

A Multi-Omics Prognostic Model Capturing Tumor Stemness and the Immune Microenvironment in Clear Cell Renal Cell Carcinoma

 and
and
Abstract
1. Introduction
2. Method and Materials
2.1. Data Collection and Processing
2.2. Identification of Different Subsets in ccRCC
2.3. Enrichment Analysis between Subgroups
2.4. Differences in Characteristics of Immune Infiltration and Response to Treatment
2.5. Characteristics of Mutation Spectrum among Subpopulations
2.6. Processing of Spatial Transcriptomics Data
2.7. Drug Susceptibility Analysis
2.8. Construction of Risk Prediction Model
2.9. Statistical Analysis
3. Results
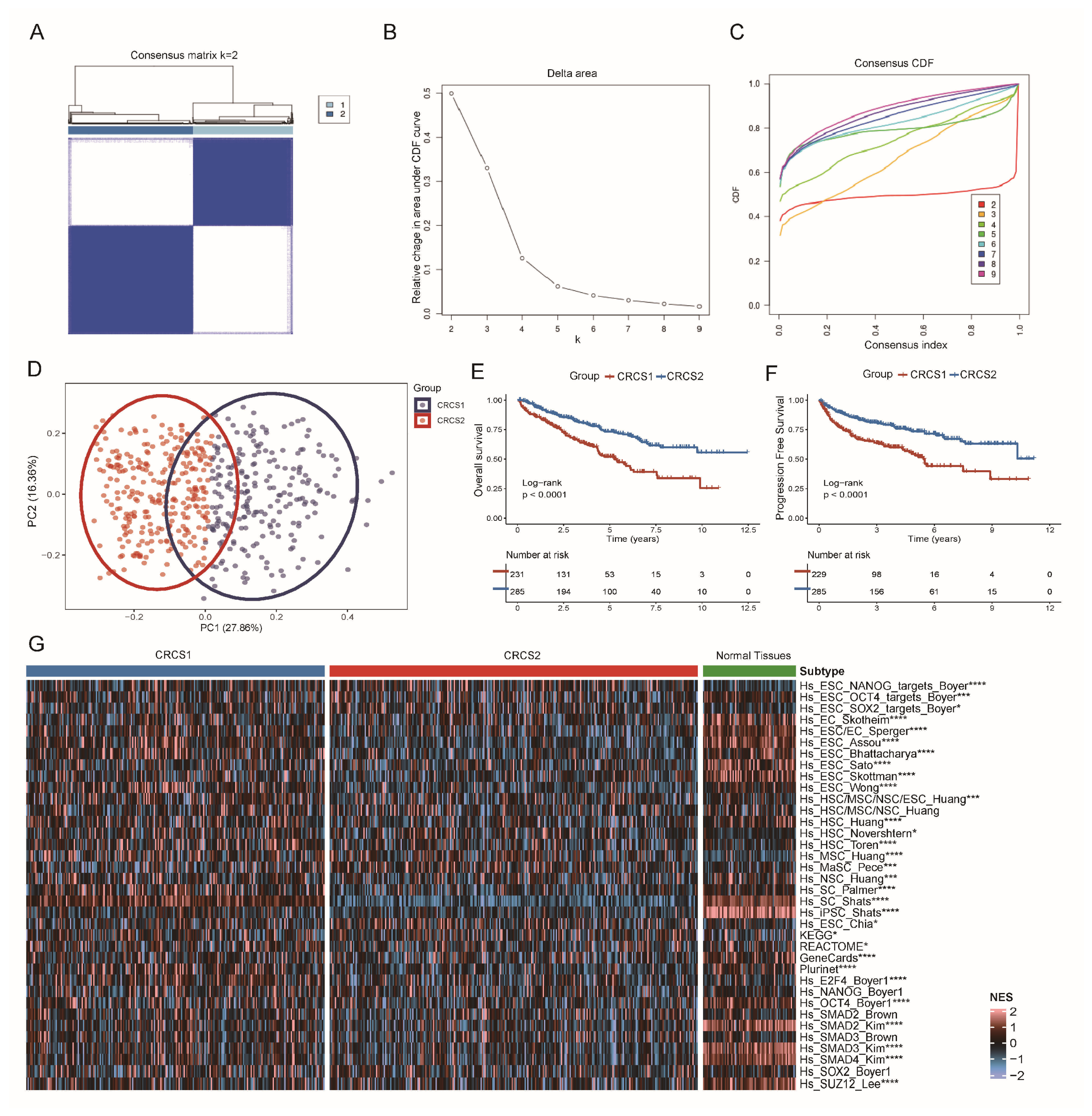
3.1. Two Clusters of ccRCC Were Identified by Clustering Analysis of Tumor Stemness Signatures
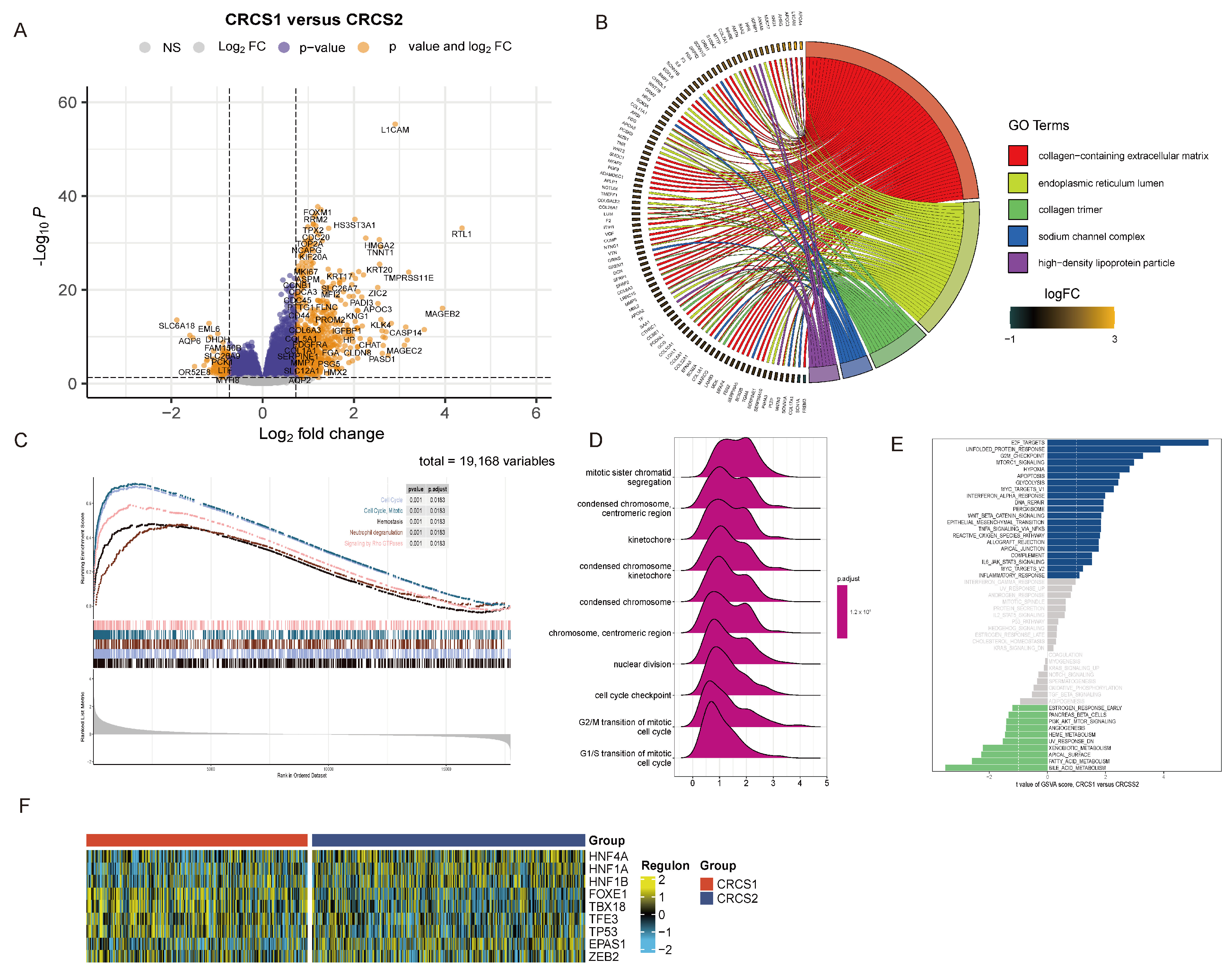
3.2. Functional Enrichment Analysis of Different Tumor Stemness Subgroups
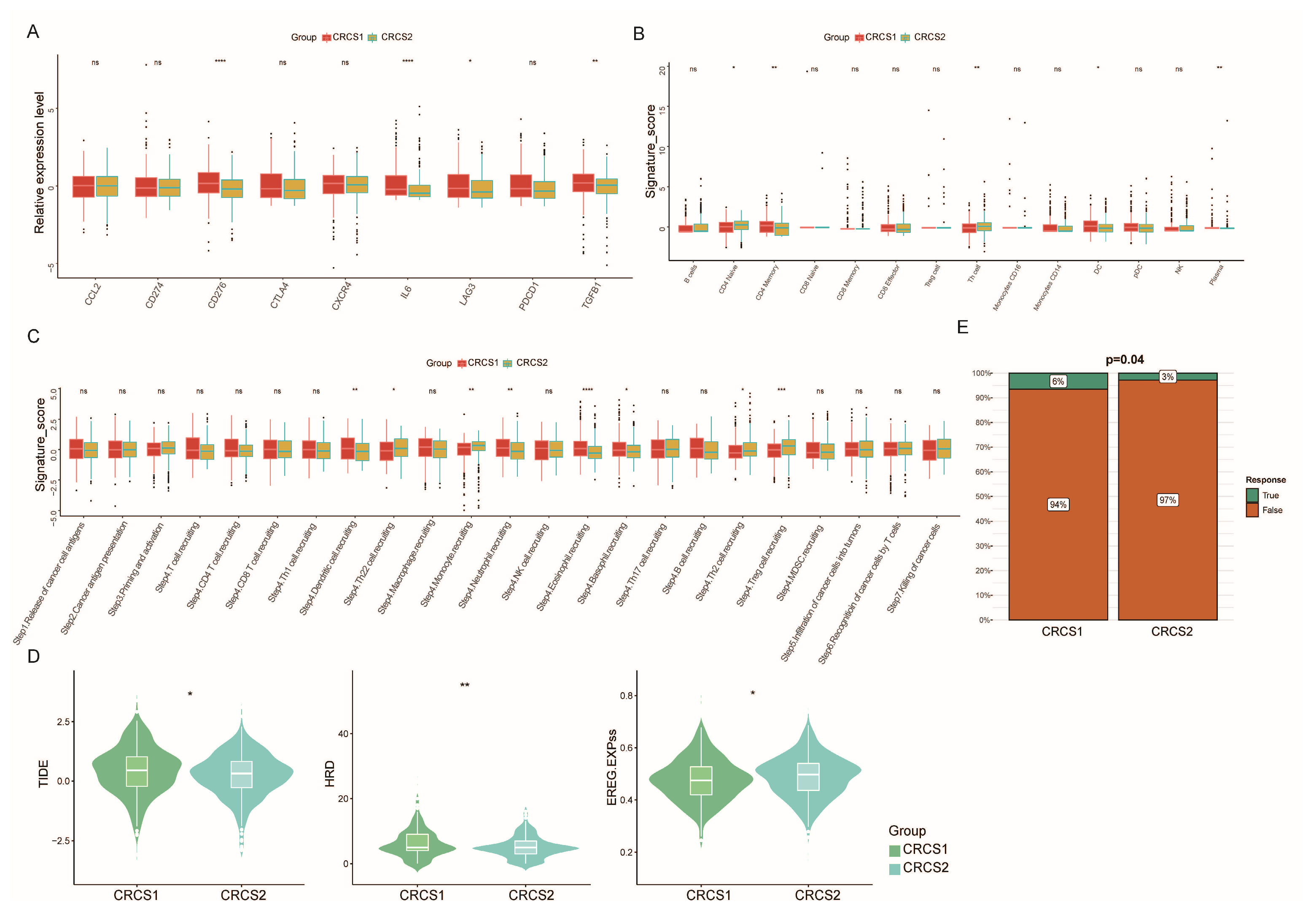
3.3. Comparison of Specific Immune Infiltration in Two Subgroups
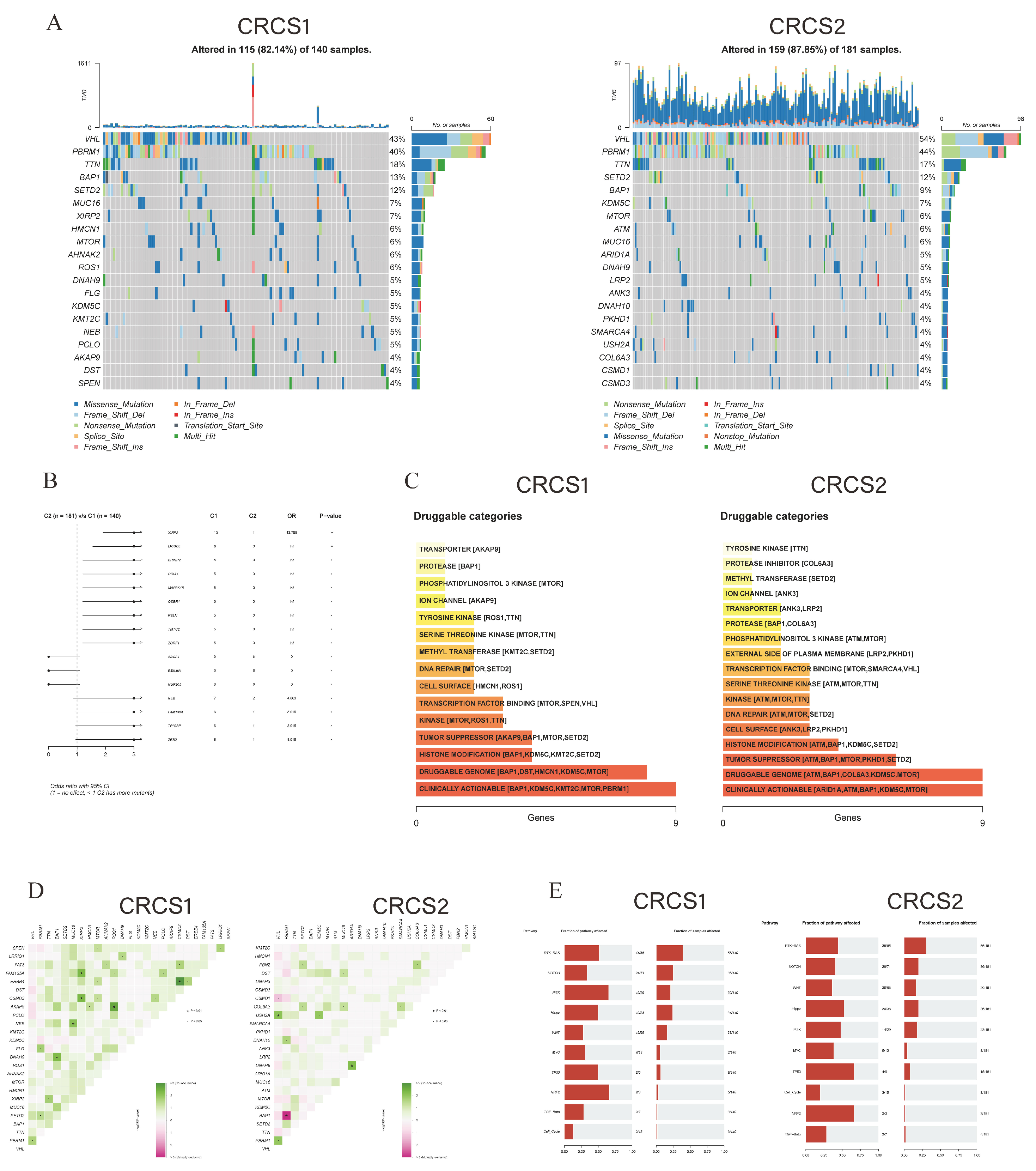
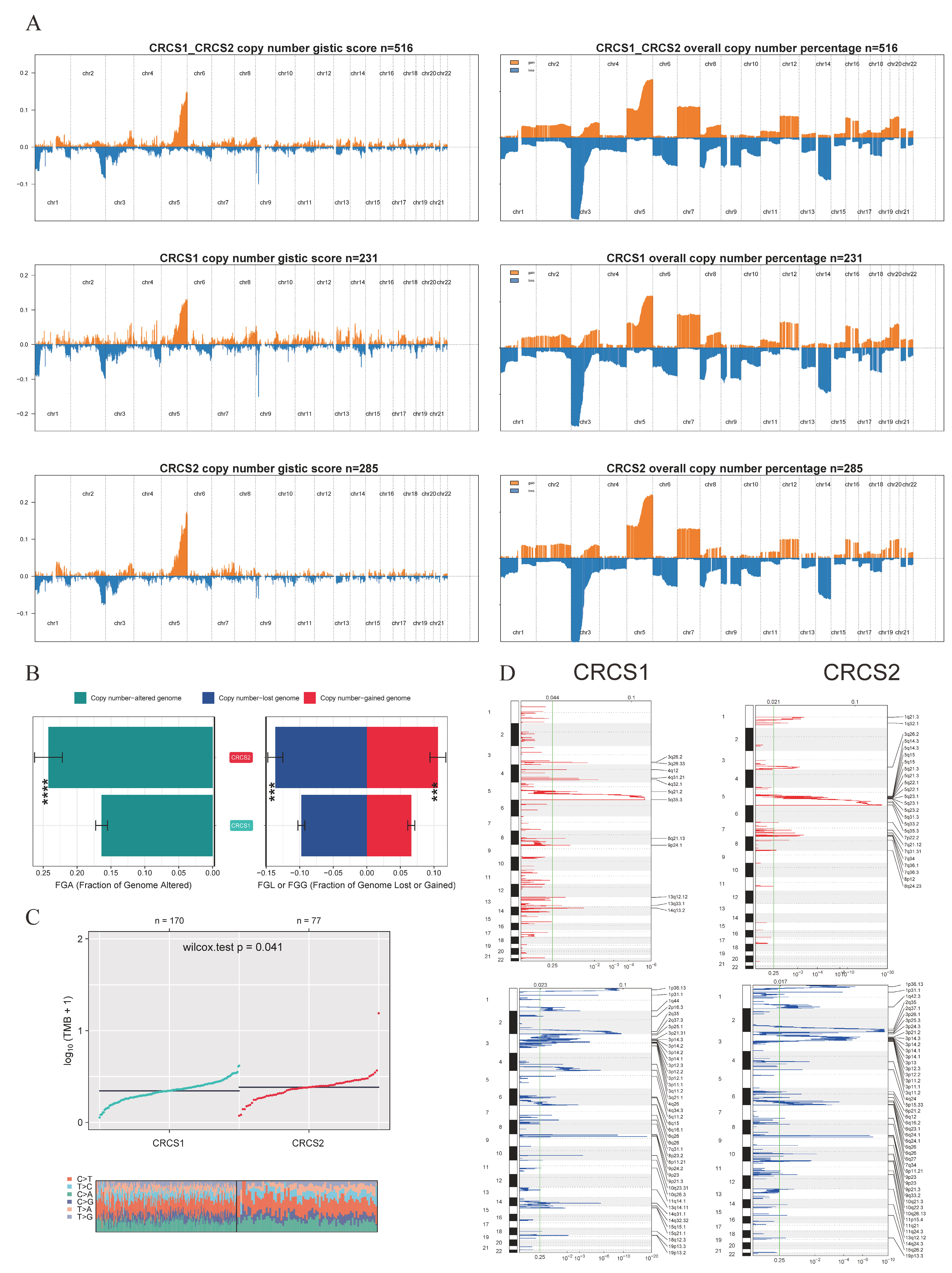
3.4. Characterization of Somatic Mutations and CNV in Two Clusters
3.5. Drug Susceptibility Profiles of Different Stemness Subsets
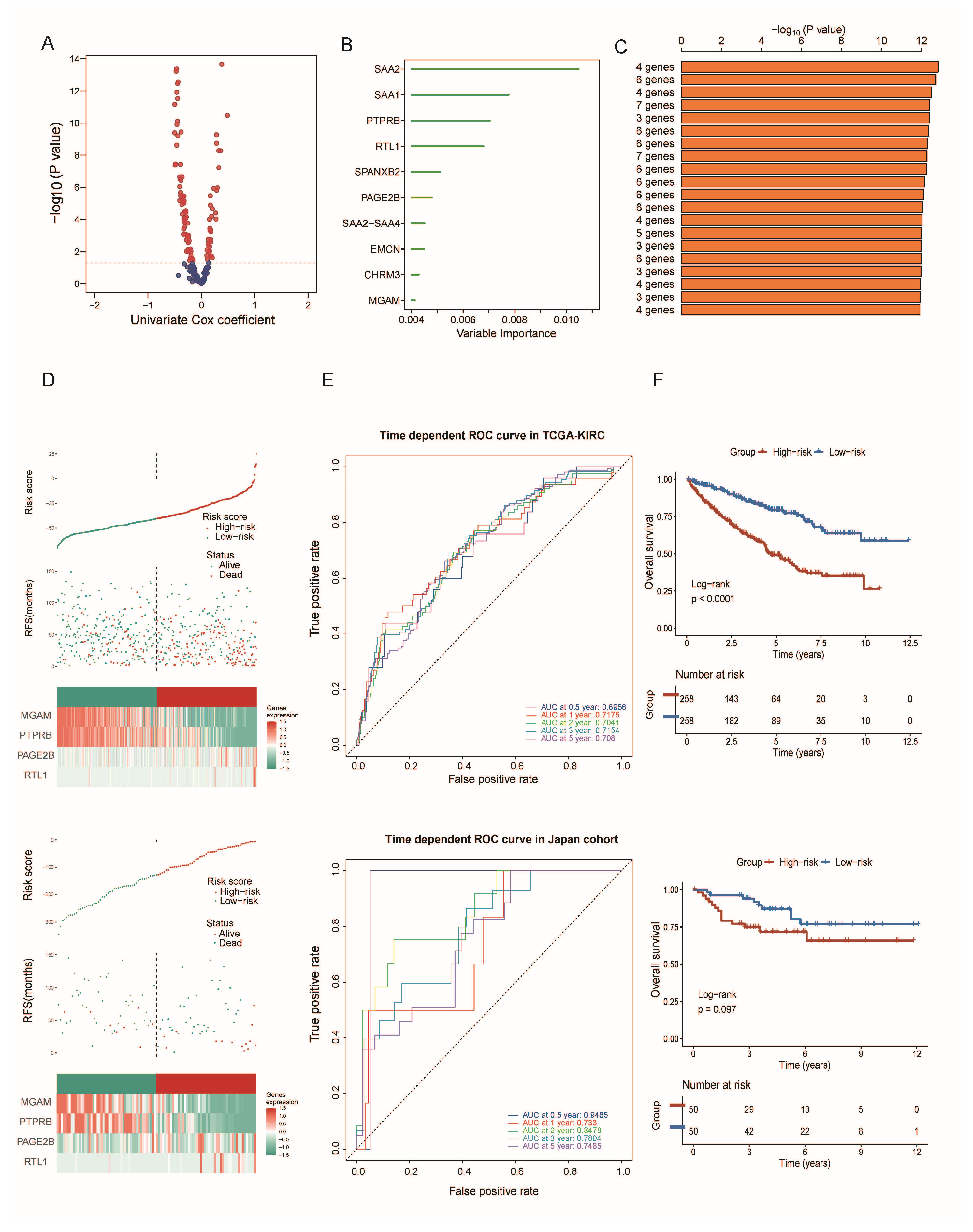
3.6. Construction and Validation of a Gene Risk Model Related to Cancer Stemness
3.7. Biological Role of SAA2 in ccRCC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hsieh, J.J.; Purdue, M.P.; Signoretti, S.; Swanton, C.; Albiges, L.; Schmidinger, M.; Heng, D.Y.; Larkin, J.; Ficarra, V. Renal cell carcinoma. Nat. Rev. Dis. Primers 2017, 3, 17009. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.; Miller, J.D.; Li, J.Z.; Russell, M.W.; Charbonneau, C. Epidemiologic and socioeconomic burden of metastatic renal cell carcinoma (mRCC): A literature review. Cancer Treat. Rev. 2008, 34, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Kotecha, R.R.; Motzer, R.J.; Voss, M.H. Towards individualized therapy for metastatic renal cell carcinoma. Nat. Rev. Clin. Oncol. 2019, 16, 621–633. [Google Scholar] [CrossRef]
- Makhov, P.; Joshi, S.; Ghatalia, P.; Kutikov, A.; Uzzo, R.G.; Kolenko, V.M. Resistance to Systemic Therapies in Clear Cell Renal Cell Carcinoma: Mechanisms and Management Strategies. Mol. Cancer Ther. 2018, 17, 1355–1364. [Google Scholar] [CrossRef]
- Jiang, A.; Li, J.; He, Z.; Liu, Y.; Qiao, K.; Fang, Y.; Qu, L.; Luo, P.; Lin, A.; Wang, L. Renal cancer: Signaling pathways and advances in targeted therapies. MedComm 2024, 5, e676. [Google Scholar] [CrossRef]
- Motzer, R.J.; Penkov, K.; Haanen, J.; Rini, B.; Albiges, L.; Campbell, M.T.; Venugopal, B.; Kollmannsberger, C.; Negrier, S.; Uemura, M.; et al. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1103–1115. [Google Scholar] [CrossRef] [PubMed]
- Braun, D.A.; Bakouny, Z.; Hirsch, L.; Flippot, R.; Van Allen, E.M.; Wu, C.J.; Choueiri, T.K. Beyond conventional immune-checkpoint inhibition—Novel immunotherapies for renal cell carcinoma. Nat. Rev. Clin. Oncol. 2021, 18, 199–214. [Google Scholar] [CrossRef]
- Haddad, A.F.; Young, J.S.; Gill, S.; Aghi, M.K. Resistance to immune checkpoint blockade: Mechanisms, counter-acting approaches, and future directions. Semin. Cancer Biol. 2022, 86, 532–541. [Google Scholar] [CrossRef]
- Gulati, S.; Martinez, P.; Joshi, T.; Birkbak, N.J.; Santos, C.R.; Rowan, A.J.; Pickering, L.; Gore, M.; Larkin, J.; Szallasi, Z.; et al. Systematic evaluation of the prognostic impact and intratumour heterogeneity of clear cell renal cell carcinoma biomarkers. Eur. Urol. 2014, 66, 936–948. [Google Scholar] [CrossRef]
- Long, Z.; Sun, C.; Tang, M.; Wang, Y.; Ma, J.; Yu, J.; Wei, J.; Ma, J.; Wang, B.; Xie, Q.; et al. Single-cell multiomics analysis reveals regulatory programs in clear cell renal cell carcinoma. Cell Discov. 2022, 8, 68. [Google Scholar] [CrossRef] [PubMed]
- Clara, J.A.; Monge, C.; Yang, Y.; Takebe, N. Targeting signalling pathways and the immune microenvironment of cancer stem cells—A clinical update. Nat. Rev. Clin. Oncol. 2020, 17, 204–232. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, I.; Miele, L. Deadly crosstalk: Notch signaling at the intersection of EMT and cancer stem cells. Cancer Lett. 2013, 341, 41–45. [Google Scholar] [CrossRef]
- Saygin, C.; Matei, D.; Majeti, R.; Reizes, O.; Lathia, J.D. Targeting Cancer Stemness in the Clinic: From Hype to Hope. Cell Stem. Cell 2019, 24, 25–40. [Google Scholar] [CrossRef]
- Nishio, M.; Otsubo, K.; Maehama, T.; Mimori, K.; Suzuki, A. Capturing the mammalian Hippo: Elucidating its role in cancer. Cancer Sci. 2013, 104, 1271–1277. [Google Scholar] [CrossRef] [PubMed]
- Pelullo, M.; Zema, S.; Nardozza, F.; Checquolo, S.; Screpanti, I.; Bellavia, D. Wnt, Notch, and TGF-beta Pathways Impinge on Hedgehog Signaling Complexity: An Open Window on Cancer. Front. Genet. 2019, 10, 711. [Google Scholar] [CrossRef]
- Wan, S.; Zhao, E.; Kryczek, I.; Vatan, L.; Sadovskaya, A.; Ludema, G.; Simeone, D.M.; Zou, W.; Welling, T.H. Tumor-associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology 2014, 147, 1393–1404. [Google Scholar] [CrossRef]
- Lee, C.G.; Das, B.; Lin, T.L.; Grimes, C.; Zhang, X.; Lavezzi, T.; Huang, L.; Cole, J.; Yau, L.; Li, L. A rare fraction of drug-resistant follicular lymphoma cancer stem cells interacts with follicular dendritic cells to maintain tumourigenic potential. Br. J. Haematol. 2012, 158, 79–90. [Google Scholar] [CrossRef]
- Yang, S.; Wang, B.; Guan, C.; Wu, B.; Cai, C.; Wang, M.; Zhang, B.; Liu, T.; Yang, P. Foxp3+IL-17+ T cells promote development of cancer-initiating cells in colorectal cancer. J. Leukoc. Biol. 2011, 89, 85–91. [Google Scholar] [CrossRef]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdottir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef]
- Kamburov, A.; Pentchev, K.; Galicka, H.; Wierling, C.; Lehrach, H.; Herwig, R. ConsensusPathDB: Toward a more complete picture of cell biology. Nucleic Acids Res. 2011, 39, D712–D717. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Fan, J.; Wang, B.; Traugh, N.; Chen, Q.; Liu, J.S.; Li, B.; Liu, X.S. TIMER: A Web Server for Comprehensive Analysis of Tumor-Infiltrating Immune Cells. Cancer Res. 2017, 77, e108–e110. [Google Scholar] [CrossRef]
- Racle, J.; Gfeller, D. EPIC: A Tool to Estimate the Proportions of Different Cell Types from Bulk Gene Expression Data. Methods Mol. Biol. 2020, 2120, 233–248. [Google Scholar]
- Mayakonda, A.; Lin, D.C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef] [PubMed]
- Jiang, A.; Bao, Y.; Wang, A.; Gong, W.; Gan, X.; Wang, J.; Bao, Y.; Wu, Z.; Liu, B.; Lu, J.; et al. Establishment of a Prognostic Prediction and Drug Selection Model for Patients with Clear Cell Renal Cell Carcinoma by Multiomics Data Analysis. Oxid. Med. Cell Longev. 2022, 2022, 3617775. [Google Scholar] [CrossRef]
- Mermel, C.H.; Schumacher, S.E.; Hill, B.; Meyerson, M.L.; Beroukhim, R.; Getz, G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011, 12, R41. [Google Scholar] [CrossRef] [PubMed]
- Meylan, M.; Petitprez, F.; Becht, E.; Bougouin, A.; Pupier, G.; Calvez, A.; Giglioli, I.; Verkarre, V.; Lacroix, G.; Verneau, J.; et al. Tertiary lymphoid structures generate and propagate anti-tumor antibody-producing plasma cells in renal cell cancer. Immunity 2022, 55, 527–541.e5. [Google Scholar] [CrossRef]
- Peng, Z.; Ye, M.; Ding, H.; Feng, Z.; Hu, K. Spatial transcriptomics atlas reveals the crosstalk between cancer-associated fibroblasts and tumor microenvironment components in colorectal cancer. J. Transl. Med. 2022, 20, 302. [Google Scholar] [CrossRef]
- Reinhold, W.C.; Sunshine, M.; Liu, H.; Varma, S.; Kohn, K.W.; Morris, J.; Doroshow, J.; Pommier, Y. CellMiner: A web-based suite of genomic and pharmacologic tools to explore transcript and drug patterns in the NCI-60 cell line set. Cancer Res. 2012, 72, 3499–3511. [Google Scholar] [CrossRef]
- Meng, J.; Jiang, A.; Lu, X.; Gu, D.; Ge, Q.; Bai, S.; Zhou, Y.; Zhou, J.; Hao, Z.; Yan, F.; et al. Multiomics characterization and verification of clear cell renal cell carcinoma molecular subtypes to guide precise chemotherapy and immunotherapy. iMeta 2023, 2, e147. [Google Scholar] [CrossRef]
- Jiang, A.; Luo, P.; Chen, M.; Fang, Y.; Liu, B.; Wu, Z.; Qu, L.; Wang, A.; Wang, L.; Cai, C. A new thinking: Deciphering the aberrance and clinical implication of copper-death signatures in clear cell renal cell carcinoma. Cell Biosci. 2022, 12, 209. [Google Scholar] [CrossRef] [PubMed]
- Jiang, A.; Song, J.; Fang, X.; Fang, Y.; Wang, Z.; Liu, B.; Wu, Z.; Qu, L.; Luo, P.; Wang, L. A novel thinking: DDR axis refines the classification of ccRCC with distinctive prognosis, multi omics landscape and management strategy. Front. Public Health 2022, 10, 1029509. [Google Scholar] [CrossRef] [PubMed]
- Xia, G.; Kageyama, Y.; Hayashi, T.; Kawakami, S.; Yoshida, M.; Kihara, K. Regulation of vascular endothelial growth factor transcription by endothelial PAS domain protein 1 (EPAS1) and possible involvement of EPAS1 in the angiogenesis of renal cell carcinoma. Cancer 2001, 91, 1429–1436. [Google Scholar] [CrossRef] [PubMed]
- Kopecka, J.; Salaroglio, I.C.; Perez-Ruiz, E.; Sarmento-Ribeiro, A.B.; Saponara, S.; Rivas, J.D.L.; Riganti, C. Hypoxia as a driver of resistance to immunotherapy. Drug Resist. Updat. 2021, 59, 100787. [Google Scholar] [CrossRef]
- Wang, B.; Zhao, Q.; Zhang, Y.; Liu, Z.; Zheng, Z.; Liu, S.; Meng, L.; Xin, Y.; Jiang, X. Targeting hypoxia in the tumor microenvironment: A potential strategy to improve cancer immunotherapy. J. Exp. Clin. Cancer Res. 2021, 40, 24. [Google Scholar] [CrossRef]
- Low, B.S.J.; Lim, C.S.; Ding, S.S.L.; Tan, Y.S.; Ng, N.H.J.; Krishnan, V.G.; Ang, S.F.; Neo, C.W.Y.; Verma, C.S.; Hoon, S. Decreased GLUT2 and glucose uptake contribute to insulin secretion defects in MODY3/HNF1A hiPSC-derived mutant beta cells. Nat. Commun. 2021, 12, 3133. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Han, Z.; Mehra, R.; Shao, W.; Cheng, M.; Feng, Q.; Ni, D.; Huang, K.; Cheng, L.; Zhang, J. Computational analysis of pathological images enables a better diagnosis of TFE3 Xp11.2 translocation renal cell carcinoma. Nat. Commun. 2020, 11, 1778. [Google Scholar] [CrossRef]
- Buttner, F.A.; Winter, S.; Stuhler, V.; Rausch, S.; Hennenlotter, J.; Fussel, S.; Zastrow, S.; Meinhardt, M.; Toma, M.; Jerónimo, C.; et al. A novel molecular signature identifies mixed subtypes in renal cell carcinoma with poor prognosis and independent response to immunotherapy. Genome Med. 2022, 14, 105. [Google Scholar] [CrossRef]
- Hah, Y.S.; Koo, K.C. Immunology and Immunotherapeutic Approaches for Advanced Renal Cell Carcinoma: A Comprehensive Review. Int. J. Mol. Sci. 2021, 22, 4452. [Google Scholar] [CrossRef]
- Lobo, N.A.; Shimono, Y.; Qian, D.; Clarke, M.F. The biology of cancer stem cells. Annu. Rev. Cell Dev. Biol. 2007, 23, 675–699. [Google Scholar] [CrossRef]
- Nguyen, L.V.; Vanner, R.; Dirks, P.; Eaves, C.J. Cancer stem cells: An evolving concept. Nat. Rev. Cancer 2012, 12, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Wu, G.; Chen, P.; Gao, L.; Chen, G.; Zhang, H. Phase separation in epigenetics and cancer stem cells. Front. Oncol. 2022, 12, 922604. [Google Scholar] [CrossRef] [PubMed]
- Chaffer, C.L.; Brueckmann, I.; Scheel, C.; Kaestli, A.J.; Wiggins, P.A.; Rodrigues, L.O.; Brooks, M.; Reinhardt, F.; Su, Y.; Polyak, K.; et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc. Natl. Acad. Sci. USA 2011, 108, 7950–7955. [Google Scholar] [CrossRef]
- Chaffer, C.L.; Marjanovic, N.D.; Lee, T.; Bell, G.; Kleer, C.G.; Reinhardt, F.; D’Alessio, A.C.; Young, R.A.; Weinberg, R.A. Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell 2013, 154, 61–74. [Google Scholar] [CrossRef]
- Seton-Rogers, S. Cancer stem cells: Easily moulded. Nat. Rev. Cancer 2013, 13, 519. [Google Scholar] [CrossRef] [PubMed]
- Roesch, A.; Fukunaga-Kalabis, M.; Schmidt, E.C.; Zabierowski, S.E.; Brafford, P.A.; Vultur, A.; Basu, D.; Gimotty, P.; Vogt, T.; Herlyn, M. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell 2010, 141, 583–594. [Google Scholar] [CrossRef]
- Chen, W.; Dong, J.; Haiech, J.; Kilhoffer, M.C.; Zeniou, M. Cancer Stem Cell Quiescence and Plasticity as Major Challenges in Cancer Therapy. Stem Cells Int. 2016, 2016, 1740936. [Google Scholar] [CrossRef]
- Deheeger, M.; Lesniak, M.S.; Ahmed, A.U. Cellular plasticity regulated cancer stem cell niche: A possible new mechanism of chemoresistance. Cancer Cell Microenviron. 2014, 1, e295. [Google Scholar]
- Zhao, L.; Lee, V.H.F.; Ng, M.K.; Yan, H.; Bijlsma, M.F. Molecular subtyping of cancer: Current status and moving toward clinical applications. Brief Bioinform. 2019, 20, 572–584. [Google Scholar] [CrossRef]
- Li, C.; Sun, Y.-D.; Yu, G.-Y.; Cui, J.-R.; Lou, Z.; Zhang, H.; Huang, Y.; Bai, C.-G.; Deng, L.-L.; Liu, P.; et al. Integrated Omics of Metastatic Colorectal Cancer. Cancer Cell 2020, 38, 734–747.e9. [Google Scholar] [CrossRef]
- Luo, H.; Zhao, Q.; Wei, W.; Zheng, L.; Yi, S.; Li, G.; Wang, W.; Sheng, H.; Pu, H.; Mo, H.; et al. Circulating tumor DNA methylation profiles enable early diagnosis, prognosis prediction, and screening for colorectal cancer. Sci. Transl. Med. 2020, 12, eaax7533. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Wang, S.-G.; Hou, Y.; Chen, Z.; Liu, L.; Li, R.; Li, N.; Zhou, L.; Yang, Y.; Wang, L.; et al. Multi-omic profiling of clear cell renal cell carcinoma identifies metabolic reprogramming associated with disease progression. Nat. Genet. 2024, 56, 442–457. [Google Scholar] [CrossRef] [PubMed]
- Rathmell, W.K.; Rumble, R.B.; Van Veldhuizen, P.J.; Al-Ahmadie, H.; Emamekhoo, H.; Hauke, R.J.; Louie, A.V.; Milowsky, M.I.; Molina, A.M.; Rose, T.L.; et al. Management of Metastatic Clear Cell Renal Cell Carcinoma: ASCO Guideline. J. Clin. Oncol. 2022, 40, 2957–2995. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Rini, B.I.; McDermott, D.F.; Aren Frontera, O.; Hammers, H.J.; Carducci, M.A.; Salman, P.; Escudier, B.; Beuselinck, B.; Amin, A.; et al. Nivolumab plus ipilimumab versus sunitinib in first-line treatment for advanced renal cell carcinoma: Extended follow-up of efficacy and safety results from a randomised, controlled, phase 3 trial. Lancet Oncol. 2019, 20, 1370–1385. [Google Scholar] [CrossRef]
- Cella, D.; Grunwald, V.; Escudier, B.; Hammers, H.J.; George, S.; Nathan, P.; Grimm, M.O.; Rini, B.I.; Doan, J.; Ivanescu, C.; et al. Patient-reported outcomes of patients with advanced renal cell carcinoma treated with nivolumab plus ipilimumab versus sunitinib (CheckMate 214): A randomised, phase 3 trial. Lancet Oncol. 2019, 20, 297–310. [Google Scholar] [CrossRef]
- Motzer, R.J.; Powles, T.; Burotto, M.; Escudier, B.; Bourlon, M.T.; Shah, A.Y.; Suárez, C.; Hamzaj, A.; Porta, C.; Hocking, C.M.; et al. Nivolumab plus cabozantinib versus sunitinib in first-line treatment for advanced renal cell carcinoma (CheckMate 9ER): Long-term follow-up results from an open-label, randomised, phase 3 trial. Lancet Oncol. 2022, 23, 888–898. [Google Scholar] [CrossRef]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Aren Frontera, O.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthélémy, P.; Porta, C.; George, S.; et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef]
- Powles, T.; Plimack, E.R.; Soulieres, D.; Waddell, T.; Stus, V.; Gafanov, R.; Nosov, D.; Pouliot, F.; Melichar, B.; Vynnychenko, I.; et al. Pembrolizumab plus axitinib versus sunitinib monotherapy as first-line treatment of advanced renal cell carcinoma (KEYNOTE-426): Extended follow-up from a randomised, open-label, phase 3 trial. Lancet Oncol. 2020, 21, 1563–1573. [Google Scholar] [CrossRef]
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef]
- Rich, J.N. Cancer stem cells: Understanding tumor hierarchy and heterogeneity. Medicine 2016, 95 (Suppl. S1), S2–S7. [Google Scholar] [CrossRef]
- Wiechert, A.; Saygin, C.; Thiagarajan, P.S.; Rao, V.S.; Hale, J.S.; Gupta, N.; Hitomi, M.; Nagaraj, A.B.; DiFeo, A.; Lathia, J.D.; et al. Cisplatin induces stemness in ovarian cancer. Oncotarget 2016, 7, 30511–30522. [Google Scholar] [CrossRef] [PubMed]
- Pattabiraman, D.R.; Weinberg, R.A. Tackling the cancer stem cells—What challenges do they pose? Nat. Rev. Drug Discov. 2014, 13, 497–512. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Cong, Y.; Wang, D.; Sun, Y.; Deng, L.; Liu, Y.; Martin-Trevino, R.; Shang, L.; McDermott, S.P.; Landis, M.D.; et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Rep. 2014, 2, 78–91. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.; Xu, M.; Hua, R.; Tan, C.; Zhang, J.; Gong, Y.; Wu, Z.; Weng, W.; Sheng, W.; Guo, W. The polycomb group protein EZH2 induces epithelial-mesenchymal transition and pluripotent phenotype of gastric cancer cells by binding to PTEN promoter. J. Hematol. Oncol. 2018, 11, 9. [Google Scholar] [CrossRef] [PubMed]
- Takeishi, S.; Nakayama, K.I. To wake up cancer stem cells, or to let them sleep, that is the question. Cancer Sci. 2016, 107, 875–881. [Google Scholar] [CrossRef]
- Zeng, X.; Zhao, H.; Li, Y.; Fan, J.; Sun, Y.; Wang, S.; Wang, Z.; Song, P.; Ju, D. Targeting Hedgehog signaling pathway and autophagy overcomes drug resistance of BCR-ABL-positive chronic myeloid leukemia. Autophagy 2015, 11, 355–372. [Google Scholar] [CrossRef]
- Yun, E.J.; Zhou, J.; Lin, C.J.; Hernandez, E.; Fazli, L.; Gleave, M.; Hsieh, J.T. Targeting Cancer Stem Cells in Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2016, 22, 670–679. [Google Scholar] [CrossRef]
- Roy, S.; Kar, M.; Roy, S.; Padhi, S.; Kumar, A.; Thakur, S.; Akhter, Y.; Gatto, G.; Banerjee, B. Inhibition of CD44 sensitizes cisplatin-resistance and affects Wnt/beta-catenin signaling in HNSCC cells. Int. J. Biol. Macromol. 2020, 149, 501–512. [Google Scholar] [CrossRef]
- Waszak, S.M.; Robinson, G.W.; Gudenas, B.L.; Smith, K.S.; Forget, A.; Kojic, M.; Garcia-Lopez, J.; Hadley, J.; Hamilton, K.V.; Indersie, E.; et al. Germline Elongator mutations in Sonic Hedgehog medulloblastoma. Nature 2020, 580, 396–401. [Google Scholar] [CrossRef]
- Carballo, G.B.; Honorato, J.R.; de Lopes, G.P.F.; Spohr, T. A highlight on Sonic hedgehog pathway. Cell Commun. Signal 2018, 16, 11. [Google Scholar] [CrossRef]
- Gong, X.; Qian, H.; Cao, P.; Zhao, X.; Zhou, Q.; Lei, J.; Yan, N. Structural basis for the recognition of Sonic Hedgehog by human Patched1. Science 2018, 361, eaas8935. [Google Scholar] [CrossRef] [PubMed]
- Dummer, R.; Guminski, A.; Gutzmer, R.; Dirix, L.; Lewis, K.D.; Combemale, P.; Herd, R.M.; Kaatz, M.; Loquai, C.; Stratigos, A.J.; et al. The 12-month analysis from Basal Cell Carcinoma Outcomes with LDE225 Treatment (BOLT): A phase II, randomized, double-blind study of sonidegib in patients with advanced basal cell carcinoma. J. Am. Acad. Dermatol. 2016, 75, 113–125.e5. [Google Scholar] [CrossRef]
- Dummer, R.; Guminksi, A.; Gutzmer, R.; Lear, J.T.; Lewis, K.D.; Chang, A.L.S.; Combemale, P.; Dirix, L.; Kaatz, M.; Kudchadkar, R.; et al. Long-term efficacy and safety of sonidegib in patients with advanced basal cell carcinoma: 42-month analysis of the phase II randomized, double-blind BOLT study. Br. J. Dermatol. 2020, 182, 1369–1378. [Google Scholar] [CrossRef]
- Chang, A.L.; Solomon, J.A.; Hainsworth, J.D.; Goldberg, L.; McKenna, E.; Day, B.M.; Weiss, G.J. Expanded access study of patients with advanced basal cell carcinoma treated with the Hedgehog pathway inhibitor, vismodegib. J. Am. Acad. Dermatol. 2014, 70, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Sekulic, A.; Migden, M.R.; Lewis, K.; Hainsworth, J.D.; Solomon, J.A.; Yoo, S.; Arron, S.T.; Friedlander, P.A.; Marmur, E.; Rudin, C.M.; et al. Pivotal ERIVANCE basal cell carcinoma (BCC) study: 12-month update of efficacy and safety of vismodegib in advanced BCC. J. Am. Acad. Dermatol. 2015, 72, 1021–1026.e8. [Google Scholar] [CrossRef]
- Berlin, J.; Bendell, J.C.; Hart, L.L.; Firdaus, I.; Gore, I.; Hermann, R.C.; Mulcahy, M.F.; Zalupski, M.M.; Mackey, H.M.; Yauch, R.L.; et al. A randomized phase II trial of vismodegib versus placebo with FOLFOX or FOLFIRI and bevacizumab in patients with previously untreated metastatic colorectal cancer. Clin. Cancer Res. 2013, 19, 258–267. [Google Scholar] [CrossRef]
- Catenacci, D.V.; Junttila, M.R.; Karrison, T.; Bahary, N.; Horiba, M.N.; Nattam, S.R.; Marsh, R.; Wallace, J.; Kozloff, M.; Rajdev, L.; et al. Randomized Phase Ib/II Study of Gemcitabine Plus Placebo or Vismodegib, a Hedgehog Pathway Inhibitor, in Patients with Metastatic Pancreatic Cancer. J. Clin. Oncol. 2015, 33, 4284–4292. [Google Scholar] [CrossRef]
- Gabai, Y.; Assouline, B.; Ben-Porath, I. Senescent stromal cells: Roles in the tumor microenvironment. Trends Cancer 2022, 9, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Zhou, P.K. DNA damage repair: Historical perspectives, mechanistic pathways and clinical translation for targeted cancer therapy. Signal Transduct. Target Ther. 2021, 6, 254. [Google Scholar]
- Liu, Y.; Lv, H.; Li, X.; Liu, J.; Chen, S.; Chen, Y.; Jin, Y.; An, R.; Yu, S.; Wang, Z. Cyclovirobuxine inhibits the progression of clear cell renal cell carcinoma by suppressing the IGFBP3-AKT/STAT3/MAPK-Snail signalling pathway. Int. J. Biol. Sci. 2021, 17, 3522–3537. [Google Scholar] [CrossRef]
- Pahwa, R.; Dubhashi, J.; Singh, A.; Jailwala, P.; Lobanov, A.; Thomas, C.J.; Ceribelli, M.; Wilson, K.; Ricketts, C.J.; Vocke, C.D.; et al. Inhibition of HSP 90 is associated with potent anti-tumor activity in Papillary Renal Cell Carcinoma. J. Exp. Clin. Cancer Res. 2022, 41, 208. [Google Scholar] [CrossRef] [PubMed]
- Ji, F.; Zhang, F.; Zhang, M.; Long, K.; Xia, M.; Lu, F.; Li, E.; Chen, J.; Li, J.; Chen, Z.; et al. Targeting the DNA damage response enhances CD70 CAR-T cell therapy for renal carcinoma by activating the cGAS-STING pathway. J. Hematol. Oncol. 2021, 14, 152. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Sekino, Y.; Li, H.-T.; Fu, G.; Yang, Z.; Zhao, S.; Gujar, H.; Zu, X.; Weisenberger, D.J.; Gill, I.S.; et al. SETD2 Deficiency Confers Sensitivity to Dual Inhibition of DNA Methylation and PARP in Kidney Cancer. Cancer Res. 2023, 83, 3813–3826. [Google Scholar] [CrossRef] [PubMed]
- Cocco, E.; Bellone, S.; El-Sahwi, K.; Cargnelutti, M.; Casagrande, F.; Buza, N.; Tavassoli, F.A.; Siegel, E.R.; Visintin, I.; Ratner, E.; et al. Serum amyloid A (SAA): A novel biomarker for uterine serous papillary cancer. Br. J. Cancer 2009, 101, 335–341. [Google Scholar] [CrossRef]
- Stone, M.L.; Lee, J.; Lee, J.W.; Coho, H.; Tariveranmoshabad, M.; Wattenberg, M.M.; Choi, H.; Herrera, V.M.; Xue, Y.; Choi-Bose, S.; et al. Hepatocytes coordinate immune evasion in cancer via release of serum amyloid A proteins. Nat. Immunol. 2024, 25, 755–763. [Google Scholar] [CrossRef]
- Wu, Q.; You, L.; Nepovimova, E.; Heger, Z.; Wu, W.; Kuca, K.; Adam, V. Hypoxia-inducible factors: Master regulators of hypoxic tumor immune escape. J. Hematol. Oncol. 2022, 15, 77. [Google Scholar]
- Zila, N.; Eichhoff, O.M.; Steiner, I.; Mohr, T.; Bileck, A.; Cheng, P.F.; Leitner, A.; Gillet, L.; Sajic, T.; Goetze, S.; et al. Proteomic Profiling of Advanced Melanoma Patients to Predict Therapeutic Response to Anti-PD-1 Therapy. Clin. Cancer Res. 2024, 30, 159–175. [Google Scholar] [CrossRef]
- Wang, Y.-T.; Chen, J.; Chang, C.-W.; Jen, J.; Huang, T.-Y.; Chen, C.-M.; Shen, R.; Liang, S.-Y.; Cheng, I.-C.; Yang, S.-C.; et al. Ubiquitination of tumor suppressor PML regulates prometastatic and immunosuppressive tumor microenvironment. J. Clin. Investig. 2017, 27, 2982–2997. [Google Scholar] [CrossRef]
- Cooley, L.S.; Rudewicz, J.; Souleyreau, W.; Emanuelli, A.; Alvarez-Arenas, A.; Clarke, K.; Falciani, F.; Dufies, M.; Lambrechts, D.; Modave, E.; et al. Experimental and computational modeling for signature and biomarker discovery of renal cell carcinoma progression. Mol. Cancer 2021, 20, 136. [Google Scholar] [CrossRef]
- Bailey, C.M.; Liu, Y.; Liu, M.; Du, X.; Devenport, M.; Zheng, P.; Liu, Y.; Wang, Y. Targeting HIF-1α abrogates PD-L1-mediated immune evasion in tumor microenvironment but promotes tolerance in normal tissues. J. Clin. Investig. 2022, 132, e150846. [Google Scholar] [CrossRef]
- Terry, S.; Engelsen, A.S.T.; Buart, S.; Elsayed, W.S.; Venkatesh, G.H.; Chouaib, S. Hypoxia-driven intratumor heterogeneity and immune evasion. Cancer Lett. 2020, 492, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Harbour, J.W.; Onken, M.D.; Roberson, E.D.; Duan, S.; Cao, L.; Worley, L.A.; Helms, C.; Bowcock, A.M. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science 2010, 330, 1410–1413. [Google Scholar] [CrossRef]
- Ho, T.H.; Kapur, P.; Joseph, R.W.; Serie, D.J.; Eckel-Passow, J.E.; Parasramka, M.; Cheville, J.C.; Wu, K.J.; Frenkel, E.; Rakheja, D.; et al. Loss of PBRM1 and BAP1 expression is less common in non-clear cell renal cell carcinoma than in clear cell renal cell carcinoma. Urol. Oncol. 2015, 33, 23.e9–23.e14. [Google Scholar] [CrossRef] [PubMed]
- da Costa, W.H.; Fares, A.F.; Bezerra, S.M.; Morini, M.A.; de Toledo Benigno, L.A.; Clavijo, D.A.; Fornazieri, L.; Rocha, M.M.; da Cunha, I.W.; de Cassio Zequi, S. Loss of BAP1 expression in metastatic tumor tissue is an event of poor prognosis in patients with metastatic clear cell renal cell carcinoma. Urol. Oncol. 2019, 37, 78–85. [Google Scholar] [CrossRef]
- Lee, C.H.; Motzer, R.; Emamekhoo, H.; Matrana, M.; Percent, I.; Hsieh, J.J.; Hussain, A.; Vaishampayan, U.; Liu, S.; McCune, S.; et al. Telaglenastat plus Everolimus in Advanced Renal Cell Carcinoma: A Randomized, Double-Blinded, Placebo-Controlled, Phase II ENTRATA Trial. Clin. Cancer Res. 2022, 28, 3248–3255. [Google Scholar] [CrossRef]
- Hutson, T.E.; Michaelson, M.D.; Kuzel, T.M.; Agarwal, N.; Molina, A.M.; Hsieh, J.J.; Vaishampayan, U.N.; Xie, R.; Bapat, U.; Ye, W.; et al. A Single-arm, Multicenter, Phase 2 Study of Lenvatinib Plus Everolimus in Patients with Advanced Non-Clear Cell Renal Cell Carcinoma. Eur. Urol. 2021, 80, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Albiges, L.; Flippot, R.; Rioux-Leclercq, N.; Choueiri, T.K. Non-Clear Cell Renal Cell Carcinomas: From Shadow to Light. J. Clin. Oncol. 2018, JCO2018792531, online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Greco, F.; Mallio, C.A. Relationship between visceral adipose tissue and genetic mutations (VHL and KDM5C) in clear cell renal cell carcinoma. Radiol. Med. 2021, 126, 645–651. [Google Scholar] [CrossRef]
- Zheng, Q.; Li, P.; Zhou, X.; Qiang, Y.; Fan, J.; Lin, Y.; Chen, Y.; Guo, J.; Wang, F.; Xue, H.; et al. Deficiency of the X-inactivation escaping gene KDM5C in clear cell renal cell carcinoma promotes tumorigenicity by reprogramming glycogen metabolism and inhibiting ferroptosis. Theranostics 2021, 11, 8674–8691. [Google Scholar] [CrossRef]
- Fernandes, F.G.; Silveira, H.C.S.; Junior, J.N.A.; da Silveira, R.A.; Zucca, L.E.; Carcano, F.M.; Sanches, A.O.N.; Neder, L.; Scapulatempo-Neto, C.; Serrano, S.V.; et al. Somatic Copy Number Alterations and Associated Genes in Clear-Cell Renal-Cell Carcinoma in Brazilian Patients. Int. J. Mol. Sci. 2021, 22, 2265. [Google Scholar] [CrossRef]
- Yuan, T.; Edelmann, D.; Fan, Z.; Alwers, E.; Kather, J.N.; Brenner, H.; Hoffmeister, M. Machine learning in the identification of prognostic DNA methylation biomarkers among patients with cancer: A systematic review of epigenome-wide studies. Artif. Intell. Med. 2023, 143, 102589. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CRCS1 | CRCS2 | p.Overall | |
|---|---|---|---|
| N = 224 | N = 282 | ||
| T: | 0.001 | ||
| T1 | 98 (43.8%) | 162 (57.4%) | |
| T2 | 28 (12.5%) | 39 (13.8%) | |
| T3 | 89 (39.7%) | 79 (28.0%) | |
| T4 | 9 (4.02%) | 2 (0.71%) | |
| N: | 0.112 | ||
| N1 | 10 (8.26%) | 5 (3.18%) | |
| NX | 111 (91.7%) | 152 (96.8%) | |
| M: | 0.560 | ||
| M1 | 41 (71.9%) | 34 (79.1%) | |
| MX | 16 (28.1%) | 9 (20.9%) | |
| grade: | 0.001 | ||
| G1 | 5 (2.23%) | 7 (2.48%) | |
| G2 | 81 (36.2%) | 136 (48.2%) | |
| G3 | 89 (39.7%) | 112 (39.7%) | |
| G4 | 46 (20.5%) | 25 (8.87%) | |
| GX | 3 (1.34%) | 2 (0.71%) | |
| stage: | 0.013 | ||
| i | 96 (42.9%) | 158 (56.0%) | |
| ii | 24 (10.7%) | 31 (11.0%) | |
| iii | 59 (26.3%) | 58 (20.6%) | |
| iv | 45 (20.1%) | 35 (12.4%) | |
| sex: | 0.061 | ||
| female | 87 (38.8%) | 86 (30.5%) | |
| male | 137 (61.2%) | 196 (69.5%) | |
| age | 60.2 (12.0) | 60.5 (12.4) | 0.769 |
| OS: | <0.001 | ||
| 0 | 127 (56.7%) | 210 (74.5%) | |
| 1 | 97 (43.3%) | 72 (25.5%) | |
| OS.time | 1210 (879) | 1560 (1018) | <0.001 |
| PFI: | <0.001 | ||
| 0 | 134 (59.8%) | 215 (76.2%) | |
| 1 | 90 (40.2%) | 67 (23.8%) | |
| PFI.time | 1004 (850) | 1373 (980) | <0.001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiong, B.; Liu, W.; Liu, Y.; Chen, T.; Lin, A.; Song, J.; Qu, L.; Luo, P.; Jiang, A.; Wang, L. A Multi-Omics Prognostic Model Capturing Tumor Stemness and the Immune Microenvironment in Clear Cell Renal Cell Carcinoma. Biomedicines 2024, 12, 2171. https://doi.org/10.3390/biomedicines12102171
Xiong B, Liu W, Liu Y, Chen T, Lin A, Song J, Qu L, Luo P, Jiang A, Wang L. A Multi-Omics Prognostic Model Capturing Tumor Stemness and the Immune Microenvironment in Clear Cell Renal Cell Carcinoma. Biomedicines. 2024; 12(10):2171. https://doi.org/10.3390/biomedicines12102171
Chicago/Turabian StyleXiong, Beibei, Wenqiang Liu, Ying Liu, Tong Chen, Anqi Lin, Jiaao Song, Le Qu, Peng Luo, Aimin Jiang, and Linhui Wang. 2024. "A Multi-Omics Prognostic Model Capturing Tumor Stemness and the Immune Microenvironment in Clear Cell Renal Cell Carcinoma" Biomedicines 12, no. 10: 2171. https://doi.org/10.3390/biomedicines12102171
APA StyleXiong, B., Liu, W., Liu, Y., Chen, T., Lin, A., Song, J., Qu, L., Luo, P., Jiang, A., & Wang, L. (2024). A Multi-Omics Prognostic Model Capturing Tumor Stemness and the Immune Microenvironment in Clear Cell Renal Cell Carcinoma. Biomedicines, 12(10), 2171. https://doi.org/10.3390/biomedicines12102171