan, W.; Thovarai, V.; Mufazalov, I.A.; Gunzer, M.; Shilovskiy, I.P.; Khaitov, M.R.; et al. Cell-Type-Specific Responses to Interleukin-1 Control Microbial Invasion and Tumor-Elicited Inflammation in Colorectal Cancer. Immunity 2019, 50, 166–180. [Google Scholar] [CrossRef] [Green Version]
- Allen, J.E.; Maizels, R.M. Diversity and Dialogue in Immunity to Helminths. Nat. Rev. Immunol. 2011, 11, 375–388. [Google Scholar] [CrossRef]
- Lloyd, C.M.; Snelgrove, R.J. Type 2 Immunity: Expanding Our View. Sci. Immunol. 2018, 3, eaat1604. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, M.; Okamura, S.; Yamaji, T.; Iwasaki, M.; Tsugane, S.; Shetty, V.; Koizumi, T. Plasma Cytokine Levels and the Presence of Colorectal Cancer. PLoS ONE 2019, 14, e0213602. [Google Scholar] [CrossRef] [Green Version]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, Density, and Location of Immune Cells within Human Colorectal Tumors Predict Clinical Outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanai, T.; Watanabe, M.; Hayashi, A.; Nakazawa, A.; Yajima, T.; Okazawa, A.; Yamazaki, M.; Ishii, H.; Hibi, T. Regulatory Effect of Interleukin-4 and Interleukin-13 on Colon Cancer Cell Adhesion. Br. J. Cancer 2000, 82, 1717–1723. [Google Scholar] [PubMed] [Green Version]
- Kantola, T.; Klintrup, K.; Väyrynen, J.P.; Vornanen, J.; Bloigu, R.; Karhu, T.; Herzig, K.-H.; Näpänkangas, J.; Mäkelä, J.; Karttunen, T.J.; et al. Stage-Dependent Alterations of the Serum Cytokine Pattern in Colorectal Carcinoma. Br. J. Cancer 2012, 107, 1729–1736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laoui, D.; Movahedi, K.; Van Overmeire, E.; Van den Bossche, J.; Schouppe, E.; Mommer, C.; Nikolaou, A.; Morias, Y.; De Baetselier, P.; Van Ginderachter, J.A. Tumor-Associated Macrophages in Breast Cancer: Distinct Subsets, Distinct Functions. Int. J. Dev. Biol. 2011, 55, 861–867. [Google Scholar] [CrossRef] [PubMed]
- Jin, Q.; Yang, H.; Jing, Z.; Hong-Hua, W.; Ben-Jing, S.; Li-Ting, W.; Li-Juan, Y.; Wei, X.; Xia, K.; Juan, W.; et al. IL4/IL4R Signaling Promotes the Osteolysis in Metastatic Bone of CRC through Regulating the Proliferation of Osteoclast Precursors. Mol. Med. 2021, 27, 152. [Google Scholar] [CrossRef]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in Inflammation, Immunity, and Disease. Cold Spring Harb. Perspect. Biol. 2014, 6, a016295. [Google Scholar] [CrossRef]
- Shiga, K.; Hara, M.; Nagasaki, T.; Sato, T.; Takahashi, H.; Sato, M.; Takeyama, H. Preoperative Serum Interleukin-6 Is a Potential Prognostic Factor for Colorectal Cancer, Including Stage II Patients. Gastroenterol. Res. Pract. 2016, 2016, 9701574. [Google Scholar] [CrossRef]
- Comstock, S.S.; Hortos, K.; Kovan, B.; McCaskey, S.; Pathak, D.R.; Fenton, J.I. Adipokines and Obesity Are Associated with Colorectal Polyps in Adult Males: A Cross-Sectional Study. PLoS ONE 2014, 9, e85939. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Liu, S.; Zhou, Y. Circulating Levels of C-Reactive Protein, Interleukin-6 and Tumor Necrosis Factor-α and Risk of Colorectal Adenomas: A Meta-Analysis. Oncotarget 2016, 7, 64371–64379. [Google Scholar] [CrossRef] [Green Version]
- Knüpfer, H.; Preiss, R. Serum Interleukin-6 Levels in Colorectal Cancer Patients—A Summary of Published Results. Int. J. Colorectal Dis. 2010, 25, 135–140. [Google Scholar] [CrossRef]
- Kakourou, A.; Koutsioumpa, C.; Lopez, D.S.; Hoffman-Bolton, J.; Bradwin, G.; Rifai, N.; Helzlsouer, K.J.; Platz, E.A.; Tsilidis, K.K. Interleukin-6 and Risk of Colorectal Cancer: Results from the CLUE II Cohort and a Meta-Analysis of Prospective Studies. Cancer Causes Control 2015, 26, 1449–1460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vainer, N.; Dehlendorff, C.; Johansen, J.S. Systematic Literature Review of IL-6 as a Biomarker or Treatment Target in Patients with Gastric, Bile Duct, Pancreatic and Colorectal Cancer. Oncotarget 2018, 9, 29820–29841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Q.; Shang, Y.; Sun, F.; Dong, X.; Niu, J.; Li, F. Interleukin-6 Promotes Epithelial-Mesenchymal Transition and Cell Invasion through Integrin β6 Upregulation in Colorectal Cancer. Oxidative Med. Cell. Longev. 2020, 2020, 8032187. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Zou, X.; Xia, W.; Gao, H.; Li, Z.; Liu, N.; Xu, Z.; Gao, C.; He, Z.; Niu, W.; et al. Integrin αvβ6 Plays a Bi-Directional Regulation Role between Colon Cancer Cells and Cancer-Associated Fibroblasts. Biosci. Rep. 2018, 38, BSR20180243. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Liu, L.; Lai, W.; Zeng, Y.; Xu, H.; Lan, Q.; Su, P.; Chu, Z. Interaction with Tumor-associated Macrophages Promotes PRL-3-induced Invasion of Colorectal Cancer Cells via MAPK Pathway-induced EMT and NF-κB Signaling-induced Angiogenesis. Oncol. Rep. 2019, 41, 2790–2802. [Google Scholar] [CrossRef]
- Franzè, E.; Marafini, I.; Troncone, E.; Salvatori, S.; Monteleone, G. Interleukin-34 Promotes Tumorigenic Signals for Colon Cancer Cells. Cell Death Discov. 2021, 7, 245. [Google Scholar] [CrossRef]
- Zhang, X.; Hu, F.; Li, G.; Li, G.; Yang, X.; Liu, L.; Zhang, R.; Zhang, B.; Feng, Y. Human Colorectal Cancer-Derived Mesenchymal Stem Cells Promote Colorectal Cancer Progression through IL-6/JAK2/STAT3 Signaling. Cell Death Dis. 2018, 9, 25. [Google Scholar] [CrossRef]
- Zhang, S.; Li, J.; Xie, P.; Zang, T.; Shen, H.; Cao, G.; Zhu, Y.; Yue, Z.; Li, Z. STAT3/c-Myc Axis-Mediated Metabolism Alternations of Inflammation-Related Glycolysis Involve with Colorectal Carcinogenesis. Rejuvenation Res. 2019, 22, 138–145. [Google Scholar] [CrossRef]
- Li, B.; Huang, C. Regulation of EMT by STAT3 in Gastrointestinal Cancer (Review). Int. J. Oncol. 2017, 50, 753–767. [Google Scholar] [CrossRef] [Green Version]
- Hendrayani, S.-F.; Al-Harbi, B.; Al-Ansari, M.M.; Silva, G.; Aboussekhra, A. The Inflammatory/cancer-Related IL-6/STAT3/NF-κB Positive Feedback Loop Includes AUF1 and Maintains the Active State of Breast Myofibroblasts. Oncotarget 2016, 7, 41974–41985. [Google Scholar] [CrossRef] [Green Version]
- Hirano, T. IL-6 in Inflammation, Autoimmunity and Cancer. Int. Immunol. 2021, 33, 127–148. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Rajput, A.; Jin, N.; Wang, J. Mechanisms of Immunosuppression in Colorectal Cancer. Cancers 2020, 12, 3850. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xu, K.; Wu, J.; Luo, C.; Li, Y.; Wu, X.; Gao, H.; Feng, G.; Yuan, B.-Z. The Changes of Th17 Cells and the Related Cytokines in the Progression of Human Colorectal Cancers. BMC Cancer 2012, 12, 418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velikova, T.V.; Miteva, L.; Stanilov, N.; Spassova, Z.; Stanilova, S.A. Interleukin-6 Compared to the Other Th17/Treg Related Cytokines in Inflammatory Bowel Disease and Colorectal Cancer. World J. Gastroenterol. 2020, 26, 1912–1925. [Google Scholar] [CrossRef]
- Dienz, O.; Rincon, M. The Effects of IL-6 on CD4 T Cell Responses. Clin. Immunol. 2009, 130, 27–33. [Google Scholar] [CrossRef] [Green Version]
- Nagasaki, T.; Hara, M.; Nakanishi, H.; Takahashi, H.; Sato, M.; Takeyama, H. Interleukin-6 Released by Colon Cancer-Associated Fibroblasts Is Critical for Tumour Angiogenesis: Anti-Interleukin-6 Receptor Antibody Suppressed Angiogenesis and Inhibited Tumour-Stroma Interaction. Br. J. Cancer 2014, 110, 469–478. [Google Scholar] [CrossRef] [Green Version]
- Tseng-Rogenski, S.S.; Hamaya, Y.; Choi, D.Y.; Carethers, J.M. Interleukin 6 Alters Localization of hMSH3, Leading to DNA Mismatch Repair Defects in Colorectal Cancer Cells. Gastroenterology 2015, 148, 579–589. [Google Scholar] [CrossRef] [Green Version]
- Tsukamoto, H.; Nishikata, R.; Senju, S.; Nishimura, Y. Myeloid-Derived Suppressor Cells Attenuate TH1 Development through IL-6 Production to Promote Tumor Progression. Cancer Immunol. Res. 2013, 1, 64–76. [Google Scholar] [CrossRef] [Green Version]
- Morre, M.; Beq, S. Interleukin-7 and Immune Reconstitution in Cancer Patients: A New Paradigm for Dramatically Increasing Overall Survival. Target. Oncol. 2012, 7, 55–68. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Zhao, L.; Wan, Y.Y.; Zhu, B. Mechanism of Action of IL-7 and Its Potential Applications and Limitations in Cancer Immunotherapy. Int. J. Mol. Sci. 2015, 16, 10267–10280. [Google Scholar] [CrossRef] [Green Version]
- Bednarz-Misa, I.; Diakowska, D.; Krzystek-Korpacka, M. Local and Systemic IL-7 Concentration in Gastrointestinal-Tract Cancers. Medicina 2019, 55, 262. [Google Scholar] [CrossRef] [Green Version]
- Najdaghi, S.; Razi, S.; Rezaei, N. An Overview of the Role of Interleukin-8 in Colorectal Cancer. Cytokine 2020, 135, 155205. [Google Scholar] [CrossRef]
- Brennan, K.; Zheng, J. Interleukin 8. In xPharm: The Comprehensive Pharmacology Reference; Enna, S.J., Bylund, D.B., Eds.; Elsevier: New York, NY, USA, 2007; pp. 1–4. ISBN 9780080552323. [Google Scholar]
- Long, X.; Ye, Y.; Zhang, L.; Liu, P.; Yu, W.; Wei, F.; Ren, X.; Yu, J. IL-8, a Novel Messenger to Cross-Link Inflammation and Tumor EMT via Autocrine and Paracrine Pathways (Review). Int. J. Oncol. 2016, 48, 5–12. [Google Scholar] [CrossRef] [Green Version]
- O’Hara, A.M.; Bhattacharyya, A.; Bai, J.; Mifflin, R.C.; Ernst, P.B.; Mitra, S.; Crowe, S.E. Tumor Necrosis Factor (TNF)-Alpha-Induced IL-8 Expression in Gastric Epithelial Cells: Role of Reactive Oxygen Species and AP Endonuclease-1/redox Factor (Ref)-1. Cytokine 2009, 46, 359–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karar, J.; Maity, A. PI3K/AKT/mTOR Pathway in Angiogenesis. Front. Mol. Neurosci. 2011, 4, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Wang, S.; Lin, Y.; Miao, Y.; Zeng, Y.; Nie, Y.; Guo, P.; Jiang, G.; Wu, J. Epithelial-Mesenchymal Transition in Cancer: Role of the IL-8/IL-8R Axis. Oncol. Lett. 2017, 13, 4577–4584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, L.; Law, B.K.; Chytil, A.M.; Brown, K.A.; Aakre, M.E.; Moses, H.L. Activation of the Erk Pathway Is Required for TGF-beta1-Induced EMT in Vitro. Neoplasia 2004, 6, 603–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, X.-T.; Dai, Z.; Song, K.; Zhang, Z.-J.; Zhou, Z.-J.; Zhou, S.-L.; Zhao, Y.-M.; Xiao, Y.-S.; Sun, Q.-M.; Ding, Z.-B.; et al. Macrophage-Secreted IL-8 Induces Epithelial-Mesenchymal Transition in Hepatocellular Carcinoma Cells by Activating the JAK2/STAT3/Snail Pathway. Int. J. Oncol. 2015, 46, 587–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, J.M.; Dominguez, C.; Hamilton, D.H.; Palena, C. The IL-8/IL-8R Axis: A Double Agent in Tumor Immune Resistance. Vaccines 2016, 4, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, L.; Jiang, N.; Zeng, J.; Wang, Y.; Cui, H. The Versatile Roles of Cancer-Associated Fibroblasts in Colorectal Cancer and Therapeutic Implications. Front. Cell Dev. Biol. 2021, 9, 733270. [Google Scholar] [CrossRef]
- Xia, W.; Chen, W.; Zhang, Z.; Wu, D.; Wu, P.; Chen, Z.; Li, C.; Huang, J. Prognostic Value, Clinicopathologic Features and Diagnostic Accuracy of Interleukin-8 in Colorectal Cancer: A Meta-Analysis. PLoS ONE 2015, 10, e0123484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conciatori, F.; Bazzichetto, C.; Sperduti, I.; Ciuffreda, L.; Falcone, I.; Bria, E.; Cognetti, F.; Milella, M. P-267 Interleukin-8 Levels as a Predictor of Colorectal Cancer Patient Prognosis. Ann. Oncol. 2021, 32, S188–S189. [Google Scholar] [CrossRef]
- Kumara, H.M.C.S.; Sutton, E.; Bellini, G.A.; Yan, X.; Cekic, V.; Gandhi, N.D.; Whelan, R.L. Plasma Interleukin-8 Levels Are Persistently Elevated for 1 Month after Minimally Invasive Colorectal Resection for Colorectal Cancer. Mol. Clin. Oncol. 2018, 8, 471–476. [Google Scholar]
- Chang, C.-M.; Lam, H.Y.P.; Hsu, H.-J.; Jiang, S.-J. Interleukin-10: A Double-Edged Sword in Breast Cancer. Tzu-Chi Med. J. 2021, 33, 203–211. [Google Scholar]
- Crawley, J.B.; Williams, L.M.; Mander, T.; Brennan, F.M.; Foxwell, B.M. Interleukin-10 Stimulation of Phosphatidylinositol 3-Kinase and p70 S6 Kinase Is Required for the Proliferative but Not the Antiinflammatory Effects of the Cytokine. J. Biol. Chem. 1996, 271, 16357–16362. [Google Scholar] [CrossRef] [Green Version]
- Hamidullah; Changkija, B.; Konwar, R. Role of Interleukin-10 in Breast Cancer. Breast Cancer Res. Treat. 2012, 133, 11–21. [Google Scholar] [CrossRef]
- Antoniv, T.T.; Ivashkiv, L.B. Interleukin-10-Induced Gene Expression and Suppressive Function Are Selectively Modulated by the PI3K-Akt-GSK3 Pathway. Immunology 2011, 132, 567–577. [Google Scholar] [CrossRef]
- Abtahi, S.; Davani, F.; Mojtahedi, Z.; Hosseini, S.V.; Bananzadeh, A.; Ghaderi, A. Dual Association of Serum Interleukin-10 Levels with Colorectal Cancer. J. Cancer Res. Ther. 2017, 13, 252–256. [Google Scholar]
- Cacev, T.; Radosević, S.; Krizanac, S.; Kapitanović, S. Influence of Interleukin-8 and Interleukin-10 on Sporadic Colon Cancer Development and Progression. Carcinogenesis 2008, 29, 1572–1580. [Google Scholar] [CrossRef] [Green Version]
- Stanilov, N.; Miteva, L.; Deliysky, T.; Jovchev, J.; Stanilova, S. Advanced Colorectal Cancer Is Associated with Enhanced IL-23 and IL-10 Serum Levels. Lab. Med. 2010, 41, 159–163. [Google Scholar] [CrossRef]
- Miteva, L.D.; Stanilov, N.S.; Deliysky, T.S.; Stanilova, S.A. Significance of −1082A/G Polymorphism of IL10 Gene for Progression of Colorectal Cancer and IL-10 Expression. Tumor Biol. 2014, 35, 12655–12664. [Google Scholar] [CrossRef] [PubMed]
- Townsend, M.H.; Felsted, A.M.; Piccolo, S.R.; Robison, R.A.; O’Neill, K.L. Metastatic Colon Adenocarcinoma Has a Significantly Elevated Expression of IL-10 Compared with Primary Colon Adenocarcinoma Tumors. Cancer Biol. Ther. 2018, 19, 913–920. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Wang, F.; Ma, C.; Hao, T.; Geng, L.; Jiang, H. Predictive Value of IL-18 and IL-10 in the Prognosis of Patients with Colorectal Cancer. Oncol. Lett. 2019, 18, 713–719. [Google Scholar] [CrossRef] [Green Version]
- Galizia, G.; Orditura, M.; Romano, C.; Lieto, E.; Castellano, P.; Pelosio, L.; Imperatore, V.; Catalano, G.; Pignatelli, C.; De Vita, F. Prognostic Significance of Circulating IL-10 and IL-6 Serum Levels in Colon Cancer Patients Undergoing Surgery. Clin. Immunol. 2002, 102, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.-Y.; Jeon, B.G.; Kim, J.-E.; Shin, R.; Ahn, H.S.; Jin, H.; Heo, S.C. Interleukin 10 Level in the Peritoneal Cavity Is a Prognostic Marker for Peritoneal Recurrence of T4 Colorectal Cancer. Sci. Rep. 2021, 11, 9212. [Google Scholar] [CrossRef]
- Gounaris, E.; Blatner, N.R.; Dennis, K.; Magnusson, F.; Gurish, M.F.; Strom, T.B.; Beckhove, P.; Gounari, F.; Khazaie, K. T-Regulatory Cells Shift from a Protective Anti-Inflammatory to a Cancer-Promoting Proinflammatory Phenotype in Polyposis. Cancer Res. 2009, 69, 5490–5497. [Google Scholar] [CrossRef] [Green Version]
- Krause, P.; Morris, V.; Greenbaum, J.A.; Park, Y.; Bjoerheden, U.; Mikulski, Z.; Muffley, T.; Shui, J.-W.; Kim, G.; Cheroutre, H.; et al. IL-10-Producing Intestinal Macrophages Prevent Excessive Antibacterial Innate Immunity by Limiting IL-23 Synthesis. Nat. Commun. 2015, 6, 7055. [Google Scholar] [CrossRef]
- Xu, D.H.; Zhu, Z.; Wakefield, M.R.; Xiao, H.; Bai, Q.; Fang, Y. The Role of IL-11 in Immunity and Cancer. Cancer Lett. 2016, 373, 156–163. [Google Scholar] [CrossRef]
- Putoczki, T.L.; Thiem, S.; Loving, A.; Busuttil, R.A.; Wilson, N.J.; Ziegler, P.K.; Nguyen, P.M.; Preaudet, A.; Farid, R.; Edwards, K.M.; et al. Interleukin-11 Is the Dominant IL-6 Family Cytokine during Gastrointestinal Tumorigenesis and Can Be Targeted Therapeutically. Cancer Cell 2013, 24, 257–271. [Google Scholar] [CrossRef] [Green Version]
- Nishina, T.; Deguchi, Y.; Ohshima, D.; Takeda, W.; Ohtsuka, M.; Shichino, S.; Ueha, S.; Yamazaki, S.; Kawauchi, M.; Nakamura, E.; et al. Interleukin-11-Expressing Fibroblasts Have a Unique Gene Signature Correlated with Poor Prognosis of Colorectal Cancer. Nat. Commun. 2021, 12, 2281. [Google Scholar] [CrossRef]
- Wei, J.; Ma, L.; Lai, Y.-H.; Zhang, R.; Li, H.; Li, C.; Lin, J. Bazedoxifene as a Novel GP130 Inhibitor for Colon Cancer Therapy. J. Exp. Clin. Cancer Res. 2019, 38, 63. [Google Scholar] [CrossRef] [PubMed]
- Mager, L.F.; Wasmer, M.-H.; Rau, T.T.; Krebs, P. Cytokine-Induced Modulation of Colorectal Cancer. Front. Oncol. 2016, 6, 96. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, D.H.; Yang, X.; Sun, Y.; Yang, C.S. Colitis-Induced IL11 Promotes Colon Carcinogenesis. Carcinogenesis 2021, 42, 557–569. [Google Scholar] [CrossRef]
- Awasthi, A.; Murugaiyan, G.; Kuchroo, V.K. Interplay between Effector Th17 and Regulatory T Cells. J. Clin. Immunol. 2008, 28, 660–670. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Xu, J.; Su, H.; Zhong, W.; Yuan, Y.; Yu, Z.; Fang, Y.; Zhou, H.; Li, C.; Huang, K. Interleukin-17 Is a Favorable Prognostic Marker for Colorectal Cancer. Clin. Transl. Oncol. 2015, 17, 50–56. [Google Scholar] [CrossRef]
- Tseng, J.-Y.; Yang, C.-Y.; Liang, S.-C.; Liu, R.-S.; Yang, S.-H.; Lin, J.-K.; Chen, Y.-M.; Wu, Y.-C.; Jiang, J.-K.; Lin, C.-H. Interleukin-17A Modulates Circulating Tumor Cells in Tumor Draining Vein of Colorectal Cancers and Affects Metastases. Clin. Cancer Res. 2014, 20, 2885–2897. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Zhao, J.; Bulek, K.; Tang, F.; Chen, X.; Cai, G.; Jia, S.; Fox, P.L.; Huang, E.; Pizarro, T.T.; et al. Inflammation Mobilizes Copper Metabolism to Promote Colon Tumorigenesis via an IL-17-STEAP4-XIAP Axis. Nat. Commun. 2020, 11, 900. [Google Scholar] [CrossRef] [Green Version]
- Pan, B.; Shen, J.; Cao, J.; Zhou, Y.; Shang, L.; Jin, S.; Cao, S.; Che, D.; Liu, F.; Yu, Y. Interleukin-17 Promotes Angiogenesis by Stimulating VEGF Production of Cancer Cells via the STAT3/GIV Signaling Pathway in Non-Small-Cell Lung Cancer. Sci. Rep. 2015, 5, 16053. [Google Scholar] [CrossRef] [Green Version]
- Gaffen, S.L. Structure and Signalling in the IL-17 Receptor Family. Nat. Rev. Immunol. 2009, 9, 556–567. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Koyama, Y.; Sanuki, R.; Mitsui, N.; Suzuki, N.; Kimura, A.; Nakajima, A.; Shimizu, N.; Maeno, M. IL-17A Stimulates the Expression of Inflammatory Cytokines via Celecoxib-Blocked Prostaglandin in MC3T3-E1 Cells. Arch. Oral Biol. 2010, 55, 679–688. [Google Scholar] [CrossRef]
- Yang, X.O.; Chang, S.H.; Park, H.; Nurieva, R.; Shah, B.; Acero, L.; Wang, Y.-H.; Schluns, K.S.; Broaddus, R.R.; Zhu, Z.; et al. Regulation of Inflammatory Responses by IL-17F. J. Exp. Med. 2008, 205, 1063–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, G.; Li, Z.; Florholmen, J.; Goll, R. Dynamic Stromal Cellular Reaction throughout Human Colorectal Adenoma-Carcinoma Sequence: A Role of TH17/IL-17A. Biomed. Pharmacother. 2021, 140, 111761. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Wu, P.; Huang, Q.; Liu, Y.; Ye, J.; Huang, J. Interleukin-17: A Promoter in Colorectal Cancer Progression. Clin. Dev. Immunol. 2013, 2013, 436307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lücke, J.; Shiri, A.M.; Zhang, T.; Kempski, J.; Giannou, A.D.; Huber, S. Rationalizing Heptadecaphobia: TH 17 Cells and Associated Cytokines in Cancer and Metastasis. FEBS J. 2021, 288, 6942–6971. [Google Scholar] [CrossRef] [PubMed]
- Thomson, A.; Bento, D.F.C.; Scurr, M.J.; Smart, K.; Somerville, M.S.; Keita, Å.V.; Gallimore, A.; Godkin, A. Prognostic Significance of Interleukin-17A-Producing Colorectal Tumour Antigen-Specific T Cells. Br. J. Cancer 2021, 124, 1552–1555. [Google Scholar] [CrossRef]
- Tong, Z.; Yang, X.O.; Yan, H.; Liu, W.; Niu, X.; Shi, Y.; Fang, W.; Xiong, B.; Wan, Y.; Dong, C. A Protective Role by Interleukin-17F in Colon Tumorigenesis. PLoS ONE 2012, 7, e34959. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Sun, X.; Zhao, X.; An, L.; Wang, Z.; Jiang, J.; Shen, W.; Yang, X.; Sun, Y. Expression and Location of IL-17A, E, F and Their Receptors in Colorectal Adenocarcinoma: Comparison with Benign Intestinal Disease. Pathol. Res. Pract. 2018, 214, 482–491. [Google Scholar] [CrossRef] [Green Version]
- Omrane, I.; Medimegh, I.; Baroudi, O.; Ayari, H.; Bedhiafi, W.; Stambouli, N.; Ferchichi, M.; Kourda, N.; Bignon, Y.-J.; Uhrhammer, N.; et al. Involvement of IL17A, IL17F and IL23R Polymorphisms in Colorectal Cancer Therapy. PLoS ONE 2015, 10, e0128911. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Yang, Z.; Wu, D.; Min, Z.; Quan, Y. Upregulation of interleukin-17F in Colorectal Cancer Promotes Tumor Invasion by Inducing Epithelial-mesenchymal Transition. Oncol. Rep. 2019, 42, 1141–1148. [Google Scholar] [CrossRef]
- Leonard, W.J.; Wan, C.-K. IL-21 Signaling in Immunity. F1000Research 2016, 5. [Google Scholar] [CrossRef]
- Stolfi, C.; Pallone, F.; Macdonald, T.T.; Monteleone, G. Interleukin-21 in Cancer Immunotherapy: Friend or Foe? Oncoimmunology 2012, 1, 351–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Jiang, X.; Zhu, J.; Yue, D.; Zhang, X.; Wang, X.; You, Y.; Wang, B.; Xu, Y.; Lu, C.; et al. IL-21/IL-21R Signaling Suppresses Intestinal Inflammation Induced by DSS through Regulation of Th Responses in Lamina Propria in Mice. Sci. Rep. 2016, 6, 31881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araki, A.; Jin, L.; Nara, H.; Takeda, Y.; Nemoto, N.; Gazi, M.Y.; Asao, H. IL-21 Enhances the Development of Colitis-Associated Colon Cancer: Possible Involvement of Activation-Induced Cytidine Deaminase Expression. J. Immunol. 2019, 202, 3326–3333. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Liu, X.; Ren, Y. Interleukin 21 Treatment in a Murine Model as a Novel Potential Cytokine Immunotherapy for Colon Cancer. Adv. Clin. Exp. Med. 2018, 27, 583–589. [Google Scholar] [CrossRef]
- Kim-Schulze, S.; Kim, H.S.; Fan, Q.; Kim, D.W.; Kaufman, H.L. Local IL-21 Promotes the Therapeutic Activity of Effector T Cells by Decreasing Regulatory T Cells within the Tumor Microenvironment. Mol. Ther. 2009, 17, 380–388. [Google Scholar] [CrossRef]
- Deng, S.; Sun, Z.; Qiao, J.; Liang, Y.; Liu, L.; Dong, C.; Shen, A.; Wang, Y.; Tang, H.; Fu, Y.-X.; et al. Targeting Tumors with IL-21 Reshapes the Tumor Microenvironment by Proliferating PD-1intTim-3-CD8+ T Cells. JCI Insight 2020, 5, e132000. [Google Scholar] [CrossRef] [Green Version]
- Shi, W.; Dong, L.; Sun, Q.; Ding, H.; Meng, J.; Dai, G. Follicular Helper T Cells Promote the Effector Functions of CD8+ T Cells via the Provision of IL-21, Which Is Downregulated due to PD-1/PD-L1-Mediated Suppression in Colorectal Cancer. Exp. Cell Res. 2018, 372, 35–42. [Google Scholar] [CrossRef]
- Wu, S.; Sun, R.; Tan, B.; Chen, B.; Zhou, W.; Gao, D.S.; Zhong, J.; Huang, H.; Jiang, J.; Lu, B. The Half-Life-Extended IL21 Can Be Combined With Multiple Checkpoint Inhibitors for Tumor Immunotherapy. Front. Cell Dev. Biol. 2021, 9, 779865. [Google Scholar] [CrossRef]
- Yang, Y.; Lv, X.; Zhan, L.; Chen, L.; Jin, H.; Tang, X.; Shi, Q.; Zou, Q.; Xiang, J.; Zhang, W.; et al. Case Report: IL-21 and Bcl-6 Regulate the Proliferation and Secretion of Tfh and Tfr Cells in the Intestinal Germinal Center of Patients with Inflammatory Bowel Disease. Front. Pharmacol. 2020, 11, 587445. [Google Scholar] [CrossRef]
- Cui, G.; Yuan, A.; Zhu, L.; Florholmen, J.; Goll, R. Increased Expression of Interleukin-21 along Colorectal Adenoma-Carcinoma Sequence and Its Predicating Significance in Patients with Sporadic Colorectal Cancer. Clin. Immunol. 2017, 183, 266–272. [Google Scholar] [CrossRef]
- De Simone, V.; Ronchetti, G.; Franzè, E.; Colantoni, A.; Ortenzi, A.; Fantini, M.C.; Rizzo, A.; Sica, G.S.; Sileri, P.; Rossi, P.; et al. Interleukin-21 Sustains Inflammatory Signals That Contribute to Sporadic Colon Tumorigenesis. Oncotarget 2015, 6, 9908–9923. [Google Scholar] [CrossRef] [PubMed]
- Kesselring, R.; Jauch, D.; Fichtner-Feigl, S. Interleukin 21 Impairs Tumor Immunosurveillance of Colitis-Associated Colorectal Cancer. Oncoimmunology 2012, 1, 537–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirchberger, S.; Royston, D.J.; Boulard, O.; Thornton, E.; Franchini, F.; Szabady, R.L.; Harrison, O.; Powrie, F. Innate Lymphoid Cells Sustain Colon Cancer through Production of Interleukin-22 in a Mouse Model. J. Exp. Med. 2013, 210, 917–931. [Google Scholar] [CrossRef] [PubMed]
- Khare, V.; Paul, G.; Movadat, O.; Frick, A.; Jambrich, M.; Krnjic, A.; Marian, B.; Wrba, F.; Gasche, C. IL10R2 Overexpression Promotes IL22/STAT3 Signaling in Colorectal Carcinogenesis. Cancer Immunol. Res. 2015, 3, 1227–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kryczek, I.; Lin, Y.; Nagarsheth, N.; Peng, D.; Zhao, L.; Zhao, E.; Vatan, L.; Szeliga, W.; Dou, Y.; Owens, S.; et al. IL-22+CD4+ T Cells Promote Colorectal Cancer Stemness via STAT3 Transcription Factor Activation and Induction of the Methyltransferase DOT1L. Immunity 2014, 40, 772–784. [Google Scholar] [CrossRef] [Green Version]
- Moniruzzaman, M.; Wang, R.; Jeet, V.; McGuckin, M.A.; Hasnain, S.Z. Interleukin (IL)-22 from IL-20 Subfamily of Cytokines Induces Colonic Epithelial Cell Proliferation Predominantly through ERK1/2 Pathway. Int. J. Mol. Sci. 2019, 20, 3468. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Wang, Z.; Liu, Y.; Mei, Z.; Wang, G.; Liang, Z.; Cui, A.; Hu, X.; Cui, L.; Yang, Y.; et al. Interleukin 22 Protects Colorectal Cancer Cells from Chemotherapy by Activating the STAT3 Pathway and Inducing Autocrine Expression of Interleukin 8. Clin. Immunol. 2014, 154, 116–126. [Google Scholar] [CrossRef]
- Wang, C.; Gong, G.; Sheh, A.; Muthupalani, S.; Bryant, E.M.; Puglisi, D.A.; Holcombe, H.; Conaway, E.A.; Parry, N.A.P.; Bakthavatchalu, V.; et al. Interleukin-22 Drives Nitric Oxide-Dependent DNA Damage and Dysplasia in a Murine Model of Colitis-Associated Cancer. Mucosal Immunol. 2017, 10, 1504–1517. [Google Scholar] [CrossRef] [Green Version]
- Ziesché, E.; Bachmann, M.; Kleinert, H.; Pfeilschifter, J.; Mühl, H. The Interleukin-22/STAT3 Pathway Potentiates Expression of Inducible Nitric-Oxide Synthase in Human Colon Carcinoma Cells. J. Biol. Chem. 2007, 282, 16006–16015. [Google Scholar] [CrossRef] [Green Version]
- Kvedaraite, E.; Lourda, M.; Ideström, M.; Chen, P.; Olsson-Åkefeldt, S.; Forkel, M.; Gavhed, D.; Lindforss, U.; Mjösberg, J.; Henter, J.-I.; et al. Tissue-Infiltrating Neutrophils Represent the Main Source of IL-23 in the Colon of Patients with IBD. Gut 2016, 65, 1632–1641. [Google Scholar] [CrossRef]
- Neurath, M.F. IL-23 in Inflammatory Bowel Diseases and Colon Cancer. Cytokine Growth Factor Rev. 2019, 45, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.S.; Yong, Z.W.E.; Wang, H.; Tan, T.Z.; Huang, R.Y.-J.; Yamamoto, D.; Inaki, N.; Hazawa, M.; Wong, R.W.; Oshima, H.; et al. Inflammatory and Mitogenic Signals Drive Interleukin 23 Subunit Alpha (IL23A) Secretion Independent of IL12B in Intestinal Epithelial Cells. J. Biol. Chem. 2020, 295, 6387–6400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiering, C.; Krausgruber, T.; Chomka, A.; Fröhlich, A.; Adelmann, K.; Wohlfert, E.A.; Pott, J.; Griseri, T.; Bollrath, J.; Hegazy, A.N.; et al. The Alarmin IL-33 Promotes Regulatory T-Cell Function in the Intestine. Nature 2014, 513, 564–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elessawi, D.F.; Alkady, M.M.; Ibrahim, I.M. Diagnostic and Prognostic Value of Serum IL-23 in Colorectal Cancer. Arab J. Gastroenterol. 2019, 20, 65–68. [Google Scholar] [CrossRef]
- Adamo, V.; Franchina, T.; Minciullo, P.L.; Pace, E.; Colonese, F.; Ricciardi, G.R.R.; Saitta, S.; Ferraro, M.; Spatari, G.; Gangemi, S. Role of Interleukin-23 Circulating Levels Increase in Resected Colorectal Cancer before and after Chemotherapy: Preliminary Data and Future Perspectives. J. Cell. Physiol. 2011, 226, 3032–3034. [Google Scholar] [CrossRef] [PubMed]
- Liew, F.Y.; Pitman, N.I.; McInnes, I.B. Disease-Associated Functions of IL-33: The New Kid in the IL-1 Family. Nat. Rev. Immunol. 2010, 10, 103–110. [Google Scholar] [CrossRef]
- Maywald, R.L.; Doerner, S.K.; Pastorelli, L.; De Salvo, C.; Benton, S.M.; Dawson, E.P.; Lanza, D.G.; Berger, N.A.; Markowitz, S.D.; Lenz, H.-J.; et al. IL-33 Activates Tumor Stroma to Promote Intestinal Polyposis. Proc. Natl. Acad. Sci. USA. 2015, 112, E2487–E2496. [Google Scholar] [CrossRef] [Green Version]
- Mertz, K.D.; Mager, L.F.; Wasmer, M.-H.; Thiesler, T.; Koelzer, V.H.; Ruzzante, G.; Joller, S.; Murdoch, J.R.; Brümmendorf, T.; Genitsch, V.; et al. The IL-33/ST2 Pathway Contributes to Intestinal Tumorigenesis in Humans and Mice. Oncoimmunology 2016, 5, e1062966. [Google Scholar] [CrossRef] [Green Version]
- Cui, G.; Qi, H.; Gundersen, M.D.; Yang, H.; Christiansen, I.; Sørbye, S.W.; Goll, R.; Florholmen, J. Dynamics of the IL-33/ST2 Network in the Progression of Human Colorectal Adenoma to Sporadic Colorectal Cancer. Cancer Immunol. Immunother. 2015, 64, 181–190. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, C.; Mahmoud, A.; Keane, J.; Murphy, C.; White, D.; Carey, S.; O’Riordain, M.; Bennett, M.W.; Brint, E.; Houston, A. An Antitumorigenic Role for the IL-33 Receptor, ST2L, in Colon Cancer. Br. J. Cancer 2016, 114, 37–43. [Google Scholar] [CrossRef] [Green Version]
- Eissmann, M.F.; Dijkstra, C.; Wouters, M.A.; Baloyan, D.; Mouradov, D.; Nguyen, P.M.; Davalos-Salas, M.; Putoczki, T.L.; Sieber, O.M.; Mariadason, J.M.; et al. Interleukin 33 Signaling Restrains Sporadic Colon Cancer in an Interferon-γ-Dependent Manner. Cancer Immunol. Res. 2018, 6, 409–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gocher, A.M.; Workman, C.J.; Vignali, D.A.A. Interferon-γ: Teammate or Opponent in the Tumour Microenvironment? Nat. Rev. Immunol. 2022, 22, 158–172. [Google Scholar] [CrossRef] [PubMed]
- Castro, F.; Cardoso, A.P.; Gonçalves, R.M.; Serre, K.; Oliveira, M.J. Interferon-Gamma at the Crossroads of Tumor Immune Surveillance or Evasion. Front. Immunol. 2018, 9, 847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slattery, M.L.; Lundgreen, A.; Bondurant, K.L.; Wolff, R.K. Interferon-Signaling Pathway: Associations with Colon and Rectal Cancer Risk and Subsequent Survival. Carcinogenesis 2011, 32, 1660–1667. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Wang, Y.; Song, Z.; Chu, J.; Qu, X. Deficiency of Interferon-Gamma or Its Receptor Promotes Colorectal Cancer Development. J. Interferon Cytokine Res. 2015, 35, 273–280. [Google Scholar] [CrossRef]
- Bernabei, P.; Coccia, E.M.; Rigamonti, L.; Bosticardo, M.; Forni, G.; Pestka, S.; Krause, C.D.; Battistini, A.; Novelli, F. Interferon-Gamma Receptor 2 Expression as the Deciding Factor in Human T, B, and Myeloid Cell Proliferation or Death. J. Leukoc. Biol. 2001, 70, 950–960. [Google Scholar] [PubMed]
- Pernis, A.; Gupta, S.; Gollob, K.J.; Garfein, E.; Coffman, R.L.; Schindler, C.; Rothman, P. Lack of Interferon Gamma Receptor Beta Chain and the Prevention of Interferon Gamma Signaling in TH1 Cells. Science 1995, 269, 245–247. [Google Scholar] [CrossRef]
- Bach, E.A.; Szabo, S.J.; Dighe, A.S.; Ashkenazi, A.; Aguet, M.; Murphy, K.M.; Schreiber, R.D. Ligand-Induced Autoregulation of IFN-Gamma Receptor Beta Chain Expression in T Helper Cell Subsets. Science 1995, 270, 1215–1218. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.-F.; Huang, Y.-X.; Wang, H.-Y.; Zhang, Y.; Bao, Y.-L.; Sun, L.-G.; Wu, Y.; Yu, C.-L.; Song, Z.-B.; Zheng, L.-H.; et al. Elucidating the Crosstalk Mechanism between IFN-Gamma and IL-6 via Mathematical Modelling. BMC Bioinform. 2013, 14, 41. [Google Scholar] [CrossRef] [Green Version]
- Messina, N.L.; Banks, K.M.; Vidacs, E.; Martin, B.P.; Long, F.; Christiansen, A.J.; Smyth, M.J.; Clarke, C.J.P.; Johnstone, R.W. Modulation of Antitumour Immune Responses by Intratumoural Stat1 Expression. Immunol. Cell Biol. 2013, 91, 556–567. [Google Scholar] [CrossRef]
- Ni, C.; Wu, P.; Zhu, X.; Ye, J.; Zhang, Z.; Chen, Z.; Zhang, T.; Zhang, T.; Wang, K.; Wu, D.; et al. IFN-γ Selectively Exerts pro-Apoptotic Effects on Tumor-Initiating Label-Retaining Colon Cancer Cells. Cancer Lett. 2013, 336, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Ivashkiv, L.B. Cross-Regulation of Signaling Pathways by Interferon-Gamma: Implications for Immune Responses and Autoimmune Diseases. Immunity 2009, 31, 539–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, I.-K.; Letterio, J.J.; Gorham, J.D. TGF-Beta 1 Inhibition of IFN-Gamma-Induced Signaling and Th1 Gene Expression in CD4+ T Cells Is Smad3 Independent but MAP Kinase Dependent. Mol. Immunol. 2007, 44, 3283–3290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Paik, P.K.; Chen, J.; Yarilina, A.; Kockeritz, L.; Lu, T.T.; Woodgett, J.R.; Ivashkiv, L.B. IFN-Gamma Suppresses IL-10 Production and Synergizes with TLR2 by Regulating GSK3 and CREB/AP-1 Proteins. Immunity 2006, 24, 563–574. [Google Scholar] [CrossRef] [Green Version]
- Becskei, A.; Grusby, M.J. Contribution of IL-12R Mediated Feedback Loop to Th1 Cell Differentiation. FEBS Lett. 2007, 581, 5199–5206. [Google Scholar] [CrossRef] [Green Version]
- Ni, L.; Lu, J. Interferon Gamma in Cancer Immunotherapy. Cancer Med. 2018, 7, 4509–4516. [Google Scholar] [CrossRef]
- Kammertoens, T.; Friese, C.; Arina, A.; Idel, C.; Briesemeister, D.; Rothe, M.; Ivanov, A.; Szymborska, A.; Patone, G.; Kunz, S.; et al. Tumour Ischaemia by Interferon-γ Resembles Physiological Blood Vessel Regression. Nature 2017, 545, 98–102. [Google Scholar] [CrossRef]
- Lu, Y.; Yang, W.; Qin, C.; Zhang, L.; Deng, J.; Liu, S.; Qin, Z. Responsiveness of Stromal Fibroblasts to IFN-Gamma Blocks Tumor Growth via Angiostasis. J. Immunol. 2009, 183, 6413–6421. [Google Scholar] [CrossRef] [Green Version]
- Takeda, K.; Nakayama, M.; Hayakawa, Y.; Kojima, Y.; Ikeda, H.; Imai, N.; Ogasawara, K.; Okumura, K.; Thomas, D.M.; Smyth, M.J. IFN-γ Is Required for Cytotoxic T Cell-Dependent Cancer Genome Immunoediting. Nat. Commun. 2017, 8, 14607. [Google Scholar] [CrossRef]
- Macias-Ceja, D.C.; Coll, S.; Bauset, C.; Seco-Cervera, M.; Gisbert-Ferrándiz, L.; Navarro, F.; Cosin-Roger, J.; Calatayud, S.; Barrachina, M.D.; Ortiz-Masia, D. IFNγ-Treated Macrophages Induce EMT through the WNT Pathway: Relevance in Crohn’s Disease. Biomedicines 2022, 10, 1093. [Google Scholar] [CrossRef]
- Jorgovanovic, D.; Song, M.; Wang, L.; Zhang, Y. Roles of IFN-γ in Tumor Progression and Regression: A Review. Biomark. Res. 2020, 8, 49. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Li, Y.; Zhang, J.; Zhang, B. PD-L1 Expression Increased by IFN-γ via JAK2-STAT1 Signaling and Predicts a Poor Survival in Colorectal Cancer. Oncol. Lett. 2020, 20, 1127–1134. [Google Scholar] [CrossRef] [PubMed]
- Lv, C.; Yuan, D.; Cao, Y. Downregulation of Interferon-γ Receptor Expression Endows Resistance to Anti-Programmed Death Protein 1 Therapy in Colorectal Cancer. J. Pharmacol. Exp. Ther. 2021, 376, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.; Deng, D.; Li, H.; Hu, X.; Shang, X.; Hou, X.; Jiang, H.; He, H. IFNγ/PD-L1 Signaling Improves the Responsiveness of Anti-PD-1 Therapy in Colorectal Cancer: An in Vitro Study. Onco Targets Ther. 2021, 14, 3051–3062. [Google Scholar] [CrossRef] [PubMed]
- Garris, C.S.; Arlauckas, S.P.; Kohler, R.H.; Trefny, M.P.; Garren, S.; Piot, C.; Engblom, C.; Pfirschke, C.; Siwicki, M.; Gungabeesoon, J.; et al. Successful Anti-PD-1 Cancer Immunotherapy Requires T Cell-Dendritic Cell Crosstalk Involving the Cytokines IFN-γ and IL-12. Immunity 2018, 49, 1148–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Chen, S.; Zeng, J. TGF-β Signaling: A Complex Role in Tumorigenesis (Review). Mol. Med. Rep. 2018, 17, 699–704. [Google Scholar] [CrossRef] [Green Version]
- Syed, V. TGF-β Signaling in Cancer. J. Cell. Biochem. 2016, 117, 1279–1287. [Google Scholar] [CrossRef]
- Derynck, R.; Zhang, Y.E. Smad-Dependent and Smad-Independent Pathways in TGF-Beta Family Signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef]
- Itatani, Y.; Kawada, K.; Sakai, Y. Transforming Growth Factor-β Signaling Pathway in Colorectal Cancer and Its Tumor Microenvironment. Int. J. Mol. Sci. 2019, 20, 5822. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Dai, P.; Hatakeyama, T.; Harada, Y.; Tanaka, H.; Yoshimura, N.; Takamatsu, T. TGF-β Signaling Regulates Pancreatic β-Cell Proliferation through Control of Cell Cycle Regulator p27 Expression. Acta Histochem. Cytochem. 2013, 46, 51–58. [Google Scholar] [CrossRef] [Green Version]
- Kiyono, K.; Suzuki, H.I.; Morishita, Y.; Komuro, A.; Iwata, C.; Yashiro, M.; Hirakawa, K.; Kano, M.R.; Miyazono, K. C-Ski Overexpression Promotes Tumor Growth and Angiogenesis through Inhibition of Transforming Growth Factor-Beta Signaling in Diffuse-Type Gastric Carcinoma. Cancer Sci. 2009, 100, 1809–1816. [Google Scholar] [CrossRef] [PubMed]
- Muthusamy, B.P.; Budi, E.H.; Katsuno, Y.; Lee, M.K.; Smith, S.M.; Mirza, A.M.; Akhurst, R.J.; Derynck, R. ShcA Protects against Epithelial-Mesenchymal Transition through Compartmentalized Inhibition of TGF-β-Induced Smad Activation. PLoS Biol. 2015, 13, e1002325. [Google Scholar] [CrossRef] [Green Version]
- Kheirelseid, E.A.H.; Miller, N.; Kerin, M.J. Molecular Biology of Colorectal Cancer: Review of the Literature. Am. J. Mol. Biol. 2013, 3, 72–80. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.-L.; Chen, Z.-Q.; Zhu, S.-L.; Liu, T.-W.; Wen, Y.; Su, Y.-S.; Xi, X.-J.; Hu, Y.; Lian, L.; Liu, F.-B. Prognostic Value of Transforming Growth Factor-Beta in Patients with Colorectal Cancer Who Undergo Surgery: A Meta-Analysis. BMC Cancer 2017, 17, 240. [Google Scholar] [CrossRef]
- Zhang, Q.; Liu, F.; Qin, L.; Liao, Z.; Song, J.; Liang, H.; Chen, X.; Zhang, Z.; Zhang, B. Characterization of TGFβ-Associated Molecular Features and Drug Responses in Gastrointestinal Adenocarcinoma. BMC Gastroenterol. 2021, 21, 284. [Google Scholar] [CrossRef]
- Moon, J.R.; Oh, S.J.; Lee, C.K.; Chi, S.G.; Kim, H.J. TGF-β1 Protects Colon Tumor Cells from Apoptosis through XAF1 Suppression. Int. J. Oncol. 2019, 54, 2117–2126. [Google Scholar] [CrossRef] [PubMed]
- Mercogliano, M.F.; Bruni, S.; Mauro, F.; Elizalde, P.V.; Schillaci, R. Harnessing Tumor Necrosis Factor Alpha to Achieve Effective Cancer Immunotherapy. Cancers 2021, 13, 564. [Google Scholar] [CrossRef]
- Ihnatko, R.; Kubes, M. TNF Signaling: Early Events and Phosphorylation. Gen. Physiol. Biophys. 2007, 26, 159–167. [Google Scholar]
- Al Obeed, O.A.; Alkhayal, K.A.; Al Sheikh, A.; Zubaidi, A.M.; Vaali-Mohammed, M.-A.; Boushey, R.; Mckerrow, J.H.; Abdulla, M.-H. Increased Expression of Tumor Necrosis Factor-α Is Associated with Advanced Colorectal Cancer Stages. World J. Gastroenterol. 2014, 20, 18390–18396. [Google Scholar] [CrossRef]
- Warsinggih; Limanu, F.; Labeda, I.; Lusikooy, R.E.; Mappincara; Faruk, M. The Relationship of Tumor Necrosis Factor Alpha Levels in Plasma toward the Stage and Differentiation Degree in Colorectal Cancer. Medicina Clínica Práctica 2021, 4, 100224. [Google Scholar] [CrossRef]
- Monteleone, G.; Pallone, F.; Stolfi, C. The Dual Role of Inflammation in Colon Carcinogenesis. Int. J. Mol. Sci. 2012, 13, 11071–11084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charkaoui, M.; Hajage, D.; Tubach, F.; Beaugerie, L.; Kirchgesner, J. Impact of Anti-Tumor Necrosis Factor Agents on the Risk of Colorectal Cancer in Patients with Ulcerative Colitis: Nationwide French Cohort Study. J. Crohn’s Colitis 2021. [Google Scholar] [CrossRef] [PubMed]
- Alkhayyat, M.; Abureesh, M.; Gill, A.; Khoudari, G.; Saleh, M.A.; Mansoor, E.; Regueiro, M. Lower Rates of Colorectal Cancer in Patients with Inflammatory Bowel Disease Using Anti-TNF Therapy. Inflamm. Bowel Dis. 2021, 27, 1052–1060. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Zhang, Z. TNF-α Promotes Colon Cancer Cell Migration and Invasion by Upregulating TROP-2. Oncol. Lett. 2018, 15, 3820–3827. [Google Scholar] [CrossRef]
- Shen, Z.; Zhou, R.; Liu, C.; Wang, Y.; Zhan, W.; Shao, Z.; Liu, J.; Zhang, F.; Xu, L.; Zhou, X.; et al. MicroRNA-105 Is Involved in TNF-α-Related Tumor Microenvironment Enhanced Colorectal Cancer Progression. Cell Death Dis. 2017, 8, 3213. [Google Scholar] [CrossRef]
- Shi, G.; Zheng, X.; Zhang, S.; Wu, X.; Yu, F.; Wang, Y.; Xing, F. Kanglaite Inhibits EMT Caused by TNF-α via NF-κΒ Inhibition in Colorectal Cancer Cells. Oncotarget 2018, 9, 6771–6779. [Google Scholar] [CrossRef] [Green Version]
- Ba, H.; Jiang, R.; Zhang, M.; Yin, B.; Wang, J.; Li, Z.; Li, B.; Zhou, X. Suppression of Transmembrane Tumor Necrosis Factor Alpha Processing by a Specific Antibody Protects Against Colitis-Associated Cancer. Front. Immunol. 2021, 12, 687874. [Google Scholar] [CrossRef]
- Barreda, D.R.; Hanington, P.C.; Belosevic, M. Regulation of Myeloid Development and Function by Colony Stimulating Factors. Dev. Comp. Immunol. 2004, 28, 509–554. [Google Scholar] [CrossRef]
- Hong, I.-S. Stimulatory versus Suppressive Effects of GM-CSF on Tumor Progression in Multiple Cancer Types. Exp. Mol. Med. 2016, 48, e242. [Google Scholar] [CrossRef] [Green Version]
- Metcalf, D. The Colony-Stimulating Factors and Cancer. Nat. Rev. Cancer 2010, 10, 425–434. [Google Scholar] [CrossRef] [Green Version]
- Sonderegger, I.; Iezzi, G.; Maier, R.; Schmitz, N.; Kurrer, M.; Kopf, M. GM-CSF Mediates Autoimmunity by Enhancing IL-6-Dependent Th17 Cell Development and Survival. J. Exp. Med. 2008, 205, 2281–2294. [Google Scholar] [CrossRef] [PubMed]
- Atanga, E.; Dolder, S.; Dauwalder, T.; Wetterwald, A.; Hofstetter, W. TNFα Inhibits the Development of Osteoclasts through Osteoblast-Derived GM-CSF. Bone 2011, 49, 1090–1100. [Google Scholar] [CrossRef] [PubMed]
- Miyashita, H.; Katayama, N.; Fujieda, A.; Shibasaki, T.; Yamamura, K.; Sugimoto, Y.; Miyata, E.; Ohishi, K.; Nishii, K.; Masuya, M.; et al. IL-4 and IL-10 Synergistically Inhibit Survival of Human Blood Monocytes Supported by GM-CSF. Int. J. Oncol. 2005, 26, 731–735. [Google Scholar] [CrossRef] [PubMed]
- Dang, P.M.-C.; Elbim, C.; Marie, J.-C.; Chiandotto, M.; Gougerot-Pocidalo, M.-A.; El-Benna, J. Anti-Inflammatory Effect of Interleukin-10 on Human Neutrophil Respiratory Burst Involves Inhibition of GM-CSF-Induced p47PHOX Phosphorylation through a Decrease in ERK1/2 Activity. FASEB J. 2006, 20, 1504–1506. [Google Scholar] [CrossRef] [PubMed]
- Kasper, S.; Kindler, T.; Sonnenschein, S.; Breitenbuecher, F.; Böhmer, F.D.; Huber, C.; Fischer, T. Cross-Inhibition of Interferon-Induced Signals by GM-CSF through a Block in Stat1 Activation. J. Interferon Cytokine Res. 2007, 27, 947–959. [Google Scholar] [CrossRef]
- Lukens, J.R.; Barr, M.J.; Chaplin, D.D.; Chi, H.; Kanneganti, T.-D. Inflammasome-Derived IL-1β Regulates the Production of GM-CSF by CD4+ T Cells and γδ T Cells. J. Immunol. 2012, 188, 3107–3115. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Zhao, Z.; Chen, Y.; Lv, Z.; Ding, X.; Wang, R.; Xiao, H.; Hou, C.; Shen, B.; Feng, J.; et al. An Epithelial-to-Mesenchymal Transition-Inducing Potential of Granulocyte Macrophage Colony-Stimulating Factor in Colon Cancer. Sci. Rep. 2017, 7, 8265. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Han, G.; Shen, B.; Li, Y. GM-CSF Facilitates the Development of Inflammation-Associated Colorectal Carcinoma. Oncoimmunology 2014, 3, e28186. [Google Scholar] [CrossRef]
- van de Laar, L.; Coffer, P.J.; Woltman, A.M. Regulation of Dendritic Cell Development by GM-CSF: Molecular Control and Implications for Immune Homeostasis and Therapy. Blood 2012, 119, 3383–3393. [Google Scholar] [CrossRef]
- Aliper, A.M.; Frieden-Korovkina, V.P.; Buzdin, A.; Roumiantsev, S.A.; Zhavoronkov, A. A Role for G-CSF and GM-CSF in Nonmyeloid Cancers. Cancer Med. 2014, 3, 737–746. [Google Scholar] [CrossRef]
- Arnold, I.C.; Artola-Boran, M.; Gurtner, A.; Bertram, K.; Bauer, M.; Frangez, Z.; Becher, B.; Kopf, M.; Yousefi, S.; Simon, H.-U.; et al. The GM-CSF-IRF5 Signaling Axis in Eosinophils Promotes Antitumor Immunity through Activation of Type 1 T Cell Responses. J. Exp. Med. 2020, 217, e20190706. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Ye, Y.; Zhang, H.; Szmitkowski, M.; Mäkinen, M.J.; Li, P.; Xia, D.; Yang, J.; Wu, Y.; Wu, H. Diagnostic and Prognostic Value of Serum Interleukin-6 in Colorectal Cancer. Medicine 2016, 95, e2502. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.-J.; Xu, J.-M.; Xu, W.-L.; Gu, D.-H.; Li, P.-W. Diagnostic Value of Interleukin-8 in Colorectal Cancer: A Case-Control Study and Meta-Analysis. World J. Gastroenterol. 2014, 20, 16334–16342. [Google Scholar] [CrossRef]
- Czajka-Francuz, P.; Francuz, T.; Cisoń-Jurek, S.; Czajka, A.; Fajkis, M.; Szymczak, B.; Kozaczka, M.; Malinowski, K.P.; Zasada, W.; Wojnar, J.; et al. Serum Cytokine Profile as a Potential Prognostic Tool in Colorectal Cancer Patients—One Center Study. Rep. Pract. Oncol. Radiother. 2020, 25, 867–875. [Google Scholar] [CrossRef]
- Razi, S.; Baradaran Noveiry, B.; Keshavarz-Fathi, M.; Rezaei, N. IL-17 and Colorectal Cancer: From Carcinogenesis to Treatment. Cytokine 2019, 116, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Feng, L.; Zhong, R.; Xia, Z.; Zhang, L.; Cui, L.; Yan, H.; Jia, X.; Zhang, Z. Icariside II Inhibits the EMT of NSCLC Cells in Inflammatory Microenvironment via down-Regulation of Akt/NF-κB Signaling Pathway. Mol. Carcinog. 2017, 56, 36–48. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Cytokine | Drug | Type of Intervention | Phase | Study Status | ClinicalTrials.Gov Identifier |
|---|---|---|---|---|---|
| TNF | INCAGN01876 | Stimulation | I/II | Completed | NCT03126110 |
| TGF | Vactosertib | Inhibition | I | Not yet recruiting | NCT05400122 |
| NIS793 | Inhibition | I | Completed | NCT02947165 | |
| AP 12009 | Inhibition | I | Completed | NCT00844064 | |
| IL-1 | Anakinra | Inhibition | II | Completed | NCT02090101 |
| CAN04 | Inhibition | I/II | Recruiting | NCT05116891 | |
| I | Recruiting | NCT03267316 | |||
| IL-7 | NT-I7 | Stimulation | I | Recruiting | NCT04054752 |
| I | Recruiting | NCT04332653 | |||
| GM-CSF | GM-CSF | Stimulation | I/II | Recruiting | NCT04929652 |
| Leukine | Stimulation | I/II | Completed | NCT00785122 | |
| Sargramostim | Stimulation | II | Completed | NCT00103142 | |
| Stimulation | II | Completed | NCT00262808 | ||
| JX-594 | Stimulation | I | Completed | NCT01469611 | |
| GVAX | Stimulation | I | Recruiting | NCT01952730 | |
| IFN-γ | IFN-γ | Stimulation | II | Completed | NCT00786643 |
| Cytokine | Receptor | Impact on Progression | TME Modulation | Main Pathways |
|---|---|---|---|---|
| IL-1β | TIR | Promotion | Metalloproteinase release [54] EMT promotion [56] CSC promotion [56] | NF-κB/miR-181a/PTEN [60] GSK-3β/Wnt/β-catenin [59] |
| IL-4 | IL-4Rα | Promotion | E-cadherin depletion [38] | ERK [75] E2F1/SP3/STAT6 [50] |
| IL-6 | IL-6R | Promotion | EMT promotion [83] CAFs stimulation [29] Angiogenesis [149] Macrophage migration [149] | JAK2/STAT3 [76] Pi3K/AKT/mTOR [87] NF-κB/STAT3 [90] ERK/MAPK [83] |
| IL-7 | IL-7Rα | Inhibition | CD4+/CD8+ T cells stimulation [99] PD-L1 depletion [99] NK stimulation [99] Tregs inhibition [100] MDSCs inhibition [100] | Apoptotic pathways through Bax and Bcl-xl proteins [49] |
| IL-8 | CXCR1 CXCR2 | Promotion | EMT promotion [108] Angiogenesis promotion [106] E-cadherin depletion [109] Neutrophil stimulation [110] TAMs stimulation [110] | PI3K/AKT/mTOR [106] RAF/MEK/ERK [108] JAK2/STAT3/Snail [109] |
| IL-10 | IL-10R1 IL-10R2 | Ambiguous | CD8+ T cells stimulation [115] APCs inhibition [102,115] Th17 lymphocytes inhibition [115] | JAK/STAT3 [117] PI3K/Akt/mTORC1 [118] SOCS3 [117] |
| IL-11 | IL-11RA | Promotion | Fibroblast stimulation [131] Epithelial cells stimulation [133] | JAK/STAT3 [132] |
| IL-17 | IL-17RA | Promotion | Angiogenesis promotion [150] MDSCs promotion [144] CD8+ T cells inhibition [144] E-cadherin depletion [150] | NF-kB/STAT3 [246] ERK/MMP 2 and 7 [246] STAT3/VEGF [246] STEAP4-XIAP [138] |
| IL-21 | IL-21R | Ambiguous | CD8+ cells promotion [152] Tregs inhibition [152] Th17 promotion [153] Th1 inhibition [153] Th2 promotion [153] | JAK/STAT3 [151] |
| IL-22 | IL-22R | Promotion | EMT promotion [247] | STAT1,3,5 [167] ERK, Akt, p38, MAPK pathways [167] DOT1L [166] |
| IL-23 | IL-23R | Promotion | Tregs inhibition [174] Th17 promotion [174] | STAT5 [30] TNF/NF-kB [173] |
| IL-33 | IL1RAP sST decoy | Ambiguous | CD4+ T cells promotion [178] Angiogenesis promotion [180] | IL-33/ST2 [53] TRAF6/NF-kB [53] MAPK/AP-1 [53] |
| IFN-γ | IFNγR1 IFNγR2 | Inhibition/ambiguous | Activation host immune surveillance [193] Upregulation the MHC molecules [193] Switch towards M1 and Th1 phenotypes [189,193] EMT promotion [201] | JAK/STAT/IRF1 [192] IL-4/STAT6 [196] Wnt/β-catenin [201] |
| TNF-α | TNFR1 TNFR2 | Ambiguous | Angiogenesis promotion [222] PD-L1 upregulation [218] Epithelial cells promotion [226] EMT inhibition [227] | TROP-2/ERK/p38 [225] NF-κB/STAT3 [90,227] Wnt/β-catenin [226] |
| TGF-β | TGFBR1 TGFBR2 | Ambiguous | Epithelial cells inhibition [207] EMT promotion [213] Angiogenesis promotion [207] | Smad [210] PI3K/AKT [210] RAS/RAF/MEK [210] MAPK [210] JNK [210] |
| GM-CSF | GM-CSFR | Ambiguous | Activation of immune response [231] EMT promotion [23,230] Tumor progression [230,238] Upregulation of VEGF signaling [230,238] Increase in CD8+ lymphocytes infiltration [242] | MAPK [240] PI3K [240] NF-kB [240] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borowczak, J.; Szczerbowski, K.; Maniewski, M.; Kowalewski, A.; Janiczek-Polewska, M.; Szylberg, A.; Marszałek, A.; Szylberg, Ł. The Role of Inflammatory Cytokines in the Pathogenesis of Colorectal Carcinoma—Recent Findings and Review. Biomedicines 2022, 10, 1670. https://doi.org/10.3390/biomedicines10071670
Borowczak J, Szczerbowski K, Maniewski M, Kowalewski A, Janiczek-Polewska M, Szylberg A, Marszałek A, Szylberg Ł. The Role of Inflammatory Cytokines in the Pathogenesis of Colorectal Carcinoma—Recent Findings and Review. Biomedicines. 2022; 10(7):1670. https://doi.org/10.3390/biomedicines10071670
Chicago/Turabian StyleBorowczak, Jędrzej, Krzysztof Szczerbowski, Mateusz Maniewski, Adam Kowalewski, Marlena Janiczek-Polewska, Anna Szylberg, Andrzej Marszałek, and Łukasz Szylberg. 2022. "The Role of Inflammatory Cytokines in the Pathogenesis of Colorectal Carcinoma—Recent Findings and Review" Biomedicines 10, no. 7: 1670. https://doi.org/10.3390/biomedicines10071670
APA StyleBorowczak, J., Szczerbowski, K., Maniewski, M., Kowalewski, A., Janiczek-Polewska, M., Szylberg, A., Marszałek, A., & Szylberg, Ł. (2022). The Role of Inflammatory Cytokines in the Pathogenesis of Colorectal Carcinoma—Recent Findings and Review. Biomedicines, 10(7), 1670. https://doi.org/10.3390/biomedicines10071670