

A Strategy for the Selection of RT-qPCR Reference Genes Based on Publicly Available Transcriptomic Datasets
 , , , ,
, , , ,  and
and {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Systematic Review of qPCR Normalizing Genes Used in Human Studies
2.2. Interrogation of Gene Expression Omnibus Datasets of Plasma Cell Dyscrasias
2.3. Primer Design
2.4. Cell Culture
2.5. Isolation of Primary Bone-Marrow-Derived Plasma Cells
2.6. RNA Extraction and cDNA Synthesis
2.7. Real-Time qPCR
2.8. Statistical Analyses
3. Results
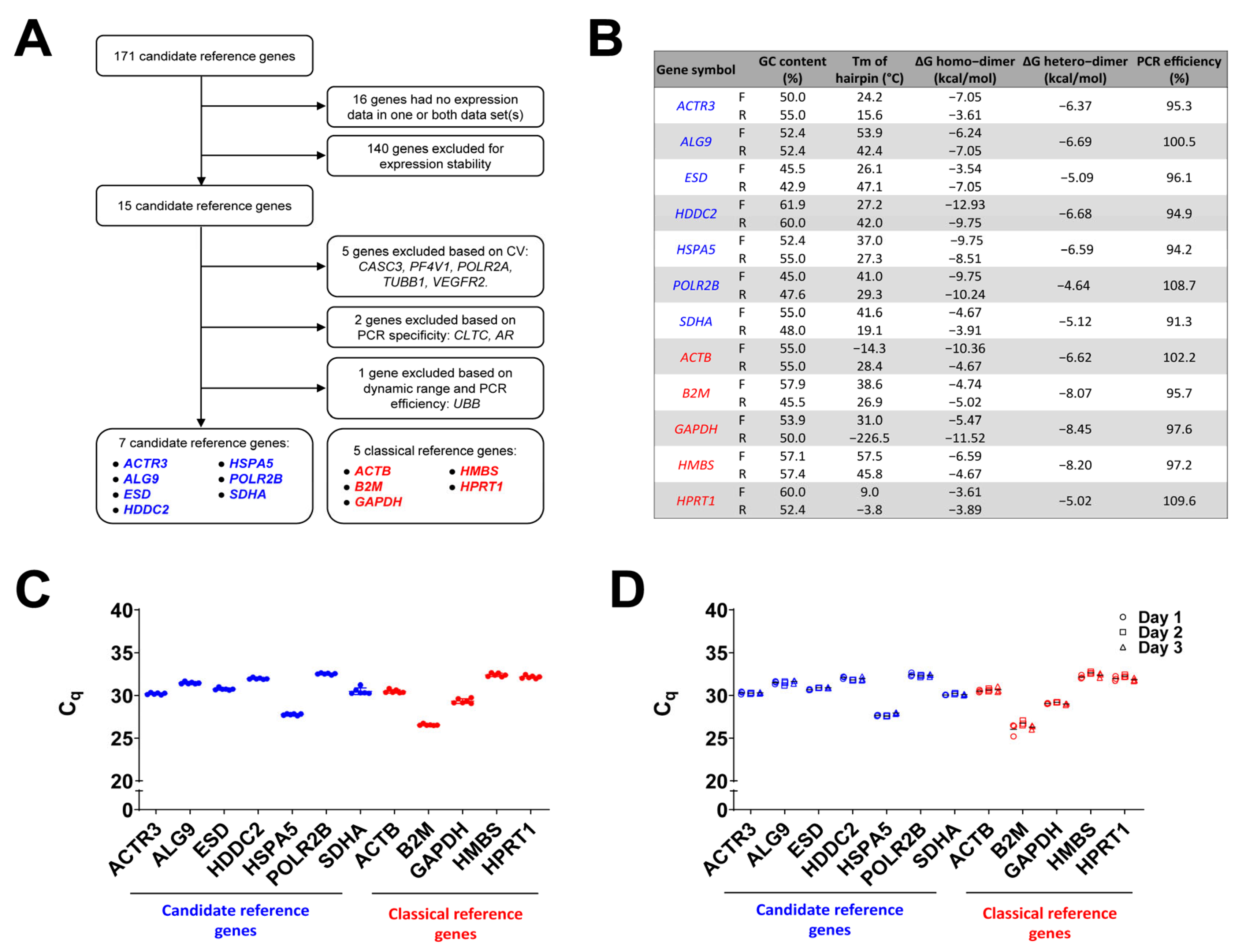
3.1. Identification of Candidate Reference Genes
3.2. Filtering of Candidate Reference Genes Based on Plasma Cell Expression Profiles
3.3. Primer Design and Validation
3.4. Expression Profiles of Investigated Genes in Primary Amyloidogenic and Control Plasma Cells
3.5. Expression Stability of Investigated Genes in Primary Amyloidogenic and Control Plasma Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huggett, J.; Dheda, K.; Bustin, S.; Zumla, A. Real-time RT-PCR normalisation; strategies and considerations. Genes. Immun. 2005, 6, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A.; Benes, V.; Garson, J.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.; et al. The need for transparency and good practices in the qPCR literature. Nat. Methods 2013, 10, 1063–1067. [Google Scholar] [CrossRef]
- Bernard, P.S.; Wittwer, C.T. Real-time PCR technology for cancer diagnostics. Clin. Chem. 2002, 48, 1178–1185. [Google Scholar] [CrossRef] [PubMed]
- Mackay, I.M.; Arden, K.E.; Nitsche, A. Real-time PCR in virology. Nucleic Acids Res. 2002, 30, 1292–1305. [Google Scholar] [CrossRef] [PubMed]
- Mackay, I.M. Real-time PCR in the microbiology laboratory. Clin. Microbiol. Infect. 2004, 10, 190–212. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A.; Mueller, R. Real-time reverse transcription PCR and the detection of occult disease in colorectal cancer. Mol. Asp. Med. 2006, 27, 192–223. [Google Scholar] [CrossRef]
- Bustin, S.A.; Mueller, R. Real-time reverse transcription PCR (qRT-PCR) and its potential use in clinical diagnosis. Clin. Sci. 2005, 109, 365–379. [Google Scholar] [CrossRef]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef]
- Dijkstra, J.R.; van Kempen, L.C.; Nagtegaal, I.D.; Bustin, S.A. Critical appraisal of quantitative PCR results in colorectal cancer research: Can we rely on published qPCR results? Mol. Oncol. 2014, 8, 813–818. [Google Scholar] [CrossRef]
- Tanaka, S.; Furukawa, T.; Plotkin, S.A. Human cytomegalovirus stimulates host cell RNA synthesis. J. Virol. 1975, 15, 297–304. [Google Scholar] [CrossRef]
- Piechaczyk, M.; Blanchard, J.M.; Marty, L.; Dani, C.; Panabieres, F.; El Sabouty, S.; Fort, P.; Jeanteur, P. Post-transcriptional regulation of glyceraldehyde-3-phosphate-dehydrogenase gene expression in rat tissues. Nucleic Acids Res. 1984, 12, 6951–6963. [Google Scholar] [CrossRef]
- Stout, J.T.; Chen, H.Y.; Brennand, J.; Caskey, C.T.; Brinster, R.L. Expression of human HPRT in the central nervous system of transgenic mice. Nature 1985, 317, 250–252. [Google Scholar] [CrossRef]
- Goidin, D.; Mamessier, A.; Staquet, M.J.; Schmitt, D.; Berthier-Vergnes, O. Ribosomal 18S RNA prevails over glyceraldehyde-3-phosphate dehydrogenase and beta-actin genes as internal standard for quantitative comparison of mRNA levels in invasive and noninvasive human melanoma cell subpopulations. Anal. Biochem. 2001, 295, 17–21. [Google Scholar] [CrossRef]
- Nazari, F.; Parham, A.; Maleki, A.F. GAPDH, beta-actin and beta2-microglobulin, as three common reference genes, are not reliable for gene expression studies in equine adipose- and marrow-derived mesenchymal stem cells. J. Anim. Sci. Technol. 2015, 57, 18. [Google Scholar] [CrossRef]
- Zhou, L.; Lim, Q.E.; Wan, G.; Too, H.P. Normalization with genes encoding ribosomal proteins but not GAPDH provides an accurate quantification of gene expressions in neuronal differentiation of PC12 cells. BMC Genom. 2010, 11, 75. [Google Scholar] [CrossRef]
- Yang, X.; Hatfield, J.T.; Hinze, S.J.; Mu, X.; Anderson, P.J.; Powell, B.C. Bone to pick: The importance of evaluating reference genes for RT-qPCR quantification of gene expression in craniosynostosis and bone-related tissues and cells. BMC Res. Notes 2012, 5, 222. [Google Scholar] [CrossRef]
- Bas, A.; Forsberg, G.; Hammarström, S.; Hammarström, M.L. Utility of the housekeeping genes 18S rRNA, β-actin and glyceraldehyde-3-phosphate-dehydrogenase for normalization in real-time quantitative reverse transcriptase-polymerase chain reaction analysis of gene expression in human T lymphocytes. Scand. J. Immunol. 2004, 59, 566–573. [Google Scholar] [CrossRef]
- Bémeur, C.; Ste-Marie, L.; Desjardins, P.; Hazell, A.S.; Vachon, L.; Butterworth, R.; Montgomery, J. Decreased β-actin mRNA expression in hyperglycemic focal cerebral ischemia in the rat. Neurosci. Lett. 2004, 357, 211–214. [Google Scholar] [CrossRef]
- Schulz, W.A.; Eickelmann, P.; Hallbrucker, C.; Sies, H.; Ha, D. Increase of β-actin mRNA upon hypotonic perfusion of perfused rat liver. FEBS Lett. 1991, 292, 264–266. [Google Scholar]
- Tricarico, C.; Pinzani, P.; Bianchi, S.; Paglierani, M.; Distante, V.; Pazzagli, M.; Bustin, S.A.; Orlando, C. Quantitative real-time reverse transcription polymerase chain reaction: Normalization to rRNA or single housekeeping genes is inappropriate for human tissue biopsies. Anal. Biochem. 2002, 309, 293–300. [Google Scholar] [CrossRef]
- Koch, I.; Weil, R.; Wolbold, R.; Brockmöller, J.; Hustert, E.; Burk, O.; Nuessler, A.; Neuhaus, P.; Eichelbaum, M.; Zanger, U. Interindividual variability and tissue-specificity in the expression of cytochrome P450 3A mRNA. Drug. Metab. Dispos. 2002, 30, 1108–1114. [Google Scholar] [CrossRef] [PubMed]
- Hampton, T.H.; Koeppen, K.; Bashor, L.; Stanton, B.A. Selection of reference genes for quantitative PCR: Identifying reference genes for airway epithelial cells exposed to Pseudomonas aeruginosa. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 319, L256–L265. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Li, Y.; Luo, D.; Liao, D.J. Pseudogenes as weaknesses of ACTB (Actb) and GAPDH (Gapdh) used as reference genes in reverse transcription and polymerase chain reactions. PLoS ONE 2012, 7, e41659. [Google Scholar] [CrossRef] [PubMed]
- Dheda, K.; Huggett, J.F.; Chang, J.S.; Kim, L.U.; Bustin, S.A.; Johnson, M.A.; Rook, G.A.; Zumla, A. The implications of using an inappropriate reference gene for real-time reverse transcription PCR data normalization. Anal. Biochem. 2005, 344, 141–143. [Google Scholar] [CrossRef]
- Piehler, A.P.; Grimholt, R.M.; Ovstebo, R.; Berg, J.P. Gene expression results in lipopolysaccharide-stimulated monocytes depend significantly on the choice of reference genes. BMC Immunol. 2010, 11, 21. [Google Scholar] [CrossRef]
- Kanadia, R.N.; Cepko, R.L. Alternative splicing produces high levels of noncoding isoforms of bHLH transcription factors during development. Genes. Dev. 2011, 25, 1344. [Google Scholar] [CrossRef]
- Alberts, B. Detection of an infectious retrovirus, XMRV, in blood cells of patients with chronic fatigue syndrome. Science 2011, 334, 1636. [Google Scholar] [CrossRef]
- Bohlenius, H.; Eriksson, S.; Parcy, F.; Nilsson, O. Retraction. Science 2007, 316, 367. [Google Scholar] [CrossRef]
- Takahashi, K.; Ikeda, N.; Nonoguchi, N.; Kajimoto, Y.; Miyatake, S.; Hagiya, Y.; Ogura, S.; Nakagawa, H.; Ishikawa, T.; Kuroiwa, T. Enhanced expression of coproporphyrinogen oxidase in malignant brain tumors: CPOX expression and 5-ALA-induced fluorescence. Neuro Oncol. 2011, 13, 1234–1243. [Google Scholar] [CrossRef]
- Sanders, R.; Bustin, S.; Huggett, J.; Mason, D. Improving the standardization of mRNA measurement by RT-qPCR. Biomol. Detect. Quantif. 2018, 15, 13–17. [Google Scholar] [CrossRef]
- Bustin, S.A.; Mueller, R.; Nolan, T. Parameters for Successful PCR Primer Design. Methods Mol. Biol. 2020, 2065, 5–22. [Google Scholar]
- Bustin, S.A.; Wittwer, C.T. MIQE: A Step Toward More Robust and Reproducible Quantitative PCR. Clin. Chem. 2017, 63, 1537–1538. [Google Scholar] [CrossRef]
- Bustin, S.A. The reproducibility of biomedical research: Sleepers awake! Biomol. Detect. Quantif. 2014, 2, 35–42. [Google Scholar] [CrossRef]
- Ceelen, L.; De Spiegelaere, W.; David, M.; De Craene, J.; Vinken, M.; Vanhaecke, T.; Rogiers, V. Critical selection of reliable reference genes for gene expression study in the HepaRG cell line. Biochem. Pharm. 2011, 81, 1255–1261. [Google Scholar] [CrossRef]
- Lowe, R.; Shirley, N.; Bleackley, M.; Dolan, S.; Shafee, T. Transcriptomics technologies. PLoS Comput. Biol. 2017, 13, e1005457. [Google Scholar] [CrossRef]
- Dai, Y.; Cao, Y.; Kohler, J.; Lu, A.; Xu, S.; Wang, H. Unbiased RNA-Seq-driven identification and validation of reference genes for quantitative RT-PCR analyses of pooled cancer exosomes. BMC Genom. 2021, 22, 27. [Google Scholar] [CrossRef]
- Alvelos, M.I.; Szymczak, F.; Castela, A.; Marin-Canas, S.; de Souza, B.M.; Gkantounas, I.; Colli, M.; Fantuzzi, F.; Cosentino, C.; Igoillo-Esteve, M.; et al. A functional genomic approach to identify reference genes for human pancreatic beta cell real-time quantitative RT-PCR analysis. Islets 2021, 13, 51–65. [Google Scholar] [CrossRef]
- Merlini, G.; Stone, M.J. Dangerous small B-cell clones. Blood 2006, 108, 2520–2530. [Google Scholar] [CrossRef]
- Paiva, B.; Martinez-Lopez, J.; Corchete, L.A.; Sanchez-Vega, B.; Rapado, I.; Puig, N.; Barrio, S.; Sanchez, M.L.; Alignani, D.; Lasa, M.; et al. Phenotypic, transcriptomic, and genomic features of clonal plasma cells in light-chain amyloidosis. Blood 2016, 127, 3035–3039. [Google Scholar] [CrossRef]
- Tiedemann, R.E.; Zhu, Y.X.; Schmidt, J.; Yin, H.; Shi, C.X.; Que, Q.; Basu, G.; Azorsa, D.; Perkins, L.M.; Braggio, E.; et al. Kinome-wide RNAi studies in human multiple myeloma identify vulnerable kinase targets, including a lymphoid-restricted kinase, GRK6. Blood 2010, 115, 1594–1604. [Google Scholar] [CrossRef]
- Chng, W.J.; Kumar, S.; Vanwier, S.; Ahmann, G.; Price-Troska, T.; Henderson, K.; Chung, T.H.; Kim, S.; Mulligan, G.; Bryant, B.; et al. Molecular dissection of hyperdiploid multiple myeloma by gene expression profiling. Cancer Res. 2007, 67, 2982–2989. [Google Scholar] [CrossRef] [PubMed]
- Lefever, S.; Pattyn, F.; Hellemans, J.; Vandesompele, J. Single-nucleotide polymorphisms and other mismatches reduce performance of quantitative PCR assays. Clin. Chem. 2013, 59, 1470–1480. [Google Scholar] [CrossRef] [PubMed]
- Arendt, B.K.; Ramirez-Alvarado, M.; Sikkink, L.A.; Keats, J.J.; Ahmann, G.J.; Dispenzieri, A.; Fonseca, R.; Ketterling, R.P.; Knudson, R.A.; Mulvihill, E.M.; et al. Biologic and genetic characterization of the novel amyloidogenic lambda light chain-secreting human cell lines, ALMC-1 and ALMC-2. Blood 2008, 112, 1931–1941. [Google Scholar] [CrossRef] [PubMed]
- Cascino, P.; Nevone, A.; Piscitelli, M.; Scopelliti, C.; Girelli, M.; Mazzini, G.; Caminito, S.; Russo, G.; Milani, P.; Basset, M.; et al. Single-molecule real-time sequencing of the M protein: Toward personalized medicine in monoclonal gammopathies. Am. J. Hematol. 2022, 97, E389–E392. [Google Scholar] [CrossRef] [PubMed]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, research0034. [Google Scholar] [CrossRef]
- Xie, F.; Xiao, P.; Chen, D.; Xu, L.; Zhang, B. miRDeepFinder: A miRNA analysis tool for deep sequencing of plant small RNAs. Plant. Mol. Biol. 2012, 80, 75–84. [Google Scholar] [CrossRef]
- Andersen, C.L.; Jensen, J.L.; Orntoft, T.F. Normalization of real-time quantitative reverse transcription-PCR data: A model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004, 64, 5245–5250. [Google Scholar] [CrossRef]
- Pfaffl, M.W.; Tichopad, A.; Prgomet, C.; Neuvians, T.P. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper--Excel-based tool using pair-wise correlations. Biotechnol. Lett. 2004, 26, 509–515. [Google Scholar] [CrossRef]
- Silver, N.; Best, S.; Jiang, J.; Thein, S.L. Selection of housekeeping genes for gene expression studies in human reticulocytes using real-time PCR. BMC Mol. Biol. 2006, 7, 33. [Google Scholar] [CrossRef]
- Kourelis, T.V.; Kumar, S.K.; Gertz, M.A.; Lacy, M.Q.; Buadi, F.K.; Hayman, S.R.; Zeldenrust, S.; Leung, N.; Kyle, R.A.; Russell, S.; et al. Coexistent multiple myeloma or increased bone marrow plasma cells define equally high-risk populations in patients with immunoglobulin light chain amyloidosis. J. Clin. Oncol. 2013, 31, 4319–4324. [Google Scholar] [CrossRef]
- Eisenberg, E.; Levanon, E.Y. Human housekeeping genes, revisited. Trends Genet. 2013, 29, 569–574. [Google Scholar] [CrossRef]
- De Jonge, H.J.; Fehrmann, R.S.; de Bont, E.S.; Hofstra, R.M.; Gerbens, F.; Kamps, W.A.; de Vries, E.G.; van der Zee, A.G.; te Meerman, G.J.; ter Elst, A. Evidence based selection of housekeeping genes. PLoS ONE 2007, 2, e898. [Google Scholar] [CrossRef]
- Scarlet, D.; Ertl, R.; Aurich, C.; Steinborn, R. The Orthology Clause in the Next Generation Sequencing Era: Novel Reference Genes Identified by RNA-seq in Humans Improve Normalization of Neonatal Equine Ovary RT-qPCR Data. PLoS ONE 2015, 10, e0142122. [Google Scholar] [CrossRef]
- Suzuki, T.; Higgins, P.J.; Crawford, D.R. Control selection for RNA quantitation. Biotechniques 2000, 29, 332–337. [Google Scholar] [CrossRef]
- Koeppen, K.; Stanton, B.A.; Hampton, T.H. ScanGEO: Parallel mining of high-throughput gene expression data. Bioinformatics 2017, 33, 3500–3501. [Google Scholar] [CrossRef]
- Eisenberg, E.; Levanon, E.Y. A-to-I RNA editing—Immune protector and transcriptome diversifier. Nat. Rev. Genet. 2018, 19, 473–490. [Google Scholar] [CrossRef]
- Nishikura, K. Functions and regulation of RNA editing by ADAR deaminases. Annu. Rev. Biochem. 2010, 79, 321–349. [Google Scholar] [CrossRef]
- Picardi, E.; D’Erchia, A.M.; Lo Giudice, C.; Pesole, G. REDIportal: A comprehensive database of A-to-I RNA editing events in humans. Nucleic Acids Res. 2017, 45, D750–D757. [Google Scholar] [CrossRef]
- Milani, P.; Merlini, G. Monoclonal IgM-related AL amyloidosis. Best. Pract. Res. Clin. Haematol. 2016, 29, 241–248. [Google Scholar] [CrossRef]
- Basset, M.; Defrancesco, I.; Milani, P.; Nuvolone, M.; Rattotti, S.; Foli, A.; Mangiacavalli, S.; Varettoni, M.; Benvenuti, P.; Cartia, C.S.; et al. Nonlymphoplasmacytic lymphomas associated with light-chain amyloidosis. Blood 2020, 135, 293–296. [Google Scholar] [CrossRef]
- Fonseca, R.; Ahmann, G.J.; Jalal, S.M.; Dewald, G.W.; Larson, D.R.; Therneau, T.M.; Gertz, M.A.; Kyle, R.A.; Greipp, P.R. Chromosomal abnormalities in systemic amyloidosis. Br. J. Haematol. 1998, 103, 704–710. [Google Scholar] [CrossRef] [PubMed]
- Hayman, S.R.; Bailey, R.J.; Jalal, S.M.; Ahmann, G.J.; Dispenzieri, A.; Gertz, M.A.; Greipp, P.R.; Kyle, R.A.; Lacy, M.Q.; Rajkumar, S.V.; et al. Translocations involving the immunoglobulin heavy-chain locus are possible early genetic events in patients with primary systemic amyloidosis. Blood 2001, 98, 2266–2268. [Google Scholar] [CrossRef] [PubMed]
- Perfetti, V.; Coluccia, A.M.; Intini, D.; Malgeri, U.; Vignarelli, M.C.; Casarini, S.; Merlini, G.; Neri, A. Translocation T(4;14)(p16.3;q32) is a recurrent genetic lesion in primary amyloidosis. Am. J. Pathol. 2001, 158, 1599–1603. [Google Scholar] [CrossRef] [PubMed]
- Bochtler, T.; Hegenbart, U.; Cremer, F.W.; Heiss, C.; Benner, A.; Hose, D.; Moos, M.; Bila, J.; Bartram, C.R.; Ho, A.D.; et al. Evaluation of the cytogenetic aberration pattern in amyloid light chain amyloidosis as compared with monoclonal gammopathy of undetermined significance reveals common pathways of karyotypic instability. Blood 2008, 111, 4700–4705. [Google Scholar] [CrossRef]
- Kim, S.Y.; Im, K.; Park, S.N.; Kim, J.A.; Yoon, S.S.; Lee, D.S. Burden of cytogenetically abnormal plasma cells in light chain amyloidosis and their prognostic relevance. Leuk. Res. 2016, 44, 45–52. [Google Scholar] [CrossRef]
- Muchtar, E.; Dispenzieri, A.; Kumar, S.K.; Ketterling, R.P.; Dingli, D.; Lacy, M.Q.; Buadi, F.K.; Hayman, S.R.; Kapoor, P.; Leung, N.; et al. Interphase fluorescence in situ hybridization in untreated AL amyloidosis has an independent prognostic impact by abnormality type and treatment category. Leukemia 2017, 31, 1562–1569. [Google Scholar] [CrossRef]
- Schoch, C.; Kohlmann, A.; Dugas, M.; Kern, W.; Schnittger, S.; Haferlach, T. Impact of trisomy 8 on expression of genes located on chromosome 8 in different AML subgroups. Genes. Chromosom. Cancer 2006, 45, 1164–1168. [Google Scholar] [CrossRef]
- Abasolo, N.; Torrell, H.; Roig, B.; Moyano, S.; Vilella, E.; Martorell, L. RT-qPCR study on post-mortem brain samples from patients with major psychiatric disorders: Reference genes and specimen characteristics. J. Psychiatr. Res. 2011, 45, 1411–1418. [Google Scholar] [CrossRef]
- Adeola, F. Normalization of Gene Expression by Quantitative RT-PCR in Human Cell Line: Comparison of 12 Endogenous Reference Genes. Ethiop. J. Health Sci. 2018, 28, 741–748. [Google Scholar] [CrossRef]
- Antonov, J.; Goldstein, D.R.; Oberli, A.; Baltzer, A.; Pirotta, M.; Fleischmann, A.; Altermatt, H.J.; Jaggi, R. Reliable gene expression measurements from degraded RNA by quantitative real-time PCR depend on short amplicons and a proper normalization. Lab. Investig. 2005, 85, 1040–1050. [Google Scholar] [CrossRef]
- Asp, J.; Brantsing, C.; Lovstedt, K.; Benassi, M.S.; Inerot, S.; Gamberi, G.; Picci, P.; Lindahl, A. Evaluation of p16 and Id1 status and endogenous reference genes in human chondrosarcoma by real-time PCR. Int. J. Oncol. 2005, 27, 1577–1582. [Google Scholar]
- Balogh, A.; Paragh, G., Jr.; Juhasz, A.; Kobling, T.; Torocsik, D.; Miko, E.; Varga, V.; Emri, G.; Horkay, I.; Scholtz, B.; et al. Reference genes for quantitative real time PCR in UVB irradiated keratinocytes. J. Photochem. Photobiol. B. 2008, 93, 133–139. [Google Scholar] [CrossRef]
- Borkowska, P.; Zielinska, A.; Paul-Samojedny, M.; Stojko, R.; Kowalski, J. Evaluation of reference genes for quantitative real-time PCR in Wharton’s Jelly-derived mesenchymal stem cells after lentiviral transduction and differentiation. Mol. Biol. Rep. 2020, 47, 1107–1115. [Google Scholar] [CrossRef]
- Brzeszczynska, J.; Brzeszczynski, F.; Samuel, K.; Morgan, K.; Morley, S.D.; Plevris, J.N.; Hayes, P.C. Validation of Reference Genes for Gene Expression Studies by RT-qPCR in HepaRG Cells during Toxicity Testing and Disease Modelling. Cells 2020, 9, 770. [Google Scholar] [CrossRef]
- Cai, J.; Li, T.; Huang, B.; Cheng, H.; Ding, H.; Dong, W.; Xiao, M.; Liu, L.; Wang, Z. The use of laser microdissection in the identification of suitable reference genes for normalization of quantitative real-time PCR in human FFPE epithelial ovarian tissue samples. PLoS ONE 2014, 9, e95974. [Google Scholar] [CrossRef]
- Ceelen, L.; De Craene, J.; De Spiegelaere, W. Evaluation of normalization strategies used in real-time quantitative PCR experiments in HepaRG cell line studies. Clin. Chem. 2014, 60, 451–454. [Google Scholar] [CrossRef]
- Chantawibul, S.; Anuwong, A.; Leelawat, K. Validation of appropriate reference genes for gene expression studies in human thyroid gland using real-time RT-PCR. J. Med. Assoc. Thai 2012, 95 (Suppl. S3), S36–S40. [Google Scholar]
- Chen, G.; Zhao, L.; Feng, J.; You, G.; Sun, Q.; Li, P.; Han, D.; Zhou, H. Validation of reliable reference genes for real-time PCR in human umbilical vein endothelial cells on substrates with different stiffness. PLoS ONE 2013, 8, e67360. [Google Scholar] [CrossRef]
- Chen, J.; Bao, Z.; Huang, Y.; Wang, Z.; Zhao, Y. Selection of Suitable Reference Genes for qPCR Gene Expression Analysis of HepG2 and L02 in Four Different Liver Cell Injured Models. Biomed. Res. Int. 2020, 2020, 8926120. [Google Scholar] [CrossRef]
- Chey, S.; Claus, C.; Liebert, U.G. Validation and application of normalization factors for gene expression studies in rubella virus-infected cell lines with quantitative real-time PCR. J. Cell. Biochem. 2010, 110, 118–128. [Google Scholar]
- Cicinnati, V.R.; Shen, Q.; Sotiropoulos, G.C.; Radtke, A.; Gerken, G.; Beckebaum, S. Validation of putative reference genes for gene expression studies in human hepatocellular carcinoma using real-time quantitative RT-PCR. BMC Cancer 2008, 8, 350. [Google Scholar] [CrossRef] [PubMed]
- Coulson, D.T.; Brockbank, S.; Quinn, J.G.; Murphy, S.; Ravid, R.; Irvine, G.B.; Johnston, J.A. Identification of valid reference genes for the normalization of RT qPCR gene expression data in human brain tissue. BMC Mol. Biol. 2008, 9, 46. [Google Scholar] [CrossRef] [PubMed]
- Curis, E.; Nepost, C.; Grillault Laroche, D.; Courtin, C.; Laplanche, J.L.; Etain, B.; Marie-Claire, C. Selecting reference genes in RT-qPCR based on equivalence tests: A network based approach. Sci. Rep. 2019, 9, 16231. [Google Scholar] [CrossRef] [PubMed]
- Curtis, K.M.; Gomez, L.A.; Rios, C.; Garbayo, E.; Raval, A.P.; Perez-Pinzon, M.A.; Schiller, P.C. EF1alpha and RPL13a represent normalization genes suitable for RT-qPCR analysis of bone marrow derived mesenchymal stem cells. BMC Mol. Biol. 2010, 11, 61. [Google Scholar] [CrossRef] [PubMed]
- Dang, W.; Zhang, X.; Ma, Q.; Chen, L.; Cao, M.; Miao, J.; Cui, Y.; Zhang, X. Selection of reference genes suitable for normalization of RT-qPCR data in glioma stem cells. Biotechniques 2020, 68, 130–137. [Google Scholar] [CrossRef] [PubMed]
- De Lazaro, I.; Kostarelos, K. Exposure to graphene oxide sheets alters the expression of reference genes used for real-time RT-qPCR normalization. Sci. Rep. 2019, 9, 12520. [Google Scholar] [CrossRef]
- De Lima, C.A.D.; de Lima, S.C.; Barbosa, A.D.; Sandrin-Garcia, P.; de Barros Pita, W.; de Azevedo Silva, J.; Crovella, S. Postmenopausal Osteoporosis reference genes for qPCR expression assays. Sci. Rep. 2019, 9, 16533. [Google Scholar] [CrossRef]
- Dean, B.; Udawela, M.; Scarr, E. Validating reference genes using minimally transformed qpcr data: Findings in human cortex and outcomes in schizophrenia. BMC Psychiatry 2016, 16, 154. [Google Scholar] [CrossRef]
- Del Pozo, T.; Gutierrez-Garcia, R.; Latorre, M.; Gonzalez, M.; Suazo, M. Identification of reference genes for quantitative real-time PCR studies in human cell lines under copper and zinc exposure. Biometals 2016, 29, 935–944. [Google Scholar] [CrossRef]
- El-Kashef, N.; Gomes, I.; Mercer-Chalmers-Bender, K.; Schneider, P.M.; Rothschild, M.A.; Juebner, M. Validation of adequate endogenous reference genes for reverse transcription-qPCR studies in human post-mortem brain tissue of SIDS cases. Forensic Sci. Med. Pathol. 2015, 11, 517–529. [Google Scholar] [CrossRef]
- Erickson, H.S.; Albert, P.S.; Gillespie, J.W.; Wallis, B.S.; Rodriguez-Canales, J.; Linehan, W.M.; Gonzalez, S.; Velasco, A.; Chuaqui, R.F.; Emmert-Buck, M.R. Assessment of normalization strategies for quantitative RT-PCR using microdissected tissue samples. Lab. Investig. 2007, 87, 951–962. [Google Scholar] [CrossRef] [PubMed]
- Fassunke, J.; Blum, M.C.; Schildhaus, H.U.; Zapatka, M.; Brors, B.; Kunstlinger, H.; Buttner, R.; Wardelmann, E.; Merkelbach-Bruse, S. qPCR in gastrointestinal stromal tumors: Evaluation of reference genes and expression analysis of KIT and the alternative receptor tyrosine kinases FLT3, CSF1-R, PDGFRB, MET and AXL. BMC Mol. Biol. 2010, 11, 100. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, E.; Cronje, M.J. Selection of suitable reference genes for quantitative real-time PCR in apoptosis-induced MCF-7 breast cancer cells. Mol. Biotechnol. 2012, 50, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Fjeldbo, C.S.; Aarnes, E.K.; Malinen, E.; Kristensen, G.B.; Lyng, H. Identification and Validation of Reference Genes for RT-qPCR Studies of Hypoxia in Squamous Cervical Cancer Patients. PLoS ONE 2016, 11, e0156259. [Google Scholar] [CrossRef]
- Fu, J.; Bian, L.; Zhao, L.; Dong, Z.; Gao, X.; Luan, H.; Sun, Y.; Song, H. Identification of genes for normalization of quantitative real-time PCR data in ovarian tissues. Acta Biochim. Biophys. Sin. 2010, 42, 568–574. [Google Scholar] [CrossRef]
- Fu, L.Y.; Jia, H.L.; Dong, Q.Z.; Wu, J.C.; Zhao, Y.; Zhou, H.J.; Ren, N.; Ye, Q.H.; Qin, L.X. Suitable reference genes for real-time PCR in human HBV-related hepatocellular carcinoma with different clinical prognoses. BMC Cancer 2009, 9, 49. [Google Scholar] [CrossRef]
- Gao, Q.; Wang, X.Y.; Fan, J.; Qiu, S.J.; Zhou, J.; Shi, Y.H.; Xiao, Y.S.; Xu, Y.; Huang, X.W.; Sun, J. Selection of reference genes for real-time PCR in human hepatocellular carcinoma tissues. J. Cancer Res. Clin. Oncol. 2008, 134, 979–986. [Google Scholar] [CrossRef]
- Gentile, A.M.; Lhamyani, S.; Coin-Araguez, L.; Oliva-Olivera, W.; Zayed, H.; Vega-Rioja, A.; Monteseirin, J.; Romero-Zerbo, S.Y.; Tinahones, F.J.; Bermudez-Silva, F.J.; et al. RPL13A and EEF1A1 Are Suitable Reference Genes for qPCR during Adipocyte Differentiation of Vascular Stromal Cells from Patients with Different BMI and HOMA-IR. PLoS ONE 2016, 11, e0157002. [Google Scholar] [CrossRef]
- Giricz, O.; Lauer-Fields, J.L.; Fields, G.B. The normalization of gene expression data in melanoma: Investigating the use of glyceraldehyde 3-phosphate dehydrogenase and 18S ribosomal RNA as internal reference genes for quantitative real-time PCR. Anal. Biochem. 2008, 380, 137–139. [Google Scholar] [CrossRef]
- Gonzalez-Bermudez, L.; Anglada, T.; Genesca, A.; Martin, M.; Terradas, M. Identification of reference genes for RT-qPCR data normalisation in aging studies. Sci. Rep. 2019, 9, 13970. [Google Scholar] [CrossRef]
- Green, T.M.; de Stricker, K.; Moller, M.B. Validation of putative reference genes for normalization of Q-RT-PCR data from paraffin-embedded lymphoid tissue. Diagn. Mol. Pathol. 2009, 18, 243–249. [Google Scholar] [CrossRef]
- Grube, S.; Gottig, T.; Freitag, D.; Ewald, C.; Kalff, R.; Walter, J. Selection of suitable reference genes for expression analysis in human glioma using RT-qPCR. J. Neurooncol 2015, 123, 35–42. [Google Scholar] [CrossRef]
- Hamalainen, H.K.; Tubman, J.C.; Vikman, S.; Kyrola, T.; Ylikoski, E.; Warrington, J.A.; Lahesmaa, R. Identification and validation of endogenous reference genes for expression profiling of T helper cell differentiation by quantitative real-time RT-PCR. Anal. Biochem. 2001, 299, 63–70. [Google Scholar] [CrossRef]
- Henn, D.; Bandner-Risch, D.; Perttunen, H.; Schmied, W.; Porras, C.; Ceballos, F.; Rodriguez-Losada, N.; Schafers, H.J. Identification of reference genes for quantitative RT-PCR in ascending aortic aneurysms. PLoS ONE 2013, 8, e54132. [Google Scholar] [CrossRef]
- Jacob, F.; Guertler, R.; Naim, S.; Nixdorf, S.; Fedier, A.; Hacker, N.F.; Heinzelmann-Schwarz, V. Careful selection of reference genes is required for reliable performance of RT-qPCR in human normal and cancer cell lines. PLoS ONE 2013, 8, e59180. [Google Scholar] [CrossRef]
- Jain, N.; Nitisa, D.; Pirsko, V.; Cakstina, I. Selecting suitable reference genes for qPCR normalization: A comprehensive analysis in MCF-7 breast cancer cell line. BMC Mol. Cell. Biol. 2020, 21, 68. [Google Scholar] [CrossRef]
- Janik, M.E.; Szwed, S.; Grzmil, P.; Kaczmarek, R.; Czerwinski, M.; Hoja-Lukowicz, D. RT-qPCR analysis of human melanoma progression-related genes—A novel workflow for selection and validation of candidate reference genes. Int. J. Biochem. Cell. Biol. 2018, 101, 12–18. [Google Scholar] [CrossRef]
- Javadirad, S.M.; Mokhtari, M.; Esfandiarpour, G.; Kolahdouzan, M. The pseudogene problem and RT-qPCR data normalization; SYMPK: A suitable reference gene for papillary thyroid carcinoma. Sci. Rep. 2020, 10, 18408. [Google Scholar] [CrossRef]
- Jung, M.; Ramankulov, A.; Roigas, J.; Johannsen, M.; Ringsdorf, M.; Kristiansen, G.; Jung, K. In search of suitable reference genes for gene expression studies of human renal cell carcinoma by real-time PCR. BMC Mol. Biol. 2007, 8, 47. [Google Scholar] [CrossRef]
- Kaszubowska, L.; Wierzbicki, P.M.; Karsznia, S.; Damska, M.; Slebioda, T.J.; Foerster, J.; Kmiec, Z. Optimal reference genes for qPCR in resting and activated human NK cells--Flow cytometric data correspond to qPCR gene expression analysis. J. Immunol. Methods 2015, 422, 125–129. [Google Scholar] [CrossRef]
- Khanna, P.; Johnson, K.L.; Maron, J.L. Optimal reference genes for RT-qPCR normalization in the newborn. Biotech. Histochem. 2017, 92, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Kidd, M.; Nadler, B.; Mane, S.; Eick, G.; Malfertheiner, M.; Champaneria, M.; Pfragner, R.; Modlin, I. GeneChip, geNorm, and gastrointestinal tumors: Novel reference genes for real-time PCR. Physiol. Genom. 2007, 30, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Kilic, Y.; Celebiler, A.C.; Sakizli, M. Selecting housekeeping genes as references for the normalization of quantitative PCR data in breast cancer. Clin. Transl. Oncol. 2014, 16, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Kirschneck, C.; Batschkus, S.; Proff, P.; Kostler, J.; Spanier, G.; Schroder, A. Valid gene expression normalization by RT-qPCR in studies on hPDL fibroblasts with focus on orthodontic tooth movement and periodontitis. Sci. Rep. 2017, 7, 14751. [Google Scholar] [CrossRef]
- Koppelkamm, A.; Vennemann, B.; Fracasso, T.; Lutz-Bonengel, S.; Schmidt, U.; Heinrich, M. Validation of adequate endogenous reference genes for the normalisation of qPCR gene expression data in human post mortem tissue. Int. J. Leg. Med. 2010, 124, 371–380. [Google Scholar] [CrossRef]
- Kozmus, C.E.; Potocnik, U. Reference genes for real-time qPCR in leukocytes from asthmatic patients before and after anti-asthma treatment. Gene 2015, 570, 71–77. [Google Scholar] [CrossRef]
- Leal, M.F.; Arliani, G.G.; Astur, D.C.; Franciozi, C.E.; Debieux, P.; Andreoli, C.V.; Smith, M.C.; Pochini, A.C.; Ejnisman, B.; Cohen, M. Comprehensive selection of reference genes for expression studies in meniscus injury using quantitative real-time PCR. Gene 2016, 584, 60–68. [Google Scholar] [CrossRef]
- Leal, M.F.; Astur, D.C.; Debieux, P.; Arliani, G.G.; Silveira Franciozi, C.E.; Loyola, L.C.; Andreoli, C.V.; Smith, M.C.; Pochini Ade, C.; Ejnisman, B.; et al. Identification of Suitable Reference Genes for Investigating Gene Expression in Anterior Cruciate Ligament Injury by Using Reverse Transcription-Quantitative PCR. PLoS ONE 2015, 10, e0133323. [Google Scholar] [CrossRef]
- Leduc, V.; Legault, V.; Dea, D.; Poirier, J. Normalization of gene expression using SYBR green qPCR: A case for paraoxonase 1 and 2 in Alzheimer’s disease brains. J. Neurosci. Methods 2011, 200, 14–19. [Google Scholar] [CrossRef]
- Leitao Mda, C.; Coimbra, E.C.; de Lima Rde, C.; Guimaraes Mde, L.; Heraclio Sde, A.; Silva Neto Jda, C.; de Freitas, A.C. Quantifying mRNA and microRNA with qPCR in cervical carcinogenesis: A validation of reference genes to ensure accurate data. PLoS ONE 2014, 9, e111021. [Google Scholar]
- Li, L.; Yan, Y.; Xu, H.; Qu, T.; Wang, B. Selection of reference genes for gene expression studies in ultraviolet B-irradiated human skin fibroblasts using quantitative real-time PCR. BMC Mol. Biol. 2011, 12, 8. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Diao, H.; Zhao, L.; Xing, Y.; Zhang, J.; Liu, N.; Yan, Y.; Tian, X.; Sun, W.; Liu, B. Identification of suitable reference genes for real-time quantitative PCR analysis of hydrogen peroxide-treated human umbilical vein endothelial cells. BMC Mol. Biol. 2017, 18, 10. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, Q.; Bai, J.; Xuan, Y.; Wang, Y. Identification of appropriate reference genes for human mesenchymal stem cell analysis by quantitative real-time PCR. Biotechnol. Lett. 2015, 37, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, Q.; Bai, J.; Yang, Y.; Zhong, L.; Wang, Y. Identification of optimal reference genes for quantitative PCR studies on human mesenchymal stem cells. Mol. Med. Rep. 2015, 11, 1304–1311. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lu, H.; Ji, Y.; Wu, S.; Yang, Y. Identification of genes for normalization of real-time RT-PCR data in placental tissues from intrahepatic cholestasis of pregnancy. Placenta 2016, 48, 133–135. [Google Scholar] [CrossRef]
- Liu, L.L.; Zhao, H.; Ma, T.F.; Ge, F.; Chen, C.S.; Zhang, Y.P. Identification of valid reference genes for the normalization of RT-qPCR expression studies in human breast cancer cell lines treated with and without transient transfection. PLoS ONE 2015, 10, e0117058. [Google Scholar] [CrossRef]
- Liu, S.; Zhu, P.; Zhang, L.; Ding, S.; Zheng, S.; Wang, Y.; Lu, F. Selection of reference genes for RT-qPCR analysis in tumor tissues from male hepatocellular carcinoma patients with hepatitis B infection and cirrhosis. Cancer Biomark. 2013, 13, 345–349. [Google Scholar] [CrossRef]
- Liu, X.; Xie, J.; Liu, Z.; Gong, Q.; Tian, R.; Su, G. Identification and validation of reference genes for quantitative RT-PCR analysis of retinal pigment epithelium cells under hypoxia and/or hyperglycemia. Gene 2016, 580, 41–46. [Google Scholar] [CrossRef]
- Lyng, M.B.; Laenkholm, A.V.; Pallisgaard, N.; Ditzel, H.J. Identification of genes for normalization of real-time RT-PCR data in breast carcinomas. BMC Cancer 2008, 8, 20. [Google Scholar] [CrossRef]
- Moermans, C.; Deliege, E.; Pirottin, D.; Poulet, C.; Guiot, J.; Henket, M.; da Silva, J.; Louis, R. Suitable reference genes determination for real-time PCR using induced sputum samples. Eur. Respir. J. 2019, 54, 1800644. [Google Scholar] [CrossRef]
- Mohelnikova-Duchonova, B.; Oliverius, M.; Honsova, E.; Soucek, P. Evaluation of reference genes and normalization strategy for quantitative real-time PCR in human pancreatic carcinoma. Dis. Mrk. 2012, 32, 203–210. [Google Scholar] [CrossRef]
- Mori, R.; Wang, Q.; Danenberg, K.D.; Pinski, J.K.; Danenberg, P.V. Both beta-actin and GAPDH are useful reference genes for normalization of quantitative RT-PCR in human FFPE tissue samples of prostate cancer. Prostate 2008, 68, 1555–1560. [Google Scholar] [CrossRef] [PubMed]
- Mossberg, K.; Svensson, P.A.; Gidlof, O.; Erlinge, D.; Jern, S.; Brogren, H. Normalization of qPCR in platelets—YWHAE a potential genericreference gene. Platelets 2016, 27, 729–734. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, T.; Okada, N.; Yoshikawa, M.; Asaka, D.; Kuboki, A.; Kojima, H.; Tanaka, Y.; Haruna, S.I. Assessment of suitable reference genes for RT-qPCR studies in chronic rhinosinusitis. Sci. Rep. 2018, 8, 1568. [Google Scholar] [CrossRef]
- Neville, M.J.; Collins, J.M.; Gloyn, A.L.; McCarthy, M.I.; Karpe, F. Comprehensive human adipose tissue mRNA and microRNA endogenous control selection for quantitative real-time-PCR normalization. Obesity 2011, 19, 888–892. [Google Scholar] [CrossRef]
- Nielsen, S.; Bassler, N.; Grzanka, L.; Swakon, J.; Olko, P.; Andreassen, C.N.; Alsner, J.; Sorensen, B.S. Optimal reference genes for normalization of qPCR gene expression data from proton and photon irradiated dermal fibroblasts. Sci. Rep. 2018, 8, 12688. [Google Scholar] [CrossRef]
- Normann, K.R.; Oystese, K.A.B.; Berg, J.P.; Lekva, T.; Berg-Johnsen, J.; Bollerslev, J.; Olarescu, N.C. Selection and validation of reliable reference genes for RT-qPCR analysis in a large cohort of pituitary adenomas. Mol. Cell. Endocrinol. 2016, 437, 183–189. [Google Scholar] [CrossRef]
- Okamura, K.; Inagaki, Y.; Matsui, T.K.; Matsubayashi, M.; Komeda, T.; Ogawa, M.; Mori, E.; Tanaka, Y. RT-qPCR analyses on the osteogenic differentiation from human iPS cells: An investigation of reference genes. Sci. Rep. 2020, 10, 11748. [Google Scholar] [CrossRef]
- O’Shaughnessy, P.J.; Monteiro, A.; Fowler, P.A. Identification of stable endogenous reference genes for real-time PCR in the human fetal gonad using an external standard technique. Mol. Hum. Reprod. 2011, 17, 620–625. [Google Scholar] [CrossRef]
- Palombella, S.; Pirrone, C.; Cherubino, M.; Valdatta, L.; Bernardini, G.; Gornati, R. Identification of reference genes for qPCR analysis during hASC long culture maintenance. PLoS ONE 2017, 12, e0170918. [Google Scholar] [CrossRef]
- Piana, C.; Wirth, M.; Gerbes, S.; Viernstein, H.; Gabor, F.; Toegel, S. Validation of reference genes for qPCR studies on Caco-2 cell differentiation. Eur. J. Pharm. Biopharm. 2008, 69, 1187–1192. [Google Scholar] [CrossRef]
- Rho, H.W.; Lee, B.C.; Choi, E.S.; Choi, I.J.; Lee, Y.S.; Goh, S.H. Identification of valid reference genes for gene expression studies of human stomach cancer by reverse transcription-qPCR. BMC Cancer 2010, 10, 240. [Google Scholar] [CrossRef]
- Ribeiro, M.A.; dos Reis, M.B.; de Moraes, L.N.; Briton-Jones, C.; Rainho, C.A.; Scarano, W.R. Defining suitable reference genes for RT-qPCR analysis on human sertoli cells after 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) exposure. Mol. Biol. Rep. 2014, 41, 7063–7066. [Google Scholar] [CrossRef]
- Riedel, G.; Rudrich, U.; Fekete-Drimusz, N.; Manns, M.P.; Vondran, F.W.; Bock, M. An extended DeltaCT-method facilitating normalisation with multiple reference genes suited for quantitative RT-PCR analyses of human hepatocyte-like cells. PLoS ONE 2014, 9, e93031. [Google Scholar] [CrossRef]
- Rohn, G.; Koch, A.; Krischek, B.; Stavrinou, P.; Goldbrunner, R.; Timmer, M. ACTB and SDHA Are Suitable Endogenous Reference Genes for Gene Expression Studies in Human Astrocytomas Using Quantitative RT-PCR. Technol. Cancer Res. Treat. 2018, 17, 1–6. [Google Scholar] [CrossRef]
- Royer, C.; Begin, A.G.; Plawinski, L.; Levesque, L.; Durrieu, M.C.; Laroche, G. Validation of reference genes for real-time PCR of cord blood mononuclear cells, differentiating endothelial progenitor cells, and mature endothelial cells. Exp. Cell. Res. 2018, 370, 389–398. [Google Scholar] [CrossRef]
- Rueda-Martinez, C.; Lamas, O.; Mataro, M.J.; Robledo-Carmona, J.; Sanchez-Espin, G.; Jimenez-Navarro, M.; Such-Martinez, M.; Fernandez, B. Selection of reference genes for quantitative real time PCR (qPCR) assays in tissue from human ascending aorta. PLoS ONE 2014, 9, e97449. [Google Scholar] [CrossRef]
- Rydbirk, R.; Folke, J.; Winge, K.; Aznar, S.; Pakkenberg, B.; Brudek, T. Assessment of brain reference genes for RT-qPCR studies in neurodegenerative diseases. Sci. Rep. 2016, 6, 37116. [Google Scholar] [CrossRef]
- Silberberg, G.; Baruch, K.; Navon, R. Detection of stable reference genes for real-time PCR analysis in schizophrenia and bipolar disorder. Anal. Biochem. 2009, 391, 91–97. [Google Scholar] [CrossRef]
- Soes, S.; Sorensen, B.S.; Alsner, J.; Overgaard, J.; Hager, H.; Hansen, L.L.; Kristensen, L.S. Identification of accurate reference genes for RT-qPCR analysis of formalin-fixed paraffin-embedded tissue from primary non-small cell lung cancers and brain and lymph node metastases. Lung Cancer 2013, 81, 180–186. [Google Scholar] [CrossRef]
- Song, W.; Zhang, W.H.; Zhang, H.; Li, Y.; Zhang, Y.; Yin, W.; Yang, Q. Validation of housekeeping genes for the normalization of RT-qPCR expression studies in oral squamous cell carcinoma cell line treated by 5 kinds of chemotherapy drugs. Cell. Mol. Biol. 2016, 62, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.C.; Ismail, M.P.; Duski, D.R.; Othman, N.H.; Bhavaraju, V.M.; Ankathil, R. Identification of Optimal Reference Genes for Normalization of RT-qPCR Data in Cancerous and Non-Cancerous Tissues of Human Uterine Cervix. Cancer Investig. 2017, 35, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Toegel, S.; Huang, W.; Piana, C.; Unger, F.M.; Wirth, M.; Goldring, M.B.; Gabor, F.; Viernstein, H. Selection of reliable reference genes for qPCR studies on chondroprotective action. BMC Mol. Biol. 2007, 8, 13. [Google Scholar] [CrossRef] [PubMed]
- Usarek, E.; Baranczyk-Kuzma, A.; Kazmierczak, B.; Gajewska, B.; Kuzma-Kozakiewicz, M. Validation of qPCR reference genes in lymphocytes from patients with amyotrophic lateral sclerosis. PLoS ONE 2017, 12, e0174317. [Google Scholar] [CrossRef] [PubMed]
- Vreeburg, R.A.; Bastiaan-Net, S.; Mes, J.J. Normalization genes for quantitative RT-PCR in differentiated Caco-2 cells used for food exposure studies. Food Funct. 2011, 2, 124–129. [Google Scholar] [CrossRef]
- Wang, H.; Yang, B.; Geng, T.; Li, B.; Dai, P.; Chen, C. Tissue-specific selection of optimal reference genes for expression analysis of anti-cancer drug-related genes in tumor samples using quantitative real-time RT-PCR. Exp. Mol. Pathol. 2015, 98, 375–381. [Google Scholar] [CrossRef]
- Wang, Q.; Ishikawa, T.; Michiue, T.; Zhu, B.L.; Guan, D.W.; Maeda, H. Stability of endogenous reference genes in postmortem human brains for normalization of quantitative real-time PCR data: Comprehensive evaluation using geNorm, NormFinder, and BestKeeper. Int. J. Leg. Med. 2012, 126, 943–952. [Google Scholar] [CrossRef]
- Weber, R.; Bertoni, A.P.; Bessestil, L.W.; Brasil, B.M.; Brum, L.S.; Furlanetto, T.W. Validation of reference genes for normalization gene expression in reverse transcription quantitative PCR in human normal thyroid and goiter tissue. Biomed. Res. Int. 2014, 2014, 198582. [Google Scholar] [CrossRef]
- Weiss, J.; Theile, D.; Haefeli, W.E. Rifampicin alters the expression of reference genes used to normalize real-time quantitative RT-PCR data. Naunyn Schmiedebergs Arch. Pharm. 2012, 385, 1025–1034. [Google Scholar] [CrossRef]
- Wisnieski, F.; Calcagno, D.Q.; Leal, M.F.; dos Santos, L.C.; Gigek Cde, O.; Chen, E.S.; Pontes, T.B.; Assumpcao, P.P.; de Assumpcao, M.B.; Demachki, S.; et al. Reference genes for quantitative RT-PCR data in gastric tissues and cell lines. World J. Gastroenterol. 2013, 19, 7121–7128. [Google Scholar] [CrossRef]
- Xie, J.; Liu, X.; Li, Y.; Liu, Y.; Su, G. Validation of RT-qPCR reference genes and determination of Robo4 expression levels in human retinal endothelial cells under hypoxia and/or hyperglycemia. Gene 2016, 585, 135–142. [Google Scholar] [CrossRef]
- Yin, W.Z.; Yang, Q.W.; Niu, K.; Ren, M.; He, D.; Song, W.Z. Validation of reference genes for the normalization of RT-qPCR expression studies on human laryngeal cancer and hypopharyngeal cancer. Eur. Rev. Med. Pharm. Sci. 2019, 23, 4199–4209. [Google Scholar]
- Zarybnicky, T.; Matouskova, P.; Ambroz, M.; Subrt, Z.; Skalova, L.; Bousova, I. The Selection and Validation of Reference Genes for mRNA and microRNA Expression Studies in Human Liver Slices Using RT-qPCR. Genes 2019, 10, 763. [Google Scholar] [CrossRef]
- Zhang, H.; Guan, Z.S.; Guan, S.H.; Yang, K.; Pan, Y.; Wu, Y.Y.; Wang, A.H.; Sun, B.B.; Hou, J.; Mu, X.X.; et al. Identification of Suitable Candidate Reference Genes for Gene Expression Analysis by RT-qPCR in Peripheral Blood Mononuclear Cells of CHB Patients. Clin. Lab. 2016, 62, 227–234. [Google Scholar] [CrossRef]
- Zhang, X.; Ding, L.; Sandford, A.J. Selection of reference genes for gene expression studies in human neutrophils by real-time PCR. BMC Mol. Biol. 2005, 6, 4. [Google Scholar] [CrossRef]
- Zhao, J.; Zhou, H.; Sun, L.; Yang, B.; Zhang, L.; Shi, H.; Zheng, Y. Selection of suitable reference genes for quantitative real-time PCR in trabecular meshwork cells under oxidative stress. Free. Radic. Res. 2017, 51, 103–111. [Google Scholar] [CrossRef]
- Zhu, X.; Zhang, L.; Hu, Y.; Zhang, J. Identification of suitable reference genes for real-time qPCR in homocysteine-treated human umbilical vein endothelial cells. PLoS ONE 2018, 13, e0210087. [Google Scholar] [CrossRef]
- Zyzynska-Granica, B.; Koziak, K. Identification of suitable reference genes for real-time PCR analysis of statin-treated human umbilical vein endothelial cells. PLoS ONE 2012, 7, e51547. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nevone, A.; Lattarulo, F.; Russo, M.; Panno, G.; Milani, P.; Basset, M.; Avanzini, M.A.; Merlini, G.; Palladini, G.; Nuvolone, M. A Strategy for the Selection of RT-qPCR Reference Genes Based on Publicly Available Transcriptomic Datasets. Biomedicines 2023, 11, 1079. https://doi.org/10.3390/biomedicines11041079
Nevone A, Lattarulo F, Russo M, Panno G, Milani P, Basset M, Avanzini MA, Merlini G, Palladini G, Nuvolone M. A Strategy for the Selection of RT-qPCR Reference Genes Based on Publicly Available Transcriptomic Datasets. Biomedicines. 2023; 11(4):1079. https://doi.org/10.3390/biomedicines11041079
Chicago/Turabian StyleNevone, Alice, Francesca Lattarulo, Monica Russo, Giada Panno, Paolo Milani, Marco Basset, Maria Antonietta Avanzini, Giampaolo Merlini, Giovanni Palladini, and Mario Nuvolone. 2023. "A Strategy for the Selection of RT-qPCR Reference Genes Based on Publicly Available Transcriptomic Datasets" Biomedicines 11, no. 4: 1079. https://doi.org/10.3390/biomedicines11041079