

The Role of mTOR in Mycobacterium tuberculosis Infection


Abstract
:1. Introduction
2. Materials and Methods
3. Pathology of M. tb
3.1. Characteristics of M. tb
3.2. Acute M. tb Infection
3.3. Latent M. tb Infection
4. Role of mTOR in the Immune System
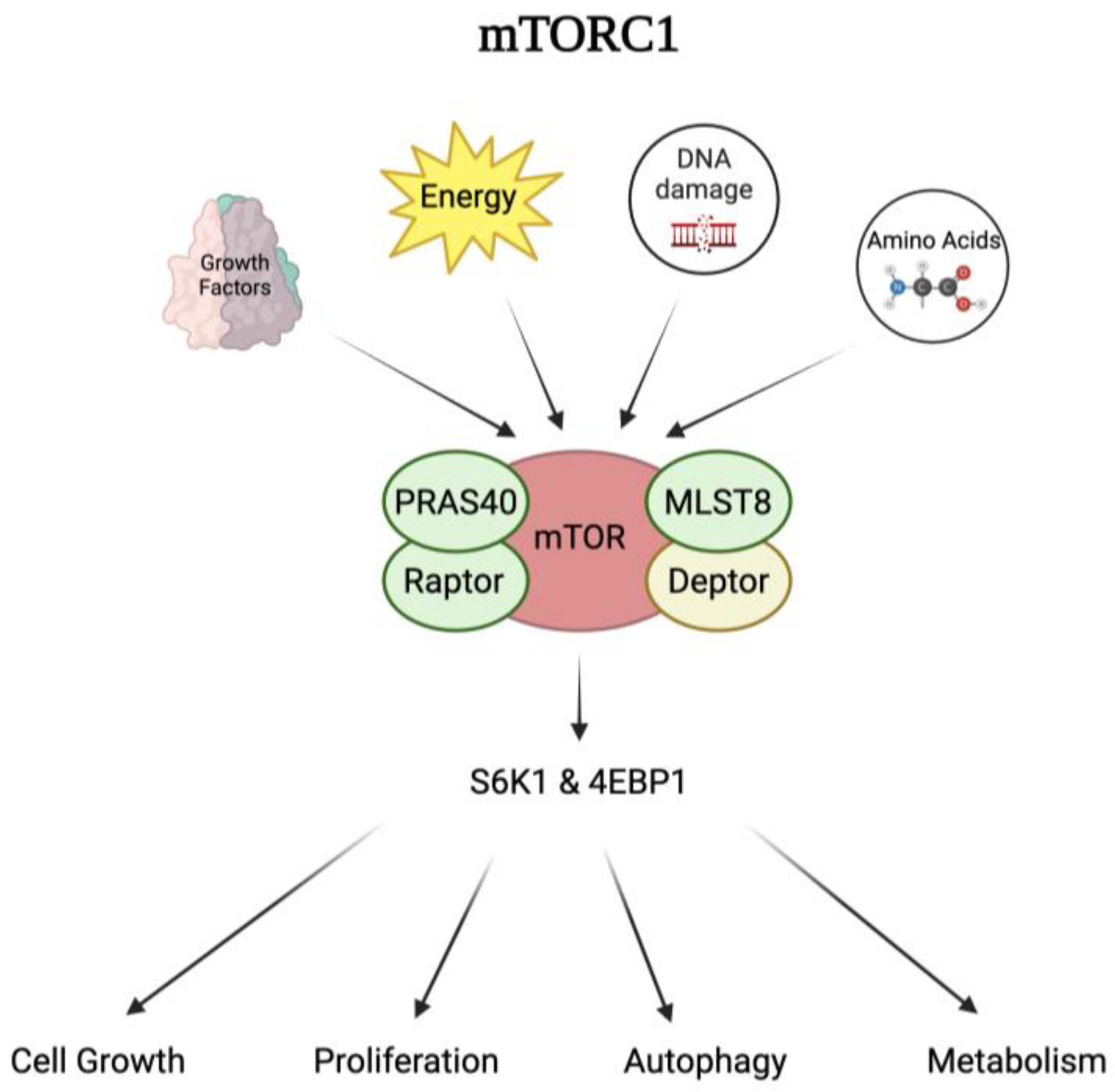
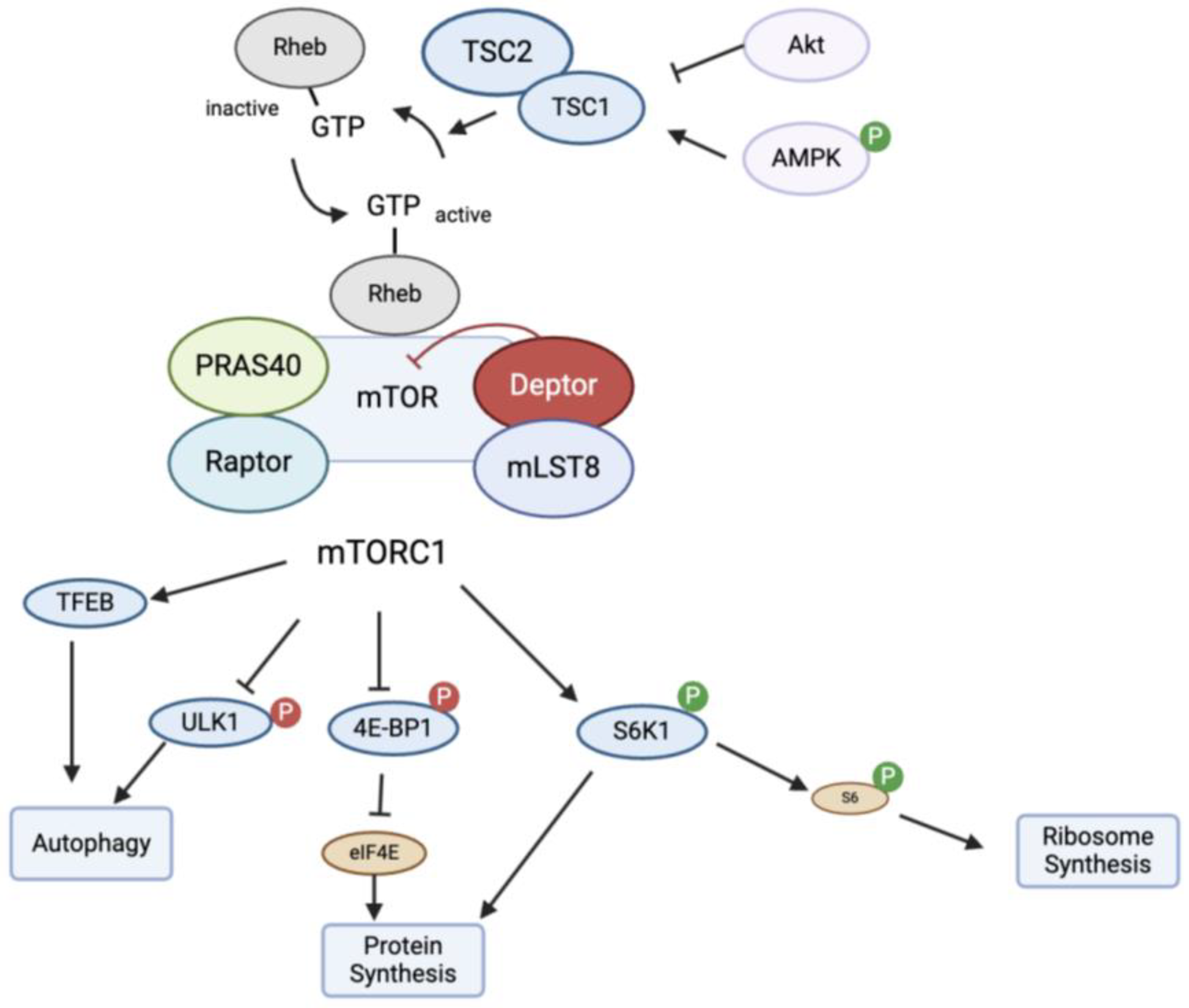
4.1. Mechanism of Action of mTOR
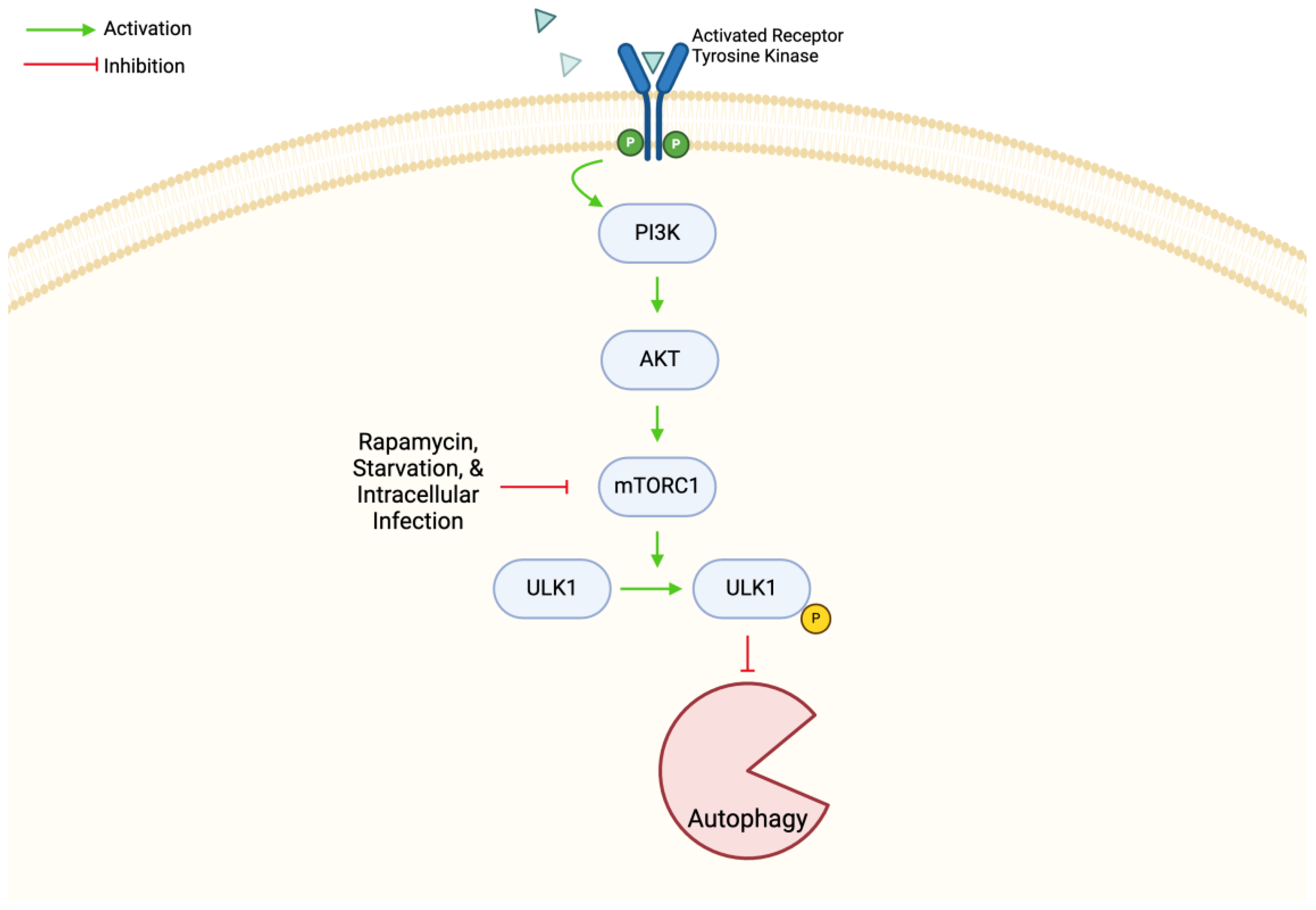
4.2. mTOR in Autophagy
5. Interplay between mTOR and M. tb Infection
6. Current and Future Therapeutics Targeting OR
6.1. Current Therapeutics
6.2. Repurposing of Existing Drugs
6.3. Potential Therapies to Explore
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Global Tuberculosis Report 2023; World Health Organization: Geneva, Switzerland, 2023.
- Oswal, N.; Lizardo, K.; Dhanyalayam, D.; Ayyappan, J.P.; Thangavel, H.; Heysell, S.K.; Nagajyothi, J.F. Host Metabolic Changes during Mycobacterium tuberculosis Infection Cause Insulin Resistance in Adult Mice. J. Clin. Med. 2022, 11, 1646. [Google Scholar] [CrossRef] [PubMed]
- Dheda, K.; Mirzayev, F.; Cirillo, D.M.; Udwadia, Z.; Dooley, K.E.; Chang, K.-C.; Omar, S.V.; Reuter, A.; Perumal, T.; Horsburgh, C.R., Jr.; et al. Multidrug-resistant tuberculosis. Nat. Rev. Dis. Prim. 2024, 10, 22. [Google Scholar] [CrossRef] [PubMed]
- Lange, C.; Chesov, D.; Heyckendorf, J.; Leung, C.C.; Udwadia, Z.; Dheda, K. Drug-resistant tuberculosis: An update on disease burden, diagnosis and treatment. Respirology 2018, 23, 656–673. [Google Scholar] [CrossRef]
- Khawbung, J.L.; Nath, D.; Chakraborty, S. Drug resistant Tuberculosis: A review. Comp. Immunol. Microbiol. Infect. Dis. 2021, 74, 101574. [Google Scholar] [CrossRef]
- Qu, M.; Zhou, X.; Li, H. BCG vaccination strategies against tuberculosis: Updates and perspectives. Hum. Vaccines Immunother. 2021, 17, 5284–5295. [Google Scholar] [CrossRef]
- Mundra, A.; Yegiazaryan, A.; Karsian, H.; Alsaigh, D.; Bonavida, V.; Frame, M.; May, N.; Gargaloyan, A.; Abnousian, A.; Venketaraman, V. Pathogenicity of Type I Interferons in Mycobacterium tuberculosis. Int. J. Mol. Sci. 2023, 24, 3919. [Google Scholar] [CrossRef]
- Nguyen, K.H.; Alcantara, C.A.; Glassman, I.; May, N.; Mundra, A.; Mukundan, A.; Urness, B.; Yoon, S.; Sakaki, R.; Dayal, S.; et al. Cutaneous Manifestations of Mycobacterium tuberculosis: A Literature Review. Pathogens 2023, 12, 920. [Google Scholar] [CrossRef]
- Delogu, G.; Sali, M.; Fadda, G. The biology of Mycobacterium tuberculosis infection. Mediterr. J. Hematol. Infect. Dis. 2013, 5, e2013070. [Google Scholar] [CrossRef]
- Alcantara, C.A.; Glassman, I.; Nguyen, K.H.; Parthasarathy, A.; Venketaraman, V. Neutrophils in Mycobacterium tuberculosis. Vaccines 2023, 11, 631. [Google Scholar] [CrossRef]
- Jamwal, S.V.; Mehrotra, P.; Singh, A.; Siddiqui, Z.; Basu, A.; Rao, K.V. Mycobacterial escape from macrophage phagosomes to the cytoplasm represents an alternate adaptation mechanism. Sci. Rep. 2016, 6, 23089. [Google Scholar] [CrossRef]
- Yu, X.; Xie, J. Roles and underlying mechanisms of ESAT-6 in the context of Mycobacterium tuberculosis–host interaction from a systems biology perspective. Cell. Signal. 2012, 24, 1841–1846. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Liu, L.; Meng, Z.; Qi, K.; Gao, X.; Feng, J.; Luo, J. Recognition of Mycobacterium tuberculosis by macrophage Toll-like receptor and its role in autophagy. Inflamm. Res. 2024, 73, 753–770. [Google Scholar] [CrossRef] [PubMed]
- Stewart, P.; Patel, S.; Comer, A.; Muneer, S.; Nawaz, U.; Quann, V.; Bansal, M.; Venketaraman, V. Role of B Cells in Mycobacterium tuberculosis Infection. Vaccines 2023, 11, 955. [Google Scholar] [CrossRef] [PubMed]
- Shanmuganathan, G.; Orujyan, D.; Narinyan, W.; Poladian, N.; Dhama, S.; Parthasarathy, A.; Ha, A.; Tran, D.; Velpuri, P.; Nguyen, K.H.; et al. Role of Interferons in Mycobacterium tuberculosis Infection. Clin. Pract. 2022, 12, 788–796. [Google Scholar] [CrossRef]
- Lin, P.L.; Plessner, H.L.; Voitenok, N.N.; Flynn, J.L. Tumor Necrosis Factor and Tuberculosis. J. Investig. Dermatol. Symp. Proc. 2007, 12, 22–25. [Google Scholar] [CrossRef] [PubMed]
- Pagán, A.J.; Lee, L.J.; Edwards-Hicks, J.; Moens, C.B.; Tobin, D.M.; Busch-Nentwich, E.M.; Pearce, E.L.; Ramakrishnan, L. mTOR-regulated mitochondrial metabolism limits mycobacterium-induced cytotoxicity. Cell 2022, 185, 3720–3738. [Google Scholar] [CrossRef]
- Davis, J.M.; Ramakrishnan, L. The role of the granuloma in expansion and dissemination of early tuberculous infection. Cell 2009, 136, 37–49. [Google Scholar] [CrossRef]
- Matta, S.K.; Kumar, D. Hypoxia and classical activation limits Mycobacterium tuberculosis survival by Akt-dependent glycolytic shift in macrophages. Cell Death Discov. 2016, 2, 16022. [Google Scholar] [CrossRef] [PubMed]
- Poladian, N.; Orujyan, D.; Narinyan, W.; Oganyan, A.K.; Navasardyan, I.; Velpuri, P.; Chorbajian, A.; Venketaraman, V. Role of NF-κB during Mycobacterium tuberculosis Infection. Int. J. Mol. Sci. 2023, 24, 1772. [Google Scholar] [CrossRef]
- Amaral, E.P.; Foreman, T.W.; Namasivayam, S.; Hilligan, K.L.; Kauffman, K.D.; Bomfim, C.C.B.; Costa, D.L.; Barreto-Duarte, B.; Gurgel-Rocha, C.; Santana, M.F.; et al. GPX4 regulates cellular necrosis and host resistance in Mycobacterium tuberculosis infection. J. Exp. Med. 2022, 219, e20220504. [Google Scholar] [CrossRef]
- Wang, J.; Wang, R.; Wang, H.; Yang, X.; Yang, J.; Xiong, W.; Wen, Q.; Ma, L. Glucocorticoids Suppress Antimicrobial Autophagy and Nitric Oxide Production and Facilitate Mycobacterial Survival in Macrophages. Sci. Rep. 2017, 7, 982. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR signaling at a glance. J. Cell Sci. 2009, 122, 3589–3594. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Peterson, T.R.; Sabatini, D.M. Regulation of the mTOR Complex 1 pathway by nutrients, growth factors, and stress. Mol. Cell 2010, 40, 310–322. [Google Scholar] [CrossRef] [PubMed]
- Foster, K.G.; Acosta-Jaquez, H.A.; Romeo, Y.; Ekim, B.; Soliman, G.A.; Carriere, A.; Roux, P.P.; Ballif, B.A.; Fingar, D.C. Regulation of mTOR Complex 1 (mTORC1) by Raptor Ser863 and Multisite Phosphorylation. J. Biol. Chem. 2010, 285, 80–94. [Google Scholar] [CrossRef] [PubMed]
- Oshiro, N.; Takahashi, R.; Yoshino, K.-I.; Tanimura, K.; Nakashima, A.; Eguchi, S.; Miyamoto, T.; Hara, K.; Takehana, K.; Avruch, J.; et al. The Proline-rich Akt Substrate of 40 kDa (PRAS40) Is a Physiological Substrate of Mammalian Target of Rapamycin Complex 1. J. Biol. Chem. 2007, 282, 20329–20339. [Google Scholar] [CrossRef]
- Peterson, T.R.; Laplante, M.; Thoreen, C.C.; Sancak, Y.; Kang, S.A.; Kuehl, W.M.; Gray, N.S.; Sabatini, D.M. DEPTOR Is an mTOR Inhibitor Frequently Overexpressed in Multiple Myeloma Cells and Required for Their Survival. Cell 2009, 137, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Long, X.; Lin, Y.; Ortiz-Vega, S.; Yonezawa, K.; Avruch, J. Rheb binds and regulates the mTOR kinase. Curr. Biol. 2005, 15, 702–713. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.M.; Blenis, J. Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 2009, 10, 307–318. [Google Scholar] [CrossRef]
- Chun, Y.; Kim, J. AMPK–mTOR Signaling and Cellular Adaptations in Hypoxia. Int. J. Mol. Sci. 2021, 22, 9765. [Google Scholar] [CrossRef]
- Zullo, A.J.; Lee, S. Mycobacterial induction of autophagy varies by species and occurs independently of mammalian target of rapamycin inhibition. J. Biol. Chem. 2012, 287, 12668–12678. [Google Scholar] [CrossRef]
- Hara, K.; Maruki, Y.; Long, X.; Yoshino, K.-I.; Oshiro, N.; Hidayat, S.; Tokunaga, C.; Avruch, J.; Yonezawa, K. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 2002, 110, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Cerni, S.; Shafer, D.; To, K.; Venketaraman, V. Investigating the role of everolimus in mTOR inhibition and autophagy promotion as a potential host-directed therapeutic target in Mycobacterium tuberculosis infection. J. Clin. Med. 2019, 8, 232. [Google Scholar] [CrossRef]
- Tatano, Y.; Shimizu, T.; Sano, C.; Tomioka, H. Roles of autophagy in killing of mycobacterial pathogens by host macrophages—Effects of some medicinal plants. Eur. J. Microbiol. Immunol. 2024, 14, 26–36. [Google Scholar] [CrossRef]
- Pan, H.; Zhong, X.-P.; Lee, S. Sustained activation of mTORC1 in macrophages increases AMPKα-dependent autophagy to maintain cellular homeostasis. BMC Biochem. 2016, 17, 14. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Sun, J.; Wang, Y.; He, W.; Wang, L.; Zheng, Y.; Wu, J.; Zhang, Y.; Jiang, X. Antimycobacterial and Anti-inflammatory Mechanisms of Baicalin via Induced Autophagy in Macrophages Infected with Mycobacterium tuberculosis. Front. Microbiol. 2017, 8, 2142. [Google Scholar] [CrossRef] [PubMed]
- Silwal, P.; Paik, S.; Kim, J.K.; Yoshimori, T.; Jo, E.-K. Regulatory mechanisms of autophagy-targeted antimicrobial therapeutics against mycobacterial infection. Front. Cell. Infect. Microbiol. 2021, 11, 633360. [Google Scholar] [CrossRef]
- Andersson, A.-M.; Andersson, B.; Lorell, C.; Raffetseder, J.; Larsson, M.; Blomgran, R. Autophagy induction targeting mTORC1 enhances Mycobacterium tuberculosis replication in HIV co-infected human macrophages. Sci. Rep. 2016, 6, 28171. [Google Scholar] [CrossRef] [PubMed]
- Strong, E.J.; Smith, K.L.J.; Saini, N.K.; Ng, T.W.; Porcelli, S.A.; Lee, S. Identification of autophagy-inhibiting factors of Mycobacterium tuberculosis by high-throughput loss-of-function screening. Infect. Immun. 2020, 88, e00269-20. [Google Scholar] [CrossRef]
- Xu, Z.; Han, X.; Ou, D.; Liu, T.; Li, Z.; Jiang, G.; Liu, J.; Zhang, J. Targeting PI3K/AKT/mTOR-mediated autophagy for tumor therapy. Appl. Microbiol. Biotechnol. 2020, 104, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Ming, S.; Song, W.; Meng, X.; Xiao, Q.; Wu, M.; Wu, Y.; Xie, H.; Zhou, J.; Zhong, H.; et al. B and T lymphocyte attenuator regulates autophagy in mycobacterial infection via the AKT/mTOR signal pathway. Int. Immunopharmacol. 2020, 91, 107215. [Google Scholar] [CrossRef]
- Zheng, Q.; Li, Z.; Zhou, Y.; Li, Y.; Gong, M.; Sun, H.; Deng, X.; Ma, Y. Heparin-Binding Hemagglutinin of Mycobacterium tuberculosis Inhibits Autophagy via Toll-like Receptor 4 and Drives M2 Polarization in Macrophages. J. Infect. Dis. 2024. [Google Scholar] [CrossRef] [PubMed]
- Weichhart, T.; Hengstschläger, M.; Linke, M. Regulation of innate immune cell function by mTOR. Nat. Rev. Immunol. 2015, 15, 599–614. [Google Scholar] [CrossRef] [PubMed]
- Sachdeva, K.; Goel, M.; Sudhakar, M.; Mehta, M.; Raju, R.; Raman, K.; Singh, A.; Sundaramurthy, V. Mycobacterium tuberculosis (Mtb) lipid mediated lysosomal rewiring in infected macrophages modulates intracellular Mtb trafficking and survival. J. Biol. Chem. 2020, 295, 9192–9210. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, G.; Ballabio, A. TFEB at a glance. J. Cell Sci. 2016, 129, 2475–2481. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Weng, S.; Luo, L.; Sun, Q.; Lin, T.; Ma, H.; He, Y.; Wu, J.; Wang, H.; Zhang, W.; et al. Mycobacterium tuberculosis hijacks host macrophages-derived interleukin 16 to block phagolysosome maturation for enhancing intracellular growth. Emerg. Microbes Infect. 2024, 13, 2322663. [Google Scholar] [CrossRef] [PubMed]
- Sachdeva, K.; Sundaramurthy, V. The Interplay of Host Lysosomes and Intracellular Pathogens. Front. Cell. Infect. Microbiol. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V. Autophagy, an immunologic magic bullet: Mycobacterium tuberculosis phagosome maturation block and how to bypass it. Futur. Microbiol. 2008, 3, 517–524. [Google Scholar] [CrossRef]
- Zhuang, L.; Yang, L.; Li, L.; Ye, Z.; Gong, W. Mycobacterium tuberculosis: Immune response, biomarkers, and therapeutic intervention. Medcomm 2024, 5, e419. [Google Scholar] [CrossRef]
- Amaral, E.P.; Riteau, N.; Moayeri, M.; Maier, N.; Mayer-Barber, K.D.; Pereira, R.M.; Lage, S.L.; Kubler, A.; Bishai, W.R.; D’império-Lima, M.R.; et al. Lysosomal Cathepsin Release Is Required for NLRP3-Inflammasome Activation by Mycobacterium tuberculosis in Infected Macrophages. Front. Immunol. 2018, 9, 1427. [Google Scholar] [CrossRef]
- O’Leary, S.; O’Sullivan, M.P.; Keane, J. IL-10 blocks phagosome maturation in Mycobacterium tuberculosis—Infected human macrophages. Am. J. Respir. Cell Mol. Biol. 2011, 45, 172–180. [Google Scholar] [CrossRef]
- Zhang, X.; Huang, T.; Peng, W.; Xie, H.; Pan, M.; Zhou, H.; Cai, B.; Wu, Y. Inhibition of the PI3K-Akt-mTOR signaling pathway in T lymphocytes in patients with active tuberculosis. Int. J. Infect. Dis. 2017, 59, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Quadir, N.; Shariq, M.; Sheikh, J.A.; Singh, J.; Sharma, N.; Hasnain, S.E.; Ehtesham, N.Z. Mycobacterium tuberculosis protein MoxR1 enhances virulence by inhibiting host cell death pathways and disrupting cellular bioenergetics. Virulence 2023, 14, 2180230. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Yi, M.; Chen, J.; Li, S.; Chen, W. Mycobacterium tuberculosis EIS gene inhibits macrophage autophagy through up-regulation of IL-10 by increasing the acetylation of histone H3. Biochem. Biophys. Res. Commun. 2016, 473, 1229–1234. [Google Scholar] [CrossRef]
- Paik, S.; Kim, K.T.; Kim, I.S.; Kim, Y.J.; Kim, H.J.; Choi, S.; Kim, H.-J.; Jo, E.-K. Mycobacterial acyl carrier protein suppresses TFEB activation and upregulates miR-155 to inhibit host defense. Front. Immunol. 2022, 13, 946929. [Google Scholar] [CrossRef] [PubMed]
- Mohareer, K.; Medikonda, J.; Vadankula, G.R.; Banerjee, S. Mycobacterial Control of Host Mitochondria: Bioenergetic and Metabolic Changes Shaping Cell Fate and Infection Outcome. Front. Cell. Infect. Microbiol. 2020, 10, 457. [Google Scholar] [CrossRef]
- Gleeson, L.E.; Sheedy, F.J.; Palsson-McDermott, E.M.; Triglia, D.; O’Leary, S.M.; O’Sullivan, M.P.; O’Neill, L.A.J.; Keane, J. Cutting Edge: Mycobacterium tuberculosis Induces Aerobic Glycolysis in Human Alveolar Macrophages That Is Required for Control of Intracellular Bacillary Replication. J. Immunol. 2016, 196, 2444–2449. [Google Scholar] [CrossRef] [PubMed]
- Paroha, R.; Wang, J.; Lee, S. PDCD4 as a marker of mTOR pathway activation and therapeutic target in mycobacterial infections. Microbiol. Spectr. 2024, 12, e0006224. [Google Scholar] [CrossRef]
- Alsayed, S.S.R.; Gunosewoyo, H. Tuberculosis: Pathogenesis, Current Treatment Regimens and New Drug Targets. Int. J. Mol. Sci. 2023, 24, 5202. [Google Scholar] [CrossRef]
- Rawat, R.; Whitty, A.; Tonge, P.J. The isoniazid-NAD adduct is a slow, tight-binding inhibitor of InhA, the Mycobacterium tuberculosis enoyl reductase: Adduct affinity and drug resistance. Proc. Natl. Acad. Sci. USA 2003, 100, 13881–13886. [Google Scholar] [CrossRef]
- Louw, G.E.; Warren, R.M.; van Pittius, N.C.G.; Leon, R.; Jimenez, A.; Hernandez-Pando, R.; McEvoy, C.R.E.; Grobbelaar, M.; Murray, M.; van Helden, P.D.; et al. Rifampicin reduces susceptibility to ofloxacin in rifampicin-resistant Mycobacterium tuberculosis through efflux. Am. J. Respir. Crit. Care Med. 2011, 184, 269–276. [Google Scholar] [CrossRef]
- Ouyang, Q.; Zhang, K.; Lin, D.; Feng, C.G.; Cai, Y.; Chen, X. Bazedoxifene Suppresses Intracellular Mycobacterium tuberculosis Growth by Enhancing Autophagy. mSphere 2020, 5, e00124-20. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sha, J.; Strong, E.; Chopra, A.K.; Lee, S. FDA-Approved Amoxapine Effectively Promotes Macrophage Control of Mycobacteria by Inducing Autophagy. Microbiol. Spectr. 2022, 10, e0250922. [Google Scholar] [CrossRef] [PubMed]
- Raien, A.; Davis, S.; Zhang, M.; Zitser, D.; Lin, M.; Pitcher, G.; Bhalodia, K.; Subbian, S.; Venketaraman, V. Effects of Everolimus in Modulating the Host Immune Responses against Mycobacterium tuberculosis Infection. Cells 2023, 12, 2653. [Google Scholar] [CrossRef]
- Ashley, D.; Hernandez, J.; Cao, R.; To, K.; Yegiazaryan, A.; Abrahem, R.; Nguyen, T.; Owens, J.; Lambros, M.; Subbian, S.; et al. Antimycobacterial Effects of Everolimus in a Human Granuloma Model. J. Clin. Med. 2020, 9, 2043. [Google Scholar] [CrossRef] [PubMed]
- Schiebler, M.; Brown, K.; Hegyi, K.; Newton, S.M.; Renna, M.; Hepburn, L.; Klapholz, C.; Coulter, S.; Obregón-Henao, A.; Tamayo, M.H.; et al. Functional drug screening reveals anticonvulsants as enhancers of mTOR-independent autophagic killing of Mycobacterium tuberculosis through inositol depletion. EMBO Mol. Med. 2015, 7, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Mahon, R.N.; Hafner, R. Immune Cell Regulatory Pathways Unexplored as Host-Directed Therapeutic Targets for Mycobacterium tuberculosis: An Opportunity to Apply Precision Medicine Innovations to Infectious Diseases. Clin. Infect. Dis. 2015, 61, S200–S216. [Google Scholar] [CrossRef] [PubMed]
- Tasneen, R.; Mortensen, D.S.; Converse, P.J.; Urbanowski, M.E.; Upton, A.; Fotouhi, N.; Nuermberger, E.; Hawryluk, N. Dual mTORC1/mTORC2 Inhibition as a Host-Directed Therapeutic Target in Pathologically Distinct Mouse Models of Tuberculosis. Antimicrob. Agents Chemother. 2021, 65, e0025321. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Kim, S.G.; Blenis, J. Rapamycin: One drug, many effects. Cell Metab. 2014, 19, 373–379. [Google Scholar] [CrossRef]
- Gonzalez-Orozco, M.; Strong, E.J.; Paroha, R.; Lee, S. Reversing BCG-mediated autophagy inhibition and mycobacterial survival to improve vaccine efficacy. BMC Immunol. 2022, 23, 43. [Google Scholar] [CrossRef]
- Abnousian, A.; Vasquez, J.; Sasaninia, K.; Kelley, M.; Venketaraman, V. Glutathione Modulates Efficacious Changes in the Immune Response against Tuberculosis. Biomedicines 2023, 11, 1340. [Google Scholar] [CrossRef]
- Cao, R.; Kolloli, A.; Kumar, R.; Owens, J.; Sasaninia, K.; Vaughn, C.; Singh, M.; Truong, E.; Kachour, N.; Beever, A.; et al. Effects of Glutathione Diminishment on the Immune Responses against Mycobacterium tuberculosis Infection. Appl. Sci. 2021, 11, 8274. [Google Scholar] [CrossRef] [PubMed]
- Kachour, N.; Beever, A.; Owens, J.; Cao, R.; Kolloli, A.; Kumar, R.; Sasaninia, K.; Vaughn, C.; Singh, M.; Truong, E.; et al. Liposomal Glutathione Helps to Mitigate Mycobacterium tuberculosis Infection in the Lungs. Antioxidants 2022, 11, 673. [Google Scholar] [CrossRef] [PubMed]
- To, K.; Cao, R.; Yegiazaryan, A.; Owens, J.; Nguyen, T.; Sasaninia, K.; Vaughn, C.; Singh, M.; Truong, E.; Medina, A.; et al. Effects of Oral Liposomal Glutathione in Altering the Immune Responses Against Mycobacterium tuberculosis and the Mycobacterium bovis BCG Strain in Individuals With Type 2 Diabetes. Front. Cell. Infect. Microbiol. 2021, 11, 657775. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Subbian, S. Harnessing the mTOR Pathway for Tuberculosis Treatment. Front. Microbiol. 2018, 9, 70. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Therapy | Target and Mechanism of Action | Outcome of Therapy Use in M. tb | Ref |
|---|---|---|---|
| Bazedoxifene | Estrogen receptor modulator, activates AMPK/mTOR signaling | Enhance autophagy through increased autophagosome formation and proteins involved with autophagy | [62] |
| Amoxapine | Anti-depressant | Enhance autophagy | [63] |
| Everolimus | mTOR inhibitor | Reduce bacterial load, decrease mycobacterial activity | [64,65] |
| Carbamazepine and Valproic acid (anticonvulsants) | Depletes myo-inositol | Stimulate autophagy, decrease bacterial load, enhance host adaptive immunity | [66] |
| Precision Medicine | Target autophagy or immune cell function | Improve host’s immune defense to decrease treatment duration and effect of infection | [67] |
| Rapamycin + CC214-2 | Partial inhibition of mTORC1 (from rapamycin) + inhibition of mTORC1 and mTORC2 (from CC214-2) | Fewer relapses of infection | [68,69] |
| Mycobacteria smegmatis pre-infection | Potentially enhance vaccination protection of Bacillus Calmette–Guérin (BCG) | Increased autophagy activation | [70] |
| Glutathione | Initiate Th1 type responses, enhance T lymphocytes | Decrease oxidative stress, improved control of infection in lung granulomas, decrease intracellular burden of M. tb | [71,72,73,74] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patel, A.; Nguyen, L.; Shea, C.; Singh, S.; Venketaraman, V. The Role of mTOR in Mycobacterium tuberculosis Infection. Biomedicines 2024, 12, 2238. https://doi.org/10.3390/biomedicines12102238
Patel A, Nguyen L, Shea C, Singh S, Venketaraman V. The Role of mTOR in Mycobacterium tuberculosis Infection. Biomedicines. 2024; 12(10):2238. https://doi.org/10.3390/biomedicines12102238
Chicago/Turabian StylePatel, Ami, Lannhi Nguyen, Christina Shea, Sunjum Singh, and Vishwanath Venketaraman. 2024. "The Role of mTOR in Mycobacterium tuberculosis Infection" Biomedicines 12, no. 10: 2238. https://doi.org/10.3390/biomedicines12102238