
Targeting Mitochondrial IDH2 Enhances Antitumor Activity of Cisplatin in Lung Cancer via ROS-Mediated Mechanism



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
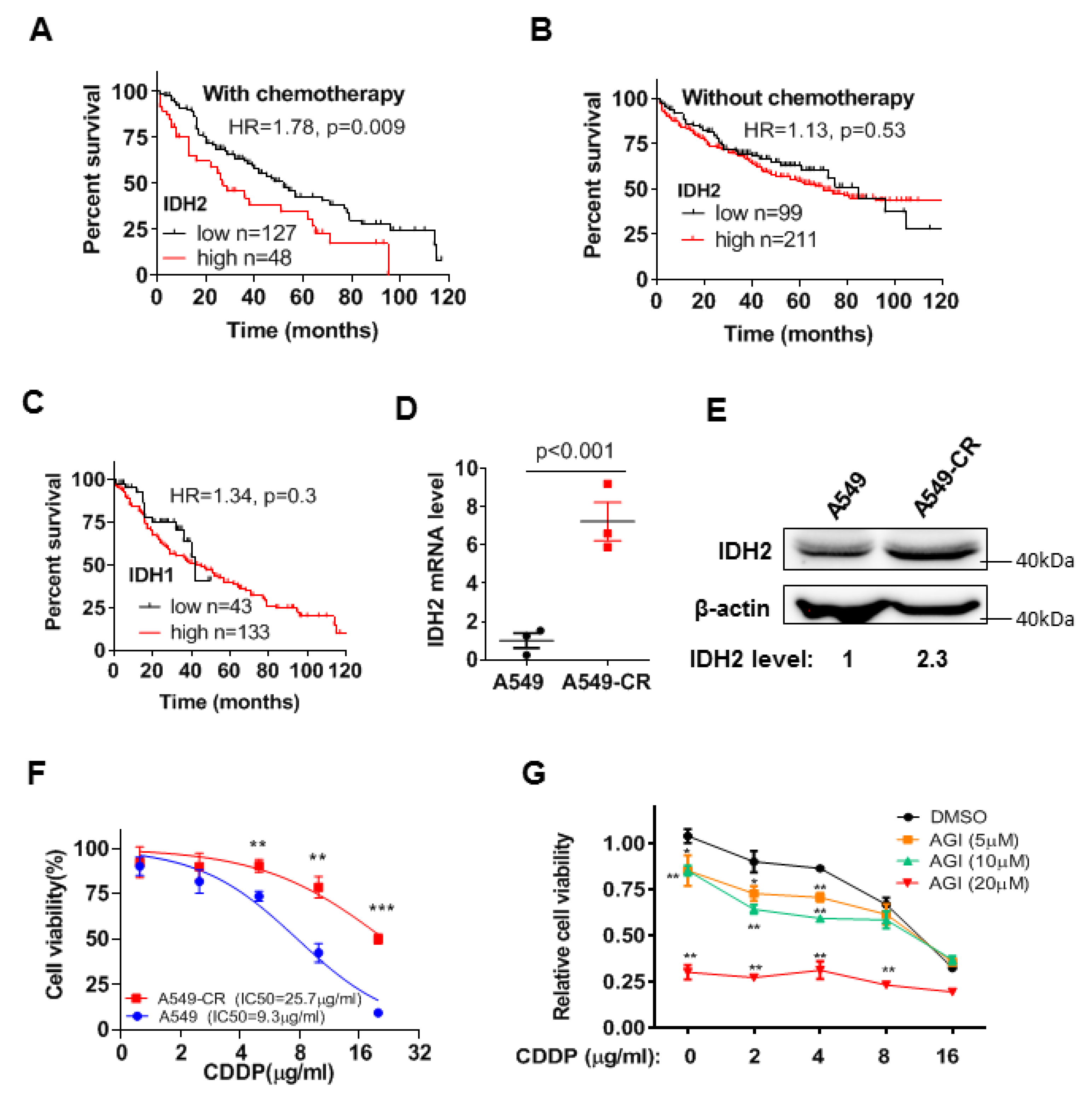
3.1. High Expression of Wild-Type IDH2 Is Correlated with Poor Survival of Lung Cancer Patients under Chemotherapy and Also Promotes Drug Resistance
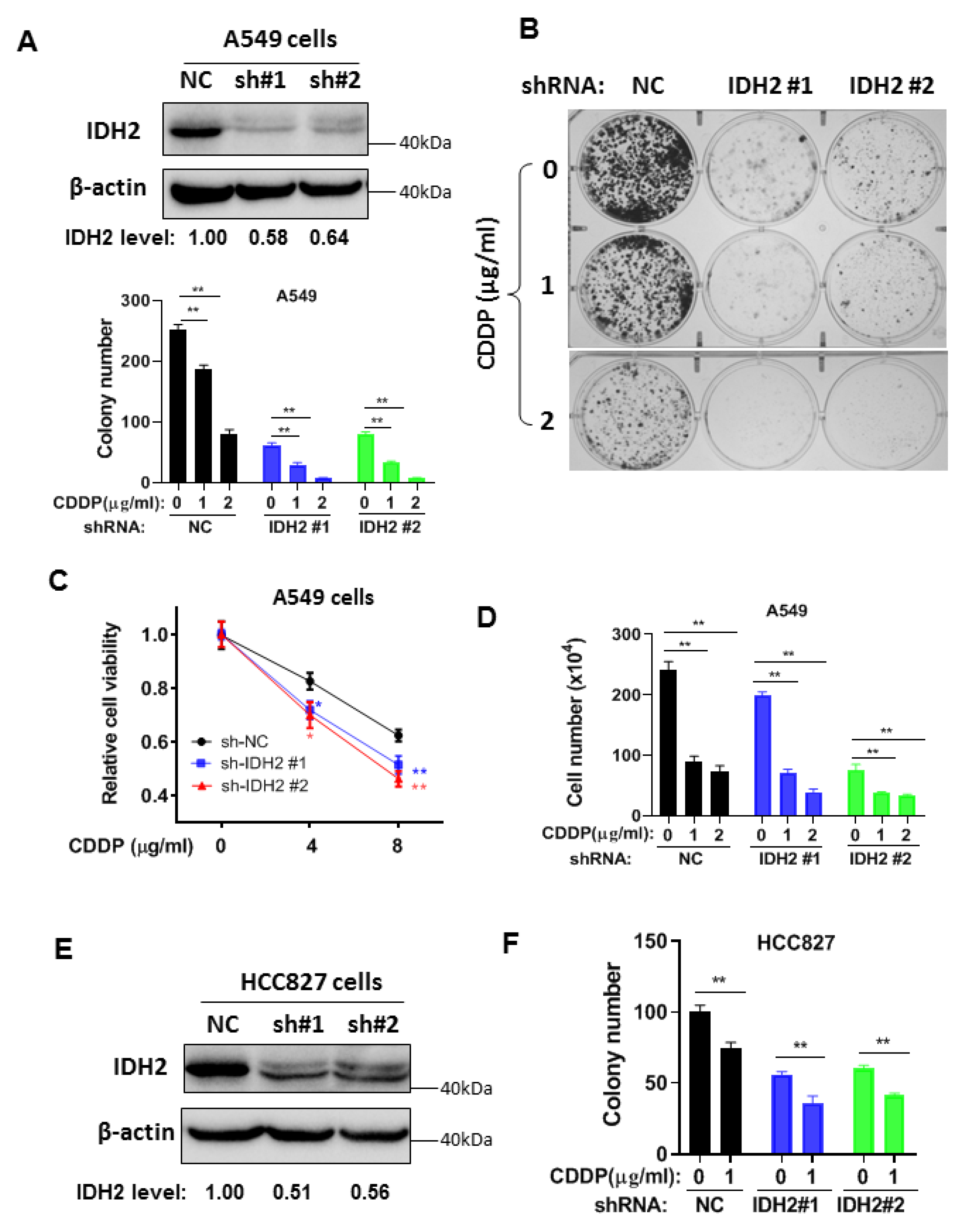
3.2. Abrogation of IDH2 Enhances the Sensitivity of Lung Cancer Cells to Cisplatin and Radiation
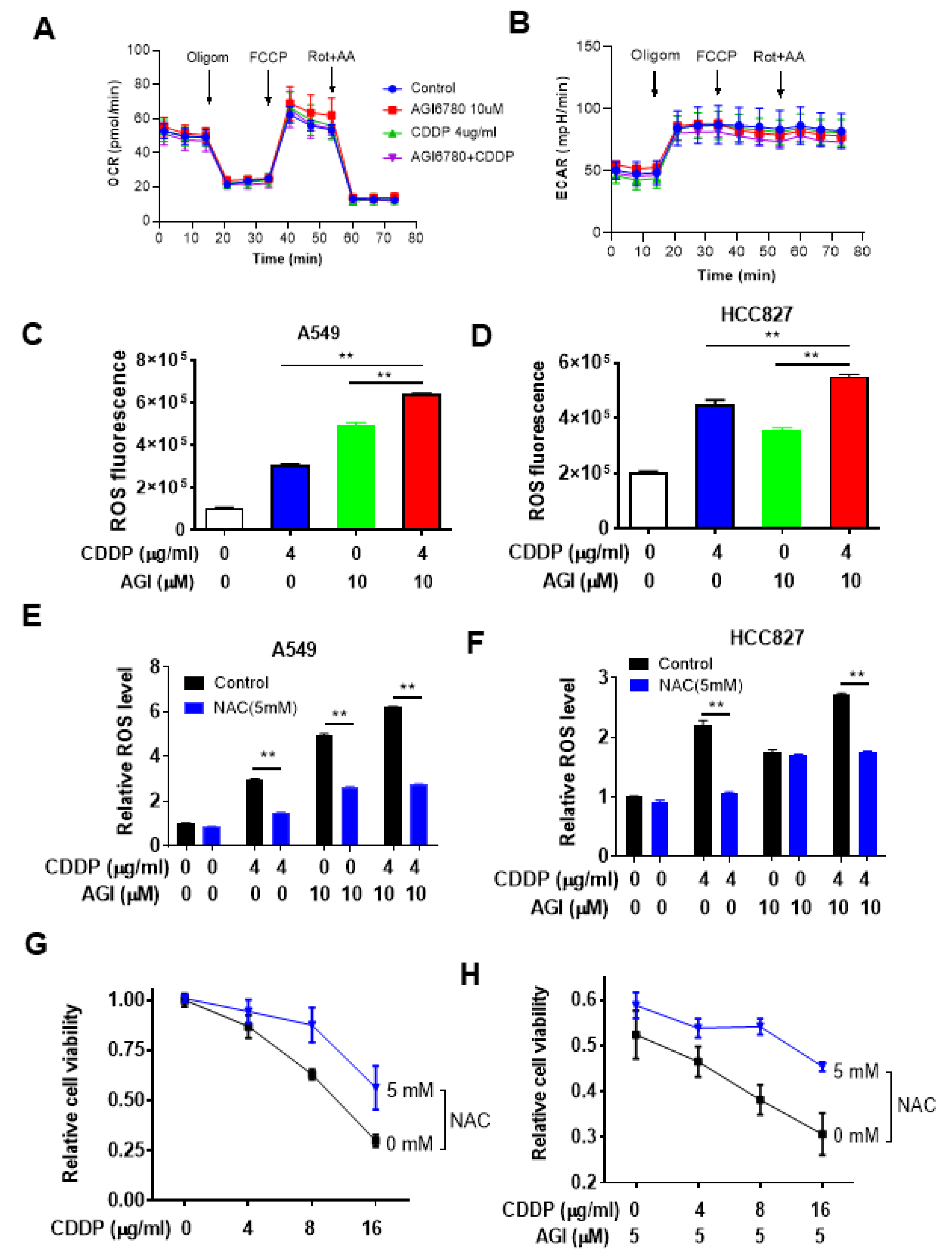
3.3. Impact of IDH2 on Mitochondrial Metabolism and ROS Generation That Affects Drug Sensitivity
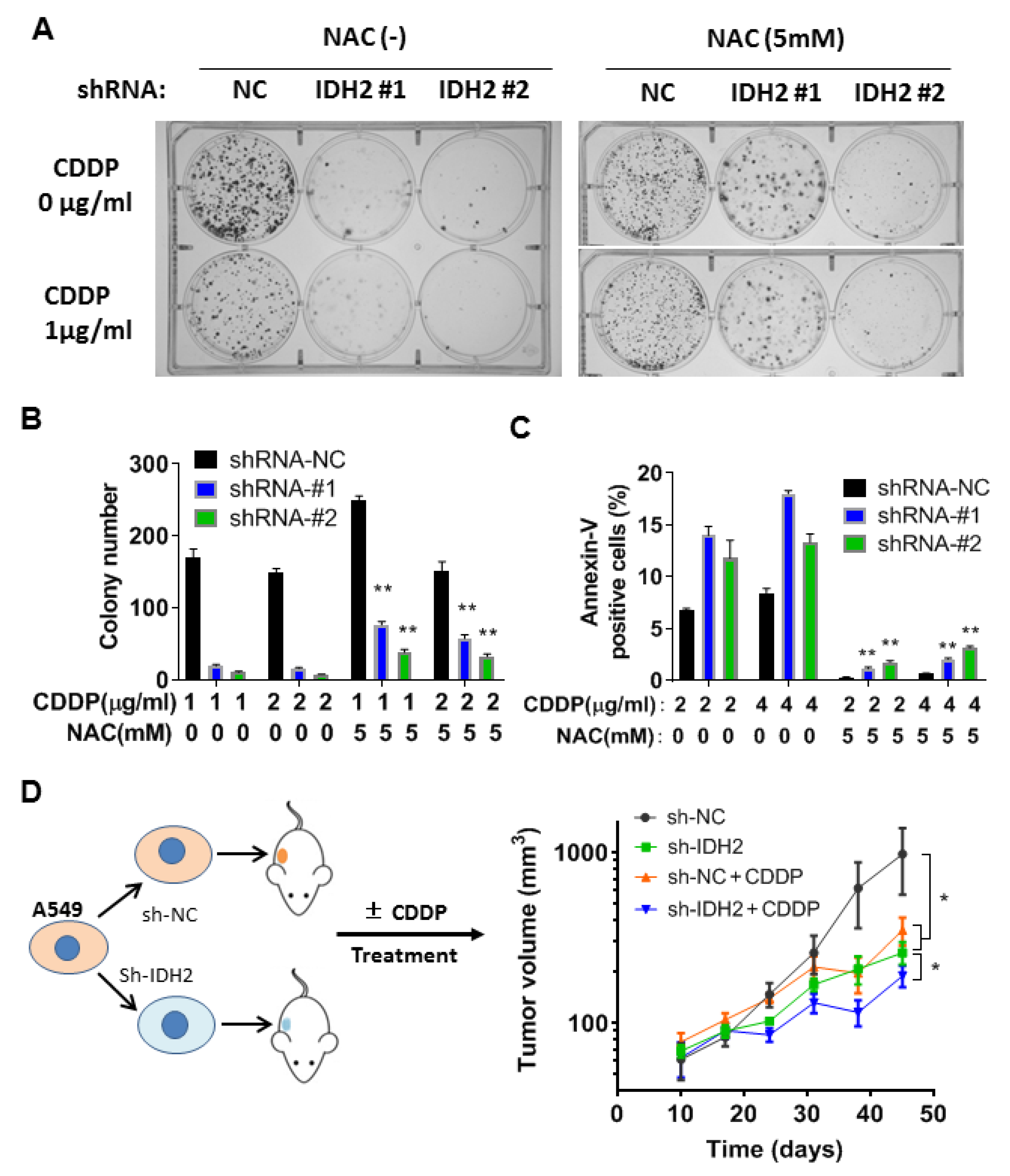
3.4. Therapeutic Effect of Cisplatin in Mice Bearing Tumor Xenografts with or without IDH2 Knockdown
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA A Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed]
- Giladi, M.; Weinberg, U.; Schneiderman, R.S.; Porat, Y.; Munster, M.; Voloshin, T.; Blatt, R.; Cahal, S.; Itzhaki, A.; Onn, A.; et al. Alternating electric fields (tumor-treating fields therapy) can improve chemotherapy treatment efficacy in non-small cell lung cancer both in vitro and in vivo. Semin. Oncol. 2014, 41, S35–S41. [Google Scholar] [CrossRef] [PubMed]
- Doroshow, D.B.; Herbst, R.S. Treatment of Advanced Non-Small Cell Lung Cancer in 2018. JAMA Oncol. 2018, 4, 569–570. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Huang, B.Y.; Yin, J.Y. Pharmacogenomics of platinum-based chemotherapy in non-small cell lung cancer: Focusing on DNA repair systems. Med. Oncol. 2017, 34, 48. [Google Scholar] [CrossRef]
- Fennell, D.A.; Summers, Y.; Cadranel, J.; Benepal, T.; Christoph, D.C.; Lal, R.; Das, M.; Maxwell, F.; Visseren-Grul, C.; Ferry, D. Cisplatin in the modern era: The backbone of first-line chemotherapy for non-small cell lung cancer. Cancer Treat. Rev. 2016, 44, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA A Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Denisenko, T.V.; Budkevich, I.N.; Zhivotovsky, B. Cell death-based treatment of lung adenocarcinoma. Cell Death Dis. 2018, 9, 117. [Google Scholar] [CrossRef]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [Green Version]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Peiris-Pages, M.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer metabolism: A therapeutic perspective. Nat. Rev. Clin. Oncol. 2017, 14, 11–31. [Google Scholar] [CrossRef] [PubMed]
- Panieri, E.; Santoro, M.M. ROS homeostasis and metabolism: A dangerous liason in cancer cells. Cell Death Dis. 2016, 7, e2253. [Google Scholar] [CrossRef] [PubMed]
- Qiao, S.; Lu, W.; Glorieux, C.; Li, J.; Zeng, P.; Meng, N.; Zhang, H.; Wen, S.; Huang, P. Wild-type IDH2 protects nuclear DNA from oxidative damage and is a potential therapeutic target in colorectal cancer. Oncogene 2021, 40, 5880–5892. [Google Scholar] [CrossRef]
- Zeng, P.; Lu, W.; Tian, J.; Qiao, S.; Li, J.; Glorieux, C.; Wen, S.; Zhang, H.; Li, Y.; Huang, P. Reductive, TCA. cycle catalyzed by wild-type IDH2 promotes acute myeloid leukemia and is a metabolic vulnerability for potential targeted therapy. J. Hematol. Oncol. 2022, 15, 30. [Google Scholar] [CrossRef]
- Short, N.J.; Konopleva, M.; Kadia, T.M.; Borthakur, G.; Ravandi, F.; DiNardo, C.D.; Daver, N. Advances in the Treatment of Acute Myeloid Leukemia: New Drugs and New Challenges. Cancer Discov. 2020, 10, 506–525. [Google Scholar] [CrossRef]
- Yu, W.; Dittenhafer-Reed, K.E.; Denu, J.M. SIRT3 protein deacetylates isocitrate dehydrogenase 2 (IDH2) and regulates mitochondrial redox status. J. Biol. Chem. 2012, 287, 14078–14086. [Google Scholar] [CrossRef]
- Zhou, L.; Wang, F.; Sun, R.; Chen, X.; Zhang, M.; Xu, Q.; Wang, Y.; Wang, S.; Xiong, Y.; Guan, K.L.; et al. SIRT5 promotes IDH2 desuccinylation and G6PD deglutarylation to enhance cellular antioxidant defense. EMBO Rep. 2016, 17, 811–822. [Google Scholar] [CrossRef]
- Wise, D.R.; Ward, P.S.; Shay, J.E.; Cross, J.R.; Gruber, J.J.; Sachdeva, U.M.; Platt, J.M.; DeMatteo, R.G.; Simon, M.C.; Thompson, C.B. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of alpha-ketoglutarate to citrate to support cell growth and viability. Proc. Natl. Acad. Sci. USA 2011, 108, 19611–19616. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Shestov, A.A.; Swain, P.; Yang, C.; Parker, S.J.; Wang, Q.A.; Terada, L.S.; Adams, N.D.; McCabe, M.T.; Pietrak, B.; et al. Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature 2016, 532, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Xu, W.; Wang, C.; Liu, F.; Guan, S.; Sun, Y.; Wang, X.; An, D.; Wen, Z.; Chen, P.; et al. The clinical significance of isocitrate dehydrogenase 2 in esophageal squamous cell carcinoma. Am. J. Cancer Res. 2017, 7, 700–714. [Google Scholar]
- Li, J.; He, Y.; Tan, Z.; Lu, J.; Li, L.; Song, X.; Shi, F.; Xie, L.; You, S.; Luo, X.; et al. Wild-type IDH2 promotes the Warburg effect and tumor growth through HIF1alpha in lung cancer. Theranostics 2018, 8, 4050–4061. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S. Cisplatin: The first metal based anticancer drug. Bioorg. Chem. 2019, 88, 102925. [Google Scholar] [CrossRef] [PubMed]
- Dilruba, S.; Kalayda, G.V. Platinum-based drugs: Past, present and future. Cancer Chemother. Pharm. 2016, 77, 1103–1124. [Google Scholar] [CrossRef]
- Amable, L. Cisplatin resistance and opportunities for precision medicine. Pharm. Res. 2016, 106, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Romani, A.M.P. Cisplatin in cancer treatment. Biochem. Pharm. 2022, 206, 115323. [Google Scholar] [CrossRef]
- Wangpaichitr, M.; Wu, C.; Li, Y.Y.; Nguyen, D.J.M.; Kandemir, H.; Shah, S.; Chen, S.; Feun, L.G.; Prince, J.S.; Kuo, M.T.; et al. Exploiting ROS and metabolic differences to kill cisplatin resistant lung cancer. Oncotarget 2017, 8, 49275–49292. [Google Scholar] [CrossRef]
- Bian, C.; Zheng, Z.; Su, J.; Wang, H.; Chang, S.; Xin, Y.; Jiang, X. Targeting Mitochondrial Metabolism to Reverse Radioresistance: An Alternative to Glucose Metabolism. Antioxidants 2022, 11, 2202. [Google Scholar] [CrossRef]
- Dohner, H.; Wei, A.H.; Lowenberg, B. Towards precision medicine for AML. Nat. Rev. Clin. Oncol. 2021, 18, 577–590. [Google Scholar] [CrossRef]
- de la Fuente, M.I. Targeting IDH1/IDH2 mutations in gliomas. Curr. Opin. Neurol. 2022, 35, 787–793. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Li, J.-j.; Lu, W.; Yang, J.; Xia, Y.; Huang, P. Targeting Mitochondrial IDH2 Enhances Antitumor Activity of Cisplatin in Lung Cancer via ROS-Mediated Mechanism. Biomedicines 2023, 11, 475. https://doi.org/10.3390/biomedicines11020475
Li H, Li J-j, Lu W, Yang J, Xia Y, Huang P. Targeting Mitochondrial IDH2 Enhances Antitumor Activity of Cisplatin in Lung Cancer via ROS-Mediated Mechanism. Biomedicines. 2023; 11(2):475. https://doi.org/10.3390/biomedicines11020475
Chicago/Turabian StyleLi, He, Jiang-jiang Li, Wenhua Lu, Jing Yang, Yunfei Xia, and Peng Huang. 2023. "Targeting Mitochondrial IDH2 Enhances Antitumor Activity of Cisplatin in Lung Cancer via ROS-Mediated Mechanism" Biomedicines 11, no. 2: 475. https://doi.org/10.3390/biomedicines11020475
APA StyleLi, H., Li, J.-j., Lu, W., Yang, J., Xia, Y., & Huang, P. (2023). Targeting Mitochondrial IDH2 Enhances Antitumor Activity of Cisplatin in Lung Cancer via ROS-Mediated Mechanism. Biomedicines, 11(2), 475. https://doi.org/10.3390/biomedicines11020475