
GLUT3 Promotes Epithelial–Mesenchymal Transition via TGF-β/JNK/ATF2 Signaling Pathway in Colorectal Cancer Cells



Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Reagents
2.3. Lentiviral shRNA-Mediated Knockdown
2.4. Quantitative Reverse Transcriptase Polymerase Chain Reaction (qRT-PCR) Analysis
2.5. Western Blot Analysis
2.6. Plasmids Cloning
2.7. Transient Transfection and the Luciferase Reporter Assay
2.8. Invasion Assay
2.9. Chromatin Immunoprecipitation Assay
2.10. Statistical Analysis
3. Results
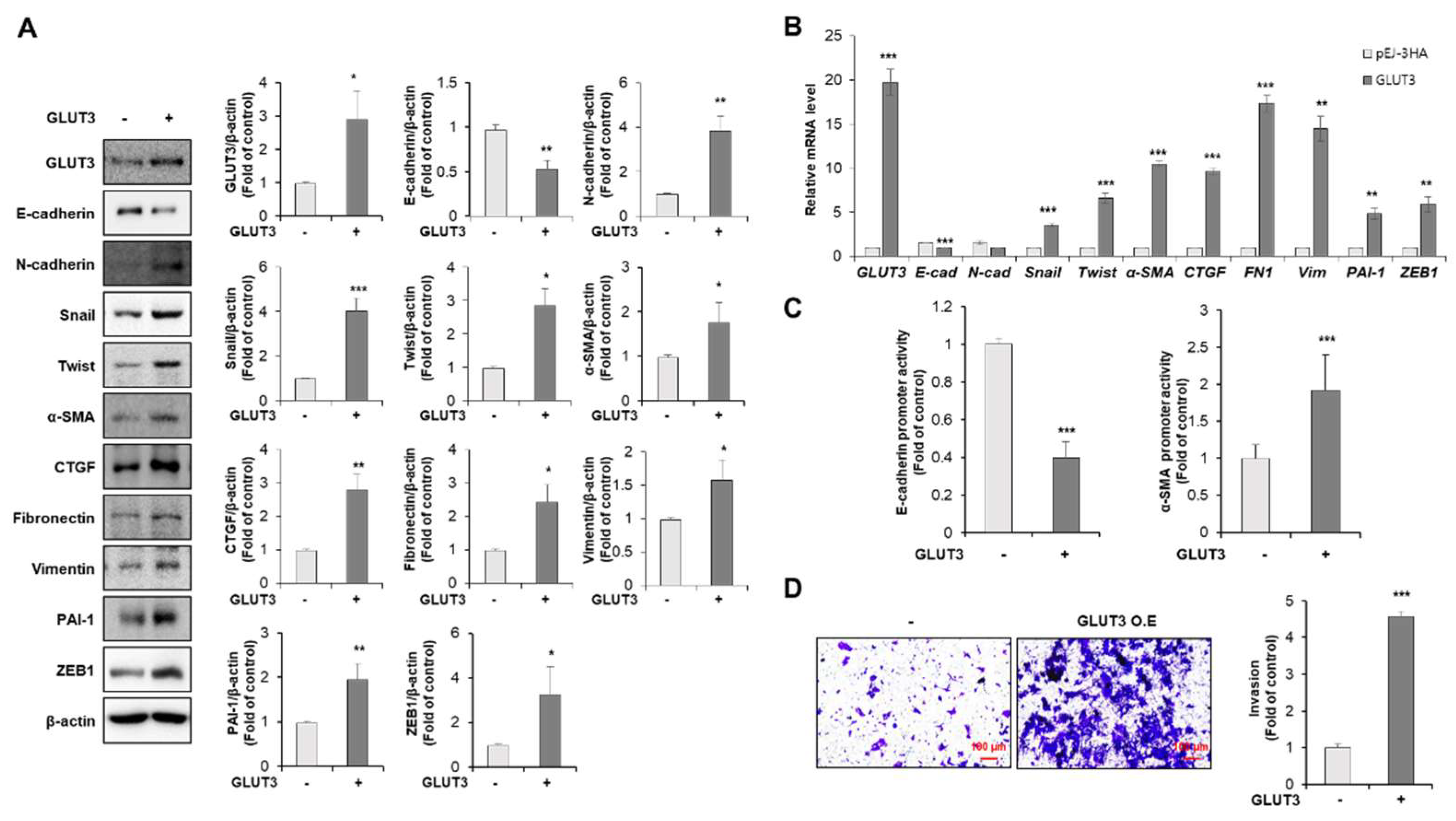
3.1. GLUT3 Regulates the Expression of EMT-Related Genes and Promotes Invasiveness in CRC Cells
3.2. GLUT3 Induces the Expression of Stemness Makers in CRC Cells
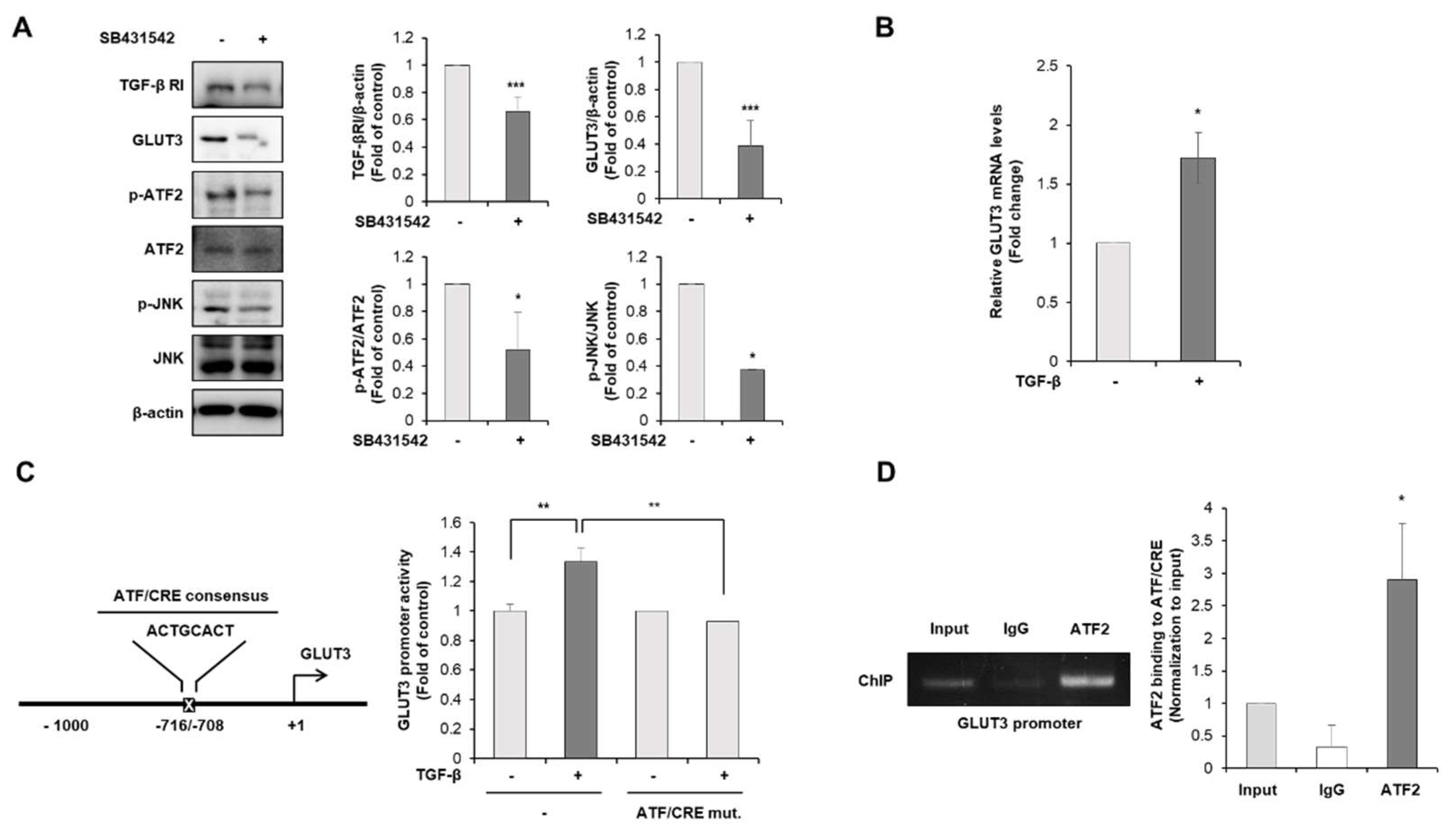
3.3. TGF-β Is the Upstream Regulator of GLUT3-Induced EMT in CRC Cells
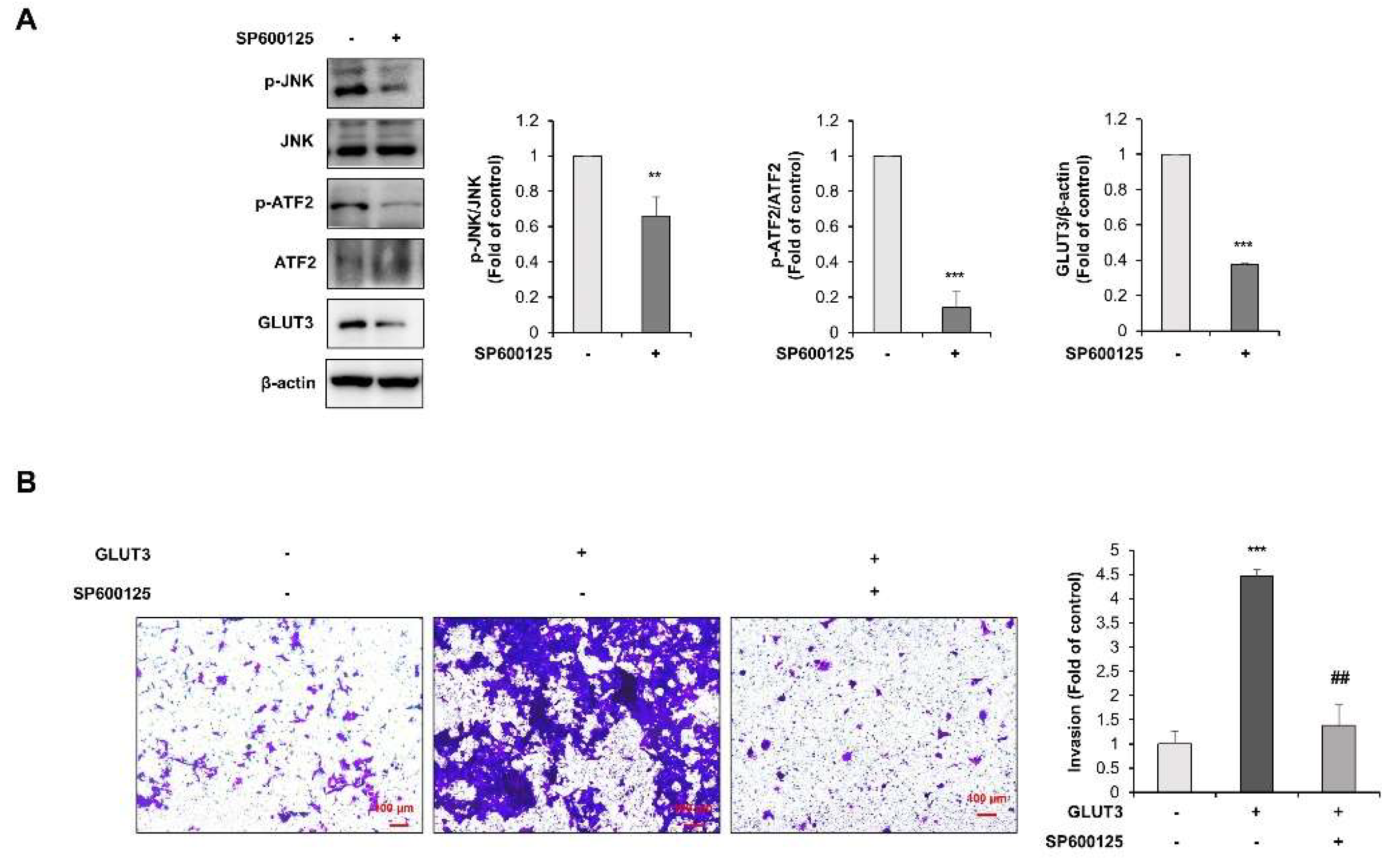
3.4. TGF-β Regulates GLUT3-Induced EMT through JNK/ATF2 Signaling Pathway in CRC Cells
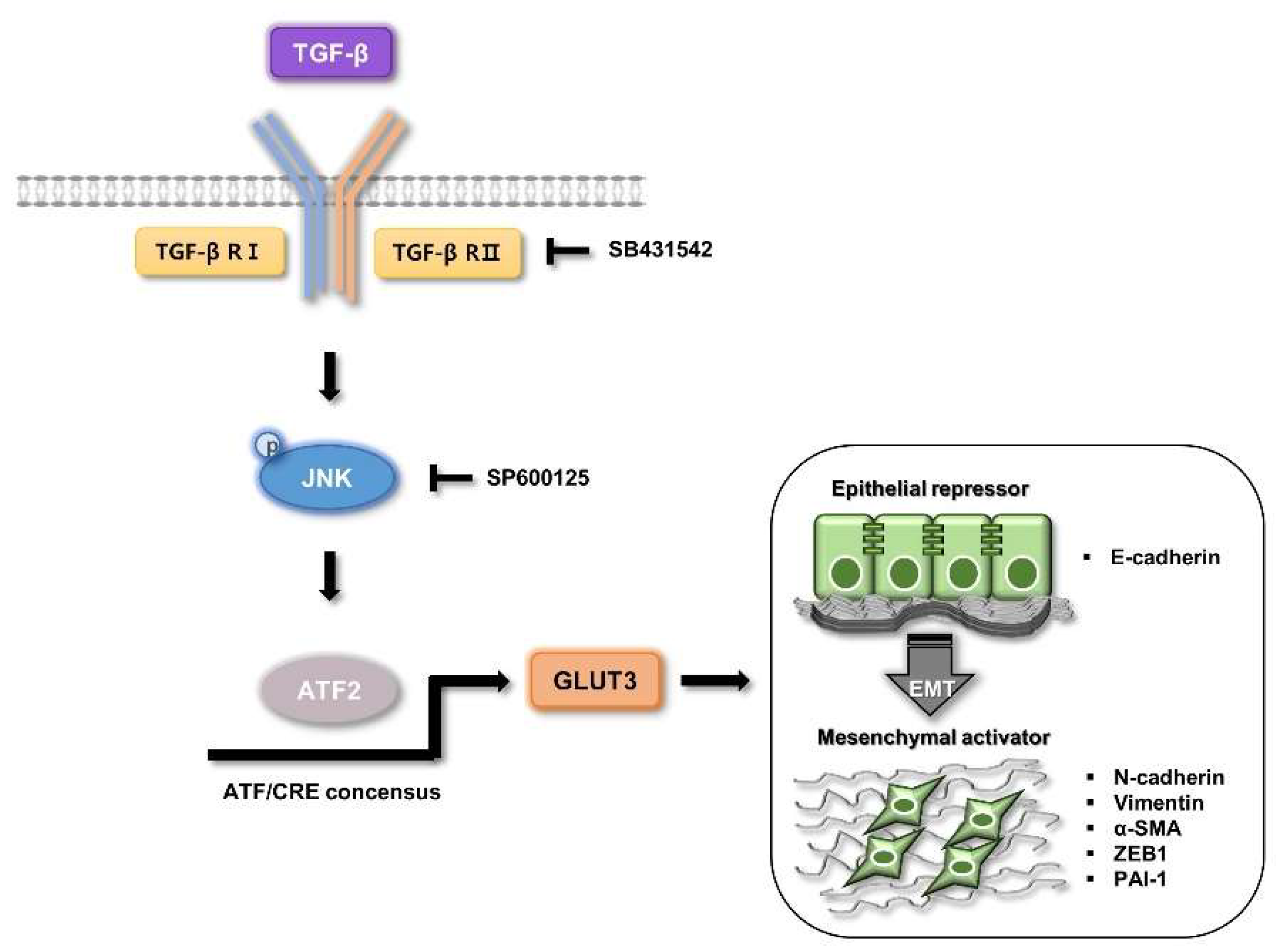
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Goding Sauer, A.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Fedewa, S.A.; Ahnen, D.J.; Meester, R.G.S.; Barzi, A.; Jemal, A. Colorectal cancer statistics, 2017. CA Cancer J. Clin. 2017, 67, 177–193. [Google Scholar] [CrossRef] [PubMed]
- Shook, D.; Keller, R. Mechanisms, mechanics and function of epithelial-mesenchymal transitions in early development. Mech. Dev. 2003, 120, 1351–1383. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. EMT: When epithelial cells decide to become mesenchymal-like cells. J. Clin. Investig. 2009, 119, 1417–1419. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.; Massagué, J. Epithelial-mesenchymal transitions: Twist in development and metastasis. Cell 2004, 118, 277–279. [Google Scholar] [CrossRef] [Green Version]
- Mittal, V. Epithelial Mesenchymal Transition in Tumor Metastasis. Annu. Rev. Pathol. 2018, 13, 395–412. [Google Scholar] [CrossRef]
- Hay, E.D. An Overview of Epithelio-Mesenchymal Transformation. Acta Anat. 1995, 154, 8–20. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Spaderna, S.; Schmalhofer, O.; Hlubek, F.; Berx, G.; Eger, A.; Merkel, S.; Jung, A.; Kirchner, T.; Brabletz, T. A transient, EMT-linked loss of basement membranes indicates metastasis and poor survival in colorectal cancer. Gastroenterology 2006, 131, 830–840. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.W.; Dang, C.V. Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res. 2006, 66, 8927–8930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Kroemer, G.; Pouyssegur, J. Tumor cell metabolism: Cancer’s Achilles’ heel. Cancer Cell 2008, 13, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Simpson, I.A.; Dwyer, D.; Malide, D.; Moley, K.H.; Travis, A.; Vannucci, S.J. The facilitative glucose transporter GLUT3: 20 years of distinction. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E242–E253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gould, G.W.; Holman, G.D. The glucose transporter family: Structure, function and tissue-specific expression. Biochem. J. 1993, 295, 329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MEDINA, R.A.; OWEN, G.I. Glucose transporters: Expression, regulation and cancer. Biol. Res. 2002, 35, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Boado, R.J.; Black, K.L.; Pardridge, W.M. Gene expression of GLUT3 and GLUT1 glucose transporters in human brain tumors. Mol. Brain Res. 1994, 27, 51–57. [Google Scholar] [CrossRef]
- Kurata, T.; Oguri, T.; Isobe, T.; Ishioka, S.; Yamakido, M. Differential expression of facilitative glucose transporter (GLUT) genes in primary lung cancers and their liver metastases. Jpn. J. Cancer Res. 1999, 90, 1238–1243. [Google Scholar] [CrossRef] [PubMed]
- Younes, M.; Brown, R.W.; Stephenson, M.; Gondo, M.; Cagle, P.T. Overexpression of Glut1 and Glut3 in stage I nonsmall cell lung carcinoma is associated with poor survival. Cancer 1997, 80, 1046–1051. [Google Scholar] [CrossRef]
- Dai, W.; Xu, Y.; Mo, S.; Li, Q.; Yu, J.; Wang, R.; Ma, Y.; Ni, Y.; Xiang, W.; Han, L.; et al. GLUT3 induced by AMPK/CREB1 axis is key for withstanding energy stress and augments the efficacy of current colorectal cancer therapies. Signal Transduct. Target. Ther. 2020, 5, 177. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Zhang, J.; Xu, Q.; Wang, B.; Yao, Y.; Sun, L.; Wang, X.; Zhou, D.; Gao, L.; Song, S.; et al. YAP promotes the proliferation and migration of colorectal cancer cells through the Glut3/AMPK signaling pathway. Oncol. Lett. 2021, 21, 312. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, S.; Huang, C.; Li, Y.; Zhao, H.; Kasim, V. Yin Yang 1 promotes the Warburg effect and tumorigenesis via glucose transporter GLUT3. Cancer Sci. 2018, 109, 2423–2434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickup, M.; Novitskiy, S.; Moses, H.L. The roles of TGFβ in the tumour microenvironment. Nat. Rev. Cancer 2013, 13, 788–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [Green Version]
- Akhurst, R.J.; Hata, A. Targeting the TGFβ signalling pathway in disease. Nat. Rev. Drug Discov. 2012, 11, 790–811. [Google Scholar] [CrossRef] [Green Version]
- Heldin, C.-H.; Miyazono, K.; ten Dijke, P. TGF-β signalling from cell membrane to nucleus through SMAD proteins. Nature 1997, 390, 465–471. [Google Scholar] [CrossRef]
- Mu, Y.; Gudey, S.K.; Landström, M. Non-Smad signaling pathways. Cell Tissue Res. 2012, 347, 11–20. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, P.; Shao, M.; Zang, X.; Zhang, J.; Mao, F.; Qian, H.; Xu, W. SALL4 activates TGF-β/SMAD signaling pathway to induce EMT and promote gastric cancer metastasis. Cancer Manag. Res. 2018, 10, 4459–4470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Lei, R.; Zhuang, X.; Li, X.; Li, G.; Lev, S.; Segura, M.F.; Zhang, X.; Hu, G. MicroRNA-182 targets SMAD7 to potentiate TGFβ-induced epithelial-mesenchymal transition and metastasis of cancer cells. Nat. Commun. 2016, 7, 13884. [Google Scholar] [CrossRef] [Green Version]
- Qu, Z.; Feng, J.; Pan, H.; Jiang, Y.; Duan, Y.; Fa, Z. Exosomes derived from HCC cells with different invasion characteristics mediated EMT through TGF-β/Smad signaling pathway. Onco. Targets Ther. 2019, 12, 6897–6905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, H.-W.; Hsu, E.-C.; Lee, S.-S.; Lang, Y.-D.; Lin, Y.-C.; Chang, C.-Y.; Lee, S.-Y.; Gu, D.-L.; Shih, J.-H.; Ho, C.-M.; et al. PSPC1 mediates TGF-β1 autocrine signalling and Smad2/3 target switching to promote EMT, stemness and metastasis. Nat. Cell Biol. 2018, 20, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.E. Non-Smad pathways in TGF-β signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef] [Green Version]
- Lamouille, S.; Derynck, R. Cell size and invasion in TGF-β–induced epithelial to mesenchymal transition is regulated by activation of the mTOR pathway. J. Cell Biol. 2007, 178, 437–451. [Google Scholar] [CrossRef] [Green Version]
- Bakin, A.V.; Tomlinson, A.K.; Bhowmick, N.A.; Moses, H.L.; Arteaga, C.L. Phosphatidylinositol 3-Kinase Function Is Required for Transforming Growth Factor β-mediated Epithelial to Mesenchymal Transition and Cell Migration*. J. Biol. Chem. 2000, 275, 36803–36810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.; Hébert, M.C.; Zhang, Y.E. TGF-β receptor-activated p38 MAP kinase mediates Smad-independent TGF-β responses. EMBO J. 2002, 21, 3749–3759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhanasekaran, D.N.; Reddy, E.P. JNK signaling in apoptosis. Oncogene 2008, 27, 6245–6251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, R.J. Signal transduction by the JNK group of MAP kinases. Cell 2000, 13–21. [Google Scholar]
- Wang, J.; Kuiatse, I.; Lee, A.V.; Pan, J.; Giuliano, A.; Cui, X. Sustained c-Jun-NH2-kinase activity promotes epithelial-mesenchymal transition, invasion, and survival of breast cancer cells by regulating extracellular signal-regulated kinase activation. Mol. Cancer Res. 2010, 8, 266–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sehgal, V.; Ram, P.T. Network Motifs in JNK Signaling. Genes Cancer 2013, 4, 409–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.; Campbell, D.; Dérijard, B.; Davis, R.J. Transcription factor ATF2 regulation by the JNK signal transduction pathway. Science 1995, 267, 389–393. [Google Scholar] [CrossRef]
- Watson, G.; Ze’ev, A.R.; Lau, E. ATF2, a paradigm of the multifaceted regulation of transcription factors in biology and disease. Pharmacol. Res. 2017, 119, 347–357. [Google Scholar] [CrossRef]
- Li, S.; Ezhevsky, S.; Dewing, A.; Cato, M.H.; Scortegagna, M.; Bhoumik, A.; Breitwieser, W.; Braddock, D.; Eroshkin, A.; Qi, J. Radiation sensitivity and tumor susceptibility in ATM phospho-mutant ATF2 mice. Genes Cancer 2010, 1, 316–330. [Google Scholar] [CrossRef] [PubMed]
- Huebner, K.; Procházka, J.; Monteiro, A.C.; Mahadevan, V.; Schneider-Stock, R. The activating transcription factor 2: An influencer of cancer progression. Mutagenesis 2019, 34, 375–389. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.-s.; Chen, C.; Wu, Z.-j.; Liu, B.; Gao, L.; Yang, Q.; Chen, W.; Chen, J.-m.; Bao, Y.; Qu, L.; et al. ATF2 predicts poor prognosis and promotes malignant phenotypes in renal cell carcinoma. J. Exp. Clin. Cancer Res. 2016, 35, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flavahan, W.A.; Wu, Q.; Hitomi, M.; Rahim, N.; Kim, Y.; Sloan, A.E.; Weil, R.J.; Nakano, I.; Sarkaria, J.N.; Stringer, B.W.; et al. Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nat. Neurosci. 2013, 16, 1373–1382. [Google Scholar] [CrossRef] [PubMed]
- Masin, M.; Vazquez, J.; Rossi, S.; Groeneveld, S.; Samson, N.; Schwalie, P.C.; Deplancke, B.; Frawley, L.E.; Gouttenoire, J.; Moradpour, D.; et al. GLUT3 is induced during epithelial-mesenchymal transition and promotes tumor cell proliferation in non-small cell lung cancer. Cancer Metab. 2014, 2, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, C.-C.; Ling, H.-H.; Chiang, M.-C.; Chung, C.-H.; Lee, W.-Y.; Chu, C.-Y.; Wu, Y.-C.; Chen, C.-H.; Lai, Y.-W.; Tsai, I.L.; et al. Metastatic Colorectal Cancer Rewrites Metabolic Program Through a Glut3-YAP-dependent Signaling Circuit. Theranostics 2019, 9, 2526–2540. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Baker, D.; ten Dijke, P. TGF-β-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis. Int. J. Mol. Sci. 2019, 20, 2767. [Google Scholar] [CrossRef] [Green Version]
- Massagué, J. TGFβ in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-β-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef]
- Hua, W.; Ten Dijke, P.; Kostidis, S.; Giera, M.; Hornsveld, M. TGFbeta-induced metabolic reprogramming during epithelial-to-mesenchymal transition in cancer. Cell. Mol. Life Sci. 2020, 77, 2103–2123. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.-J.; Wagner, S.; Liu, F.; O’Reilly, M.A.; Robbins, P.D.; Green, M.R. Retinoblastoma gene product activates expression of the human TGF-β2 gene through transcription factor ATF-2. Nature 1992, 358, 331–334. [Google Scholar] [CrossRef] [PubMed]
- Landström, M. The TAK1–TRAF6 signalling pathway. Int. J. Biochem. Cell Biol. 2010, 42, 585–589. [Google Scholar] [CrossRef]
- Mali, A.V.; Joshi, A.A.; Hegde, M.V.; Kadam, S.S. Enterolactone modulates the ERK/NF-κB/snail signaling pathway in triple-negative breast cancer cell line MDA-MB-231 to revert the TGF-β-induced epithelial-mesenchymal transition. Cancer Biol. Med. 2018, 15, 137–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, F.; Wang, H.; Chen, E.; Bian, E.; Xu, Y.; Ji, X.; Yang, Z.; Hua, X.; Zhang, Y.; Zhao, B. LncRNA-ATB promotes TGF-β-induced glioma cells invasion through NF-κB and P38/MAPK pathway. J. Cell. Physiol. 2019, 234, 23302–23314. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Zhang, X.-J.; Zou, H.; Zhang, Y.-Y.; Xia, J.-W.; Zhang, P.; Zhang, Y.-Z.; Li, J.; Dong, L.; Wumaier, G.; et al. PTPL1 suppresses lung cancer cell migration via inhibiting TGF-β1-induced activation of p38 MAPK and Smad 2/3 pathways and EMT. Acta Pharmacol. Sin. 2021, 42, 1280–1287. [Google Scholar] [CrossRef]
- Jilany Khan, G.; Gao, Y.; Gu, M.; Wang, L.; Khan, S.; Naeem, F.; Semukunzi, H.; Roy, D.; Yuan, S.; Sun, L. TGF-β1 causes EMT by regulating N-Acetyl glucosaminyl transferases via downregulation of non muscle myosin II-A through JNK/P38/PI3K pathway in lung cancer. Curr. Cancer Drug Targets 2018, 18, 209–219. [Google Scholar] [CrossRef]
- Yang, L.; Yu, Y.; Xiong, Z.; Chen, H.; Tan, B.; Hu, H. Downregulation of SEMA4C Inhibit Epithelial-Mesenchymal Transition (EMT) and the Invasion and Metastasis of Cervical Cancer Cells via Inhibiting Transforming Growth Factor-beta 1 (TGF-β1)-Induced Hela cells p38 Mitogen-Activated Protein Kinase (MAPK) Activation. Med. Sci. Monit. 2020, 26, e918123. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, N.; Gao, R.; Zhu, Y.; Zhang, Z.; Xu, X.; Wang, J.; Li, Z.; Liu, X.; Li, Z.; et al. TGF-β1 induced fascin1 expression facilitates the migration and invasion of kidney carcinoma cells through ERK and JNK signaling pathways. Biochem. Biophys. Res. Commun. 2018, 501, 913–919. [Google Scholar] [CrossRef]
- Sundqvist, A.; Voytyuk, O.; Hamdi, M.; Popeijus, H.E.; Bijlsma-van der Burgt, C.; Janssen, J.; Martens, J.W.M.; Moustakas, A.; Heldin, C.-H.; ten Dijke, P.; et al. JNK-Dependent cJun Phosphorylation Mitigates TGFβ- and EGF-Induced Pre-Malignant Breast Cancer Cell Invasion by Suppressing AP-1-Mediated Transcriptional Responses. Cells 2019, 8, 1481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santibañez, J.F. JNK mediates TGF-β1-induced epithelial mesenchymal transdifferentiation of mouse transformed keratinocytes. FEBS Lett. 2006, 580, 5385–5391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Mao, H.; Nie, J.; Chen, W.; Yang, Q.; Dong, X.; Yu, X. Transforming growth factor β1 induces epithelial–mesenchymal transition by activating the JNK–Smad3 pathway in rat peritoneal mesothelial cells. Perit. Dial. Int. 2008, 28, 88–95. [Google Scholar] [CrossRef]
- Fu, H.; Hu, Z.; Wen, J.; Wang, K.; Liu, Y. TGF-beta promotes invasion and metastasis of gastric cancer cells by increasing fascin1 expression via ERK and JNK signal pathways. Acta Biochim. Biophys. Sin. 2009, 41, 648–656. [Google Scholar] [CrossRef] [Green Version]
- Hocevar, B.A.; Brown, T.L.; Howe, P.H. TGF-β induces fibronectin synthesis through a c-Jun N-terminal kinase-dependent, Smad4-independent pathway. EMBO J. 1999, 18, 1345–1356. [Google Scholar] [CrossRef] [Green Version]
- Pant, I.; Rao, S.G.; Kondaiah, P. Role of areca nut induced JNK/ATF2/Jun axis in the activation of TGF-β pathway in precancerous Oral Submucous Fibrosis. Sci. Rep. 2016, 6, 34314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weigert, C.; Sauer, U.; Brodbeck, K.; Pfeiffer, A.; Häring, H.U.; Schleicher, E.D. AP-1 proteins mediate hyperglycemia-induced activation of the human TGF-β1 promoter in mesangial cells. J. Am. Soc. Nephrol. 2000, 11, 2007–2016. [Google Scholar] [CrossRef]
- van Dam, H.; Castellazzi, M. Distinct roles of Jun: Fos and Jun: ATF dimers in oncogenesis. Oncogene 2001, 20, 2453–2464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kallunki, T.; Deng, T.; Hibi, M.; Karin, M. c-Jun can recruit JNK to phosphorylate dimerization partners via specific docking interactions. Cell 1996, 87, 929–939. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Bergami, P.; Lau, E.; Ronai, Z.E. Emerging roles of ATF2 and the dynamic AP1 network in cancer. Nat. Rev. Cancer 2010, 10, 65–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Liu, Z.; Guo, K. The effect of JDP2 and ATF2 on the epithelial-mesenchymal transition of human pancreatic cancer cell lines. Pathol. Oncol. Res. 2012, 18, 571–577. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Gene | Primer Sequence | |
|---|---|---|---|
| Human (qRT-PCR) | 18S rRNA | Forward | GCAATTATTCCCCATGAACG |
| Reverse | GGCCTCACTAAACCATCCAA | ||
| GLUT3 | Forward | TTGCTCTTCCCCTCCGCTGC | |
| Reverse | ACCGTGTGCCTGCCCTTCAA | ||
| CDH1 (E-cadherin) | Forward | TCC CCG GCC AGC CAT | |
| Reverse | GCA GAG CCA AGA GGA GAC C | ||
| CDH2 (N-cadherin) | Forward | GAG GCT TCT GGT GAA ATC GC | |
| Reverse | AGA AGA GGC TGT CCT TCA TGC | ||
| Snail | Forward | GCTGCAGGACTCTAATCCAGA | |
| Reverse | ATCTCCGGAGGTGGGATG | ||
| Twist | Forward | GGCATCACTATGGACTTTCTCTATT | |
| Reverse | GGCCAGTTTGATCCCAGTATT | ||
| α-SMA | Forward | CAGTGGAATGCAGTGGAAGA | |
| Reverse | AGGGAAGCTGAAAGCTGAAG | ||
| CTGF | Forward | AGG ATG TGC ATT CTC CAG CC | |
| Reverse | GCC ACA AGC TGT CCA GTC TA | ||
| Fibronectin1 | Forward | GAACTATGATGCCGACCAGAA | |
| Reverse | GGTTGTGCAGATTTCCTCGT | ||
| Vimentin | Forward | GCT TCA GAG AGA GGA AGC CG | |
| Reverse | AAG GTC AAG ACG TGC CAG AG | ||
| Bmi1 | Forward | TGAAGATAGAGGAGAGGTTGC | |
| Reverse | CTGCTGGGCATCGTAAGTAT | ||
| Nanog | Forward | GTCCCGGTCAAGAAACAGAA | |
| Reverse | TGCGTCACACCATTGCTATT | ||
| OCT3/4 | Forward | ATTCAGCCAAACGACCATCT | |
| Reverse | ACACTCGGACCACATCCTTC | ||
| PAI-1 | Forward | GACTCGTGAAGTCAGCCTGAAAC | |
| Reverse | GACTCGTGAAGTCAGCCTGAAAC | ||
| ZEB1 | Forward | GGCATACACCTACTCAACTACGG | |
| Reverse | TGGGCGGTGTAGAATCAGAGTC | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, M.-Y.; Lee, D.-Y.; Yun, S.-M.; Kim, E.-H. GLUT3 Promotes Epithelial–Mesenchymal Transition via TGF-β/JNK/ATF2 Signaling Pathway in Colorectal Cancer Cells. Biomedicines 2022, 10, 1837. https://doi.org/10.3390/biomedicines10081837
Song M-Y, Lee D-Y, Yun S-M, Kim E-H. GLUT3 Promotes Epithelial–Mesenchymal Transition via TGF-β/JNK/ATF2 Signaling Pathway in Colorectal Cancer Cells. Biomedicines. 2022; 10(8):1837. https://doi.org/10.3390/biomedicines10081837
Chicago/Turabian StyleSong, Moon-Young, Da-Young Lee, Sun-Mi Yun, and Eun-Hee Kim. 2022. "GLUT3 Promotes Epithelial–Mesenchymal Transition via TGF-β/JNK/ATF2 Signaling Pathway in Colorectal Cancer Cells" Biomedicines 10, no. 8: 1837. https://doi.org/10.3390/biomedicines10081837
APA StyleSong, M.-Y., Lee, D.-Y., Yun, S.-M., & Kim, E.-H. (2022). GLUT3 Promotes Epithelial–Mesenchymal Transition via TGF-β/JNK/ATF2 Signaling Pathway in Colorectal Cancer Cells. Biomedicines, 10(8), 1837. https://doi.org/10.3390/biomedicines10081837