Abstract
Head and neck squamous cell carcinoma (HNSCC) is one of the most common malignancies globally, representing a significant public health problem with a poor prognosis. The development of efficient therapeutic strategies for HNSCC prevention and treatment is urgently needed. The PI3K/AKT/mTOR (PAM) signaling pathway is a highly conserved transduction network in eukaryotic cells that promotes cell survival, growth, and cycle progression. Dysfunction in components of this pathway, such as hyperactivity of PI3K, loss of PTEN function, and gain-of-function mutations in AKT, are well-known drivers of treatment resistance and disease progression in cancer. In this review, we discuss the major mutations and dysregulations in the PAM signaling pathway in HNSCC. We highlight the results of clinical trials involving inhibitors targeting the PAM signaling pathway as a strategy for treating HNSCC. Additionally, we examine the primary mechanisms of resistance to drugs targeting the PAM pathway and potential therapeutic strategies.
1. Introduction
Head and neck squamous cell carcinoma (HNSCC), arising in the oral cavity, larynx, and pharynx, is one of the most common malignancies, ranked sixth worldwide, and affects 600,000 patients annually [1]. HNSCC has a poor prognosis, with only 40–50% surviving more than 5 years [2]. Smoking and alcohol consumption are recognized risk factors for the development of HNC. Meanwhile, viral infection is considered a risk factor associated with the development of HNC subgroups. Infection with specific types of human papillomavirus (HPV) is highly correlated with oropharyngeal cancer, with HPV16 being the most common genotype [3,4]. Currently, surgery, radiation, and chemotherapy are the main therapies for HNSCC. However, these nonselective treatments are not tolerated well with severe systemic complications. While surgery or radiation therapy can often lead to a cure for most patients with early-stage HNSCC, those facing aggressive forms of the disease or those in advanced stages, which account for approximately two-thirds of new diagnoses, tend to have a higher likelihood of recurrence, with a 50% overall survival rate over five years [5]. There are only three targeted drugs approved by the FDA: the epidermal growth factor receptor (EGFR) antibody cetuximab and the programmed death receptor-1 (PD-1) antibodies pembrolizumab and nivolumab [6,7,8]. Cetuximab or pembrolizumab may cause life-threatening thromboembolism [9]. The risk of venous or pulmonary embolism increases approximately 1.5 times with cetuximab or pembrolizumab treatment [9]. Cisplatin is the agent suggested for HNSCC to use in the adjuvant setting [10], which can be associated with significant side effects including kidney disease, blood or bone marrow problems. A new, more targeted therapy is needed to improve the current treatment plan for HNSCC. Understanding the gene alterations involved in HNSCC formation and progression would help develop novel precision therapeutic options. Precision medicine focuses on tailoring treatment plans for patients. One approach for precision medicine is to predict the behavior of a type of tumor based on big omics data analysis, which involves characterizing each tumor’s molecular and genetic features through multiple omics analyses of tumor samples [11]. Institutions worldwide contribute to the sharing of big omics data on tumors through international projects. The other approach is to develop personalized preclinical platforms using in vitro and in vivo techniques to test the behavior and drug sensitivity of samples derived from patients [11]. Deep sequencing of the genomic landscape of HNSCC reveals the diversity of genetic alterations in this malignancy. Although many specific molecules are altered in each tumor, they all involve only a few driving signaling pathways. Among these, the PAM pathway is most frequently activated, playing a central role in cancer development and progression [12]. In turn, targeting the PAM pathway may represent a precision treatment approach for HNSCC. It should be noted that dysregulation of the PAM pathway is common in both HPV-positive and -negative HNSCC [13]. Viral oncogenes such as HPV E6 and E7 can release regulation of EGFR/PI3K/Akt/mTOR and increase PIK3CA gene mutations [14]. Targeted therapy targeting the PAM pathway is a promising treatment strategy for HPV-positive and HPV-negative HNSCC.
Phosphoinositide 3-kinases (PI3Ks) are a family of enzymes that play crucial roles in various cellular functions, including cell growth, proliferation, and differentiation, which in turn are integral to cancer development and progression. It has been reported that the most mutated and amplified oncogene in human cancers, including HNSCC, is PI3K catalytic subunit alpha isoform (PIK3CA), the gene that programs for the p110a isoform of PI3K [15]. PI3K is supposed to be regulated by the tumor suppressor phosphatase and tensin homolog (PTEN). So an inactivating mutation or loss of PTEN can induce the formation of cancer by releasing the suppression of the PI3K pathway [16]. It has also been reported that other effectors like protein kinase B (Akt) and mammalian target of rapamycin (mTOR), which are downstream of PI3K, also play important roles in carcinogenesis. Together, the members of the PI3K/Akt/mTOR axis function as a group to interact with the regulation of several other signaling molecules in HNSCC [17]. The PAM pathway is upregulated in over 90% of HNSCC cases [18]. Upregulated PAM signaling would worsen radiotherapy resistance and cytostatic drug resistance [8]. As a result, developing pharmacologic inhibitors targeting the PAM axis has potential benefits for HNSCC patients, which have later been reported in several initial preclinical and clinical studies.
1.1. The PAM Signaling Pathway Composition and Function
The PAM signaling cascade is critical to cell survival, growth, proliferation, angiogenesis, transcription, and apoptosis [19] (Figure 1). Various molecules, including glucose, insulin, and many cytokines and growth factors, can initiate PAM signaling [20].
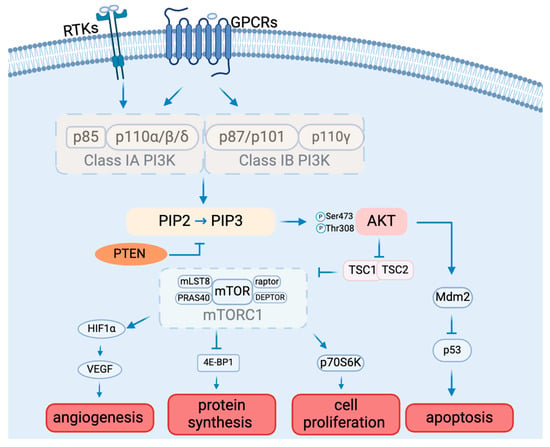
Figure 1.
The PAM signaling pathway composition and function.
1.2. PI3Ks
There are three classes of PI3Ks: class I, class II, and class III, classified by structure and substrate preference [21]. Class I PI3Ks can be activated directly by cell surface receptors, which are best studied among all classes. Class I PI3Ks are divided into subclasses IA and IB based on their mode of activation [21]. The difference is that one can be activated by RTKs, G protein-coupled receptors, and the small G protein RAS (Class IA), and the other one can only be activated by G-protein-coupled receptors (Class 1B) [22]. Class IA PI3Ks are heterodimers made up of a p110 catalytic subunit (p110α, p110β, or p110δ) and a p85 regulatory subunit (p85α, p85β, p85γ, p55α, or p50α). Class IB PI3K consists of a p110γ catalytic subunit and a p101 or p87 regulatory subunit. [23]. P110α and p110β are widely expressed in multiple tissues. In contrast, p110δ and p110γ are mainly found in leukocytes [24,25]. Cell surface receptor activation results in the tyrosine phosphorylation of cell surface receptors. The p85 regulatory subunit of class IA PI3Ks binds directly to tyrosine receptors on cell membranes. The activated PI3K consequently catalyzes the conversion of PIP2 to PIP3 [26]. PIP3 functions as a second messenger, controlling numerous downstream signaling pathways [26].
1.3. AKT
Akt is the main molecule downstream of the PI3K signaling pathway, and it is a serine/threonine protein kinase with three isoforms, Akt1, Akt2, and Akt3, which are encoded by PKBα, PKBβ, and PKBγ, respectively [27,28]. Akt requires phosphorylation for activation. AKT1 is extensively expressed in numerous tissues. While AKT2 is found mostly in insulin-sensitive tissues and at low levels in other tissues, AKT3 is solely expressed in the brain and testis [29]. The distinct tissue expression patterns of the various AKT subtypes point to their crucial involvement in the preservation of physiological processes in various organs or tissues [30]. The three AKT subtypes have extremely similar three-dimensional structures made up of three distinct functional domains [31]. The N-terminal pleckstrin homology (PH) domain interacts with PI3K and launches recruitment to the plasma membrane, conformational change, and subsequent phosphorylation [32]. The central kinase catalytic domain shares a high degree of homology with the enzymatic activity regions of protein kinase A (PKA) and protein kinase C (PKC) [33]. Moreover, AKT activation requires the phosphorylation of Thr308, which is situated in this domain. For full activation of AKT, Ser473 needs to be in the regulatory region of the C-terminal AKT domain [34,35]. Akt, once activated, engages with numerous downstream targets implicated in fostering cell survival, growth, proliferation, angiogenesis, metabolism, and migration [36,37,38].
1.4. mTOR
The mechanistic target of rapamycin (mTOR) is a protein kinase that is a specific target of the natural compound rapamycin. It is also referred to as RAFT1 (rapamycin and FKBP12 target), RAPT 1 (rapamycin target 1), SEP (sirolimus effector protein), or FKBP12-rapamcyin-associated protein (FRAP) [39]. This 289 kDa serine/threonine kinase is a member of the PI3K-related protein kinase (PIKK) family because its C-terminus is highly homologous to the catalytic domain of PI3K [39]. Mammalian target of rapamycin complex 1 (mTORC1) and mammalian target of rapamycin complex 2 (mTORC2) are two physically and functionally different complexes that are formed by mTOR.
mTORC1 mainly regulates cell growth and metabolism [40]. mTORC1 consists of mTOR (mammalian target of rapamycin), raptor (regulatory-associated protein of mTOR), mLST8 (mammalian lethal with SEC13 protein 8), and two negative regulators, PRAS40 (proline-rich AKT substrate 40 kDa) and DEPTOR (DEP domain-containing mTOR-interacting protein), and is inhibited by rapamycin. mTORC2 mainly controls cell proliferation and survival, which is composed of mTOR (mammalian target of rapamycin), Rictor (rapamycin-insensitive companion of mTOR), mLST8 (mammalian lethal with SEC13 protein 8), SIN1 (mammalian stress-activated protein kinase-interacting protein), PRR5 (protein observed with Rictor 1), and Protor2 [39]. mTORC2 can indeed be activated by growth factors such as insulin and IGF-1, ultimately leading to the phosphorylation and activation of Akt by phosphorylating Ser473 as part of the insulin/IGF-1 signaling pathway [41]. As a downstream molecule of AKT, phosphorylated AKT activates mTORC1 by phosphorylating the tuberous sclerosis complex (TSC). In addition to controlling mTOR activity, the AKT/TSC1-TSC2 signaling pathway also controls cell division and growth. mTORC1 activation requires TSC2’s GTPase activity and function of inhibiting the small GTPase Rheb [42]. Once TSC2 is phosphorylated by AKT, it can no longer inhibit mTORC1. The activated mTOR can further induce angiogenesis by activating hypoxia-inducible factor 1 alpha (HIF-1α), which can further activate vascular endothelial growth factor (VEGF), the important factor for angiogenesis [43]. The monoclonal antibody medication bevacizumab targets multiple cancers by inhibiting VEGF [44]. mTOR can also affect protein synthesis by inhibiting factor 4E-binding protein 1 (4E-BP1) [45,46] and cell proliferation by activating ribosomal protein S6 kinase beta-1 (S6K1), also known as p70S6 kinase (p70S6K) [47]. 4E-BP1 and S6K1 may also be involved in the regulation of H1F-1α [43].
1.5. Other AKT Target Proteins
The PI3K/AKT signaling pathway adversely regulates the transcription factors known as forkhead box O (FOXO), which are thought to have an inhibitory influence on cell proliferation. By downregulating FOXO and activating AKT and other targets, PI3K signaling controls the growth of cells [27].
Glycogen synthase kinase-3 (GSK-3) is another important molecule downstream of AKT, and it is a serine/threonine protease. There are two subtypes of GSK-3: GSK-3beta and GSK-3alpha, with 97% sequence identity in the catalytically active areas of these two kinds. Furthermore, GSK-3beta and GSK-3alpha are widely expressed in cells and organs and share similar biological characteristics [48]. GSK-3beta has been shown in recent research to be able to phosphorylate a wide range of endogenous substrates, including many proteins and transcription factors that are important in metabolism. Thus, GSK-3beta is essential for cell proliferation, development, carcinogenesis, and glucose homeostasis control [49,50,51].
Mdm2 is another substrate of AKT. When AKT phosphorylates Mdm2, it leads to the downregulation of p53 [52]. This reduction in p53 levels promotes autophagy. Thus, the activation of the PI3K/AKT signaling pathway and its phosphorylation of downstream Mdm2 may play a key role in the mechanism of autophagy-induced apoptosis [53].
1.6. PTEN
Numerous variables regulate the PI3K/AKT/mTOR signal transduction pathway. The opposing process of PIP3 production is catalyzed by the tumor suppressor PTEN, which changes PIP3 into PIP2 [54]. PTEN hinders cell division and disrupts cellular metabolism by downregulating the PI3K/AKT/mTOR pathway, so PTEN activity inhibition triggers AKT and related pathways [55]. By modifying AKT activity, PTEN has a significant impact on glucose homeostasis regulation [56].
2. The Roles of the PAM Pathway in Head and Neck Cancer
The PAM pathway is a highly conserved signal transduction network in eukaryotic cells that promotes cell growth, survival, and cycle progression [57]. The malfunction of PAM signal transduction may be a risk factor for the development of cancer [38]. The PAM pathway abnormality is found in about half of malignancies [57]. Furthermore, treatment resistance in cancer is often a result of hyperactivation of the PAM pathway [57,58].
2.1. PI3K Mutations in HNSCC
The most often found mutant oncogene throughout tumor lineages is the PIK3CA activating mutation, which encodes the p110α catalytic subunit of PI3K. This mutations not only present in HNSCC [59], but also detected in gastric cancer [60], gallbladder cancer [61], and melanoma [62]. Eighty percent of the alterations reported by the HNSCC were missense mutations in PIK3CA [63]. The p.(E545K) mutation, which results from a single nucleotide change (c.1633 G > A) and substitutes lysine (K) for the amino acid (AA) glutamic acid (E) at position 545 in the PIK3CA gene, was the most prevalent missense mutation [63]. Then came the p.(E542K) mutation, which is caused by a single nucleotide alteration (c.1624 G > A) and causes an AA substitution of a K for an E at position 542. These two mutations are in the helical domain of PIK3CA. In the kinase domain, the most common mutation was p.(H1047R), which results from a single nucleotide change (c.3140A > G), leading to the substitution of histidine (H) at position 1047 with arginine (R) [63]. In HNSCC, PIK3CA messenger RNA (mRNA) overexpression is a common occurrence. Mutations and copy number gains (0.5–11%) are also seen in several PI3K regulatory subunits and other PI3K isoforms (PIK3CB, PIK3CD, and PIK3CG) [64].
Hyperactivity of PI3K plays a significant role in the development and progression of many cancers [65]. The hyperactivity of PI3K promotes the production of VEGF, which stimulates normal and tumor angiogenesis [66]. By phosphorylating Ser1177, activated AKT causes eNOS distribution in the vascular endothelium, which in turn leads blood vessels to produce nitric oxide (NO). This phenomenon aids in the regulation of vascular processes like angiogenesis, vascular remodeling, and vasodilation [67]. Moreover, significant quantities of HIF-1α are expressed when AKT activation occurs. HIF-1α is a crucial regulator of angiogenesis that can increase the expression of VEGF and other angiogenic factors, thus fostering angiogenesis. HIF-1α could encourage endothelial cell migration and proliferation and enhance vascular permeability, plasma protein exosmosis, and cellulose scaffold formation to aid in endothelial cell migration and offer support for vascular growth. HIF-1α could also induce the proteolytic enzyme system, which breaks down the extracellular matrix and encourages angiogenesis [68]. Angiogenesis plays an important role in the growth, metastasis, and lethality of tumors.
Hyperactivity of the PI3K and PI3K/AKT/mTOR signaling pathways, also through the following methods, plays a significant role in encouraging tumor invasion and metastasis. Activated AKT facilitates the invasion of tumors and increases Mdm2 expression. Mdm2 is the primary negative regulator of p53, promoting its degradation and thereby inactivating its tumor-suppressing function [69]. It also facilitates the spreading of tumors. It has been revealed that AKT1, a downstream PI3K molecule, and actin polarization promotion control the invasion and metastasis of breast cancer cells. Palladin is an actin-related protein that plays a key role in cell migration, controls the organization of the actin system, and contributes to the formation of the cytoskeleton. AKT1 can phosphorylate palladin’s Ser507, which controls the invasion and metastasis of cancer cells [70]. Cell invasion and metastasis are encouraged by the activation of matrix metalloproteinases (MMPs), a class of proteolytic enzymes that take part in the extracellular matrix’s degradation. MMP-2 and MMP-9 are two of the 23 different types of MMPs that are known to be essential for cell invasion. PI3K/AKT/mTOR can break down the extracellular matrix, increase the expression of MMP-2 at the mRNA and protein levels, and increase the invasion and metastasis of cancer cells [71].
2.2. AKT Mutations in HNSCC
Activating mutations or amplifications of AKT upstream factors, such as in PTEN, PIK3CA, growth factor, or cytokine receptors, typically cause changes in AKT activity by enhancing the expression and activity of one, two, or all three isoforms of AKT [72]. Genes encoding one of the three isoforms of the oncogenic protein AKT—which is known to be involved in controlling cell survival, proliferation, growth, apoptosis, and glycogen metabolism [73]—have been found to harbor gain-of-function missense mutations and amplification [74].
The overall pooled prevalence of AKT mutations in head and neck cancer was 2% [63]. The most frequent mutation was found in the hotspot p.(E17K), which is caused by a single nucleotide change (c.49 G > A) and leads to the AA substitution of a K for an E at position 17 [63].
2.3. MTOR Mutations in HNSCC
To start cell division, mTOR encourages cyclin D1 to attach to cyclin-dependent kinase (CDK). Elevated expression of cyclin D1 can abbreviate the cell cycle, hasten the development of cancer, and cause the cell cycle to move from the G1 to the S phase. Furthermore, mTOR controls the production of biological macromolecules, including lipids, proteins, and nucleotides, which supply the building blocks needed for the proliferation of cancer cells [71].
According to the meta-analysis published in 2021, the overall pooled prevalence of MTOR mutations in head and neck cancer was 3% [63]. The population with the highest frequency of MTOR mutations was Asia (4.1%), Europe (2.3%), and North America (1.7%). Missense mutations were more frequent, but none of the mutations have been reported more than once [63].
2.4. PTEN Mutations in HNSCC
The tumor suppressor PTEN plays a major role in PI3K/PIP3 signal termination by dephosphorylating PIP3 and converting it back to PIP2. Consequently, PTEN functions as a crucial negative regulator of the PAM pathway, which influences cell growth and survival; on the other hand, PTEN absence releases the inhibition of this intracellular signaling [75]. In both primary and metastatic breast cancer, PTEN loss typically results in hyperactivation of the PAM pathway, which in turn promotes cell proliferation [76,77]. Resistance to anticancer treatment is associated with both PTEN downregulation and PAM pathway activation [78].
Numerous malignancies include mutations that cause PTEN to lose its function [79,80]. The prevalence of the PTEN mutation in head and neck cancer was reported at 4% by a meta-analysis including 4785 samples described in 57 articles [63]. According to this meta-analysis, forty percent of PTEN mutations are missense and 11% are nonsense mutations. In head and neck cancer, the most frequent mutations were p.(Q171X) and the single nucleotide alteration c.511C > T [63] (Figure 2).
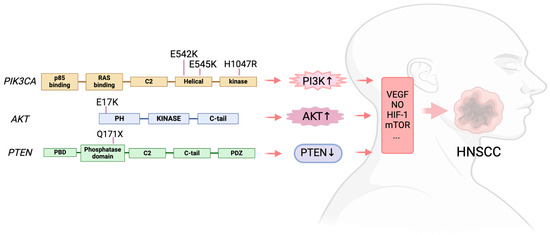
Figure 2.
The PAM signaling mutations in HNSCC.
3. Targeting the PAM Axis in Head and Neck Cancer
Recent characterization of molecular alterations in HNSCC has revealed that the PI3K/mTOR signaling pathway is the most frequently dysregulated pathway in this type of cancer [81]. Consequently, using targeted molecular therapies to reduce mTOR pathway activity could have anticancer effects in HNSCC.
3.1. PI3K Inhibitors
PI3K is a prominent pharmacological target for cancer treatment along the PAM pathway because of its remarkable correlation between hyperactivity and the evolution of human tumors, the production of boosted tumor microvessels, and a rise in the number of invasive cancer cells. Even though the Food and Drug Administration (FDA) has authorized several inhibitors, concerns about toxicity, sensitivity indicators, and resistance development persist [82]. It is noteworthy that PI3K inhibitors may be divided into three primary categories: pan-PI3K inhibitors (pan-PI3Ki), isoform-specific PI3K inhibitors (IS PI3Ki), and dual PI3K/mTOR inhibitors (dual PI3K/mTORi) [83]. All PI3K class I isoforms’ catalytic activity is suppressed by pan-PI3Ki. Therefore, regardless of the kind of PI3K gene or PTEN changes involved, these medications are often beneficial in cancers that produce large levels of PIP3.
Pan-PI3Ki may have a wider spectrum of action since it includes several molecular targets, but there is a growing danger of both on- and off-target toxicity [84]. Numerous pan-PI3Ki have been studied in clinical settings. Buparlisib (BKM120) is an orally bioavailable pan-PI3K inhibitor [84]. As a potent pan-class I PI3K inhibitor, buparlisib targets the ATP binding site of the p110 kinase domain. Its inhibitory effectiveness is significantly reduced against class IB p110γ but equally effective on class IA isoforms of p110α, β, and δ [85]. When taken orally, buparlisib is quickly absorbed, and the amount in the serum rises in direct proportion to the dosage [86]. Preclinical findings further suggested that its anticancer efficacy was due to an antiangiogenic impact [85] and the reduction of microtubular dynamics [87]. In cell lines with wild-type PIK3CA as well as mutant forms carrying any hotspot mutation of E542K, E545K, or H1047R, buparlisib decreased PI3K activity in vivo [88]. The most frequent adverse effects in a phase I dose-escalation trial for advanced solid tumors were tiredness, rash, altered liver function, and changes in glucose metabolism [89]. Results of the phase III trial about buparlisib in combination with chemotherapy (paclitaxel) compared to paclitaxel alone in patients with recurrent or metastatic head and neck squamous cell carcinoma are awaited (NCT04338399) (Table 1). Another pan-PI3Ki, PX-866, binds to Lys in the ATP catalytic site to irreversibly inhibit class I PI3K [90]. In xenograft models of human HNSCC, PX-866 prevented the growth of tumors. Two of these cases included PIK3CA gene amplification, while the other involved E545K [91]. Nevertheless, PX-866’s clinical studies did not yield encouraging outcomes [92]. Copanlisib, another pan-PI3Ki, combined with cetuximab demonstrated limited efficacy and unfavorable toxicity in HNSCC patients (NCT02822482) [93].

Table 1.
Clinical trials targeting the PAM signaling pathway in head and neck cancer.
A more favorable side effect profile is possible with IS PI3Ki, a selective inhibitor of PI3Kα that has anticancer efficacy without affecting other isoforms of PI3K. Just three IS PI3Ki—alpelisib, duvelisib, and idelalisib—have received FDA approval. Alpelisib is a specific inhibitor of PI3Kα, which is the product of frequently mutated PIK3CA [94]. Alpelisib inhibits wild-type PI3Kα and PI3Kα with common PI3KCA mutations, such as E545K or H1047R, more potently than PI3Kδ or PI3Kγ [95]. Preclinical evidence also revealed that cancer cells with PIK3CA mutations are more susceptible to alpelisib’s PI3K inhibition. Reliable preclinical models are crucial for studying relevant molecular mechanisms and selecting the final interventions for clinical research. These models include immortalized-cell and primary tumor cultures in monolayer or 3D and animal models [96]. Preclinical models of HNSCC, which can be used to analyze the biological roles of genetic variants and altered gene expression, will be highly valuable for translating molecular discoveries into enhanced clinical care [97]. An extensive panel of cancer cell lines was used for an in vitro pharmacologic sensitivity screen, and the results showed that sensitivity to alpelisib was positively correlated with the presence of the PIK3CA mutation, amplification, or copy number increase. This finding was further supported by an in vivo investigation employing mouse models [95]. HNSCC cell lines with the PIK3CA H1047R mutation were more vulnerable to the antiproliferative impact of alepelisib than were cell lines with the wild-type PIK3CA [98]. The PIK3CA mutation was the best predictive factor that corresponded with a good response to alpelisib in another in vivo study, regardless of the location where it was found [95]. PI3K inhibitors are being investigated in conjunction with other targeted medicines since compensatory hyperactivation of PIK3CA is one of the main reasons for treatment resistance. As for duvelisib, several clinical trials are underway to test it for the treatment of advanced HNSCC (NCT05057247 and NCT04193293) (Table 1). Idelalisib, also known as CAL-101, has been approved by the FDA as the first-in-class PI3K inhibitor for hematological cancer treatment [99], but the possibility of using idelalisib in HNSCC remains unestablished. Currently, there are no clinical trials exploring the efficacy and risks of Idelalisib for head and neck cancer.
3.2. AKT Inhibitor
MK-2206, GDC0068, and D-21266 are the three AKT inhibitors that have been employed in HNSCC studies.
MK-2206 is an orally active, allosteric inhibitor of AKT that prohibits membrane recruitment and phosphorylation of AKT [100]. MK-2206 treatment is adequate to prevent HNSCC migration and chemotaxis in vitro. Treatment with MK-2206 improves survival in an orthotopic model by reducing the size of the main tumor and cervical metastases [101]. However, the phase II clinical trials of MK-2206 in HNSCC were terminated due to the limited activity observed [102].
Ipatasertib (GDC0068) is an orally administered, ATP-competitive, highly selective inhibitor of AKT [103]. It suppresses downstream signaling and raises AKT phosphorylation. Ipatasertib is now being evaluated alone (NCT05172258) or in combination with chemotherapy (NCT05172245) for the treatment of HNSCC in phase I or II trials.
Perifelosine (D-21266) is an oral alkylphospholipid that prevents AKT phosphorylation [104]. Preclinical research using a rat mammary cancer model revealed that perifosine had antitumor properties [105]. Phase I trials of perifosine show that when administered in accordance with a loading and maintenance schedule, perifosine can safely maintain drug concentrations that are close to those attained in preclinical models, showing indications of antitumor activity [106]. The toxicities observed included gastrointestinal toxicity, arthralgia, and fatigue. The phase II trial of perifosine in HNSCC patients reveals that the doses and schedule used lack single-agent activity [104].
3.3. mTOR Inhibitor
The first-generation mTOR inhibitor rapamycin (sirolimus) has been extensively studied and is known to form a complex with FKBP12, inhibiting the activity of mTORC1. In the context of head and neck cancer (HNC), several Phase I and II trials have evaluated the safety and efficacy of rapamycin in patients with stage II-IVA HNSCC. For instance, in a trial involving 16 patients, rapamycin was well tolerated, with 25% of patients showing RECIST responses, and downstream proteins such as AKT, S6, 4EBP, and Ki67 exhibited promising effects (NCT01195922) [107]. However, another trial involving four HNC patients, despite the combination regimen with bevacizumab, showed no objective responses by RECIST criteria, although there was a reduction in tumor size [108]. Overall, while some studies did not show objective responses, the long-term use of mTOR inhibitors like rapamycin, especially in combination regimens, demonstrated tolerability and potential efficacy in HNC patients, suggesting promising effects with careful adverse effects management [109].
The second-generation mTOR inhibitor everolimus (EVE), particularly everolimus (EVE), has been evaluated through a comprehensive review of several Phase I and II clinical trials [109,110,111,112,113] encompassing patients with stage two to four HNC. Of these trials, most were completed successfully, while two were terminated due to adverse effects when combined with other therapies (NCT01009346, NCT01057277) [109].
Among the completed trials, two did not demonstrate significant benefits. For instance, in the NCT01051791 Phase II study, where EVE was administered to recurrent/metastatic HNC patients, adverse effects led to discontinuation in 33% of patients, and the results failed to improve clinical benefits, with no objective responses observed and a clinical benefit rate (CBR) of 28%, along with median progression-free survival (PFS) and overall survival (OS) of 1.5 and 4.5 months, respectively [112].
Similarly, in the NCT00942734 Phase II study, the addition of EVE to the erlotinib regimen did not show significant benefits in 35 platinum-resistant/metastatic HNC patients, despite tolerable adverse effects. The results indicated a median PFS of 11.9 weeks and a median OS of 10.25 months [110]. However, four trials did show clinical benefits. For example, in the NCT01283334 Phase I + II study, EVE combined with cetuximab and carboplatin yielded encouraging results, with a 61.5% objective response rate (all partial responses) and a PFS of 8.15 months [111]. The NCT01333085 Phase I + II Study, using the CAPRA regimen (everolimus, carboplatin, and paclitaxel) as induction therapy, showed promising results in 50 locally advanced HNC patients, with a 79% overall response rate and encouraging translational results indicating a direct effect of EVE in tumors. Furthermore, the NCT00935961 Phase I study using EVE combined with cisplatin and docetaxel in advanced HNC patients demonstrated a progression-free survival rate of 87.5% at 1 year and 76.6% at 2 years [113]. Similarly, the NCT00858663 Phase I study employing EVE with cisplatin and radiotherapy in stage 2-4 HNC patients showed a two-year progression-free survival of 85% and a two-year overall survival of 92% [114].
Despite the two trials that did not yield significant beneficial results, whether using EVE as monotherapy or in combination with erlotinib, the potential of EVE as part of combined therapy in HNC remains promising. The positive outcomes observed in trials combining EVE with various agents, such as cetuximab, carboplatin, paclitaxel, docetaxel, and radiotherapy, indicate the potential for EVE as a component of combined therapy options in HNC, although further investigation is warranted to delineate optimal treatment strategies.
3.4. The PAM Pathway Inhibitor Resistance
Because the PAM pathway may not be fully inhibited, may reactivate, or other prosurvival pathways may be activated, the PAM pathway inhibitors may be ineffective [115,116]. When a single node in the PAM pathway, such as mTOR, is inhibited, a negative feedback loop can be released, which often activates AKT in response and ends up with a compensatory activation [116]. When inhibiting downstream signaling molecules of PAM, such as mTOR, simultaneous inhibition of upstream signaling molecules may help avoid compensatory activation and resistance (Figure 3). When compared to mTOR blockade alone, inhibition of both PI3K and mTOR may improve antitumor effectiveness [117,118]. The drugs SF1126, dactolisib (NVP-BEZ235), voxtalisib (XL765), gedatolisib (PKI-587), and GSK1059615 are examples of dual PI3K/mTOR inhibitors (PI3K/mTORi) [65,119]. It is currently unclear what effect SF1126, dactolisib, and voxtalisib have on head and neck cancer. Relevant clinical studies on SF1126 have been completed, and the results are being organized (NCT0264412). Gedatolisib is currently being used in phase I clinical trials exploring the efficacy for HNSCC (NCT03065062). GSK1059615 was reported to induce antitumor activity, possibly by provoking the programmed necrosis pathway in vitro and in a mouse model [120].
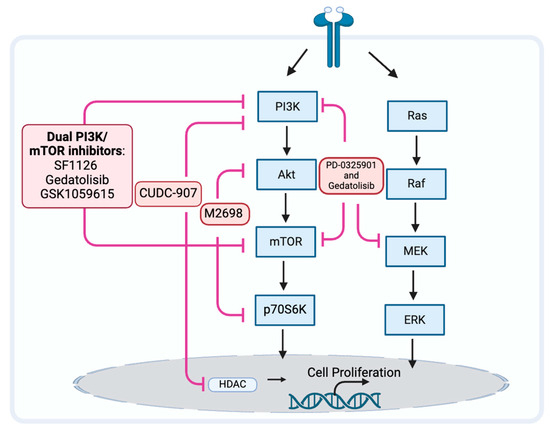
Figure 3.
Strategy may prevent tumor resistance to PAM pathway inhibitors.
M2698 is an oral dual inhibitor of p70S6K and AKT, which blocks p70S6K to inhibit the PAM pathway while simultaneously targeting AKT to overcome the compensatory feedback loop [121]. A clinical investigation including patients with advanced cancer [122] and mouse models [121] indicated this potential effect of p70S6K/AKT dual inhibition. Currently, there are no reported studies on the use of this dual inhibitor for HNSCC.
Modulation of the PAM pathway signaling or contemporaneous changes in other pathways that activate parallel tumor signaling pathways can activate compensatory prosurvival programs and result in PAM pathway inhibitor resistance [122,123]. By concurrently inhibiting alternative pathways, cancer cells that were previously susceptible to PI3K inhibition can now evade treatment resistance by downregulating other signaling proteins.
There is a known connection between the PI3K/AKT/mTOR and RAS/RAF/MEK/ERK pathways at several nodes, and this interaction may result in a potential pathway convergence for the development of therapeutic combinations [124]. MEK inhibitor PD-0325901 overcomes resistance to PI3K/mTOR inhibitor gedatolisib (PF-05212384) and potentiates antitumor effects in HNSCC cell lines [125]. Further research is needed to confirm the effects and side effects for patients with HNSCC.
The epidermal growth factor receptor (EGFR) family is a subclass of RTK proteins and consists of four members: EGFR (ErbB1, HER1), ErbB2 (HER2), ErbB3 (HER3), and ErbB4 (HER4). Patients with cancer that overexpresses HER2 now have a considerably better prognosis thanks to anti-HER2 treatments. Constitutive stimulation of the PI3K pathway is one mechanism of resistance to anti-HER2 treatments. The therapeutic drawbacks of single-agent anti-HER2 treatment may be addressed by combination therapy, including HER2 and PAM inhibitors [126].
Histone deacetylases (HDACs) are epigenetic modifiers that play crucial roles in many key biological functions. HDAC inhibitors like CUDC-907, which inhibit HDAC and the PI3K pathway, are a relatively new emerging class of anticancer drugs [127]. Concurrent inhibition of HDAC can also downregulate other signaling proteins and bypass treatment resistance in cancer cells that are resistant to PI3K inhibition due to alternative pathway activation. The simultaneous targeted inhibition of HDACs and PI3Ks with their respective inhibitors in combination showed synergistic therapeutic efficacy. An in vitro discovery showing that the administration of an HDAC inhibitor effectively overcomes mTOR inhibitor resistance in lymphoma cells provided evidence for the potential advantages of dual inhibition [128]. Additionally, combined inhibition of PI3K and HDAC reverses platinum drug resistance by preventing DNA repair and multidrug resistance transporters, according to an in vivo investigation [129]. Combination inhibition of HDAC with dual PI3K-mTOR inhibitors possesses an antitumor effect against HNSCC in vivo [130]. The evaluation of the benefits and side effects for patients with HNSCC requires additional investigation.
The combination of PAM inhibitors reduces the occurrence of resistance and holds greater potential for treating HNSCC. However, it is crucial to determine the optimal combination for each patient. Personalized preclinical platforms to test gene expression profiles and drug sensitivity of patient samples may help in selecting the appropriate combination. Additionally, special attention must be given to managing the toxicity of innovative combination approaches.
4. Conclusions
Targeting the PAM signaling pathways as a precision therapeutic approach in HNSCC has been extensively studied in experimental models and clinical trials, as the significance of the PAM signaling pathways in HNSCC has been well-documented. Moreover, there is now a justification for combining the PAM with different routes, targeting itself and alternative pathways to increase their antitumor efficaciousness. Overall, we anticipate that the creation of innovative PAM co-targeting techniques may result in long-lasting effects and cancer remission, extending the survival and improving the quality of life of HNSCC patients.
Author Contributions
Q.J. conceived the idea, drafted and edited the manuscript, and approved the final draft submitted. J.X. edited the manuscript and approved the final draft submitted. Y.-C.H. edited the manuscript and approved the final draft submitted. N.L.K. edited the manuscript and approved the final draft submitted. Y.Z. edited the manuscript and approved the final draft submitted. L.H. edited the manuscript and approved the final draft submitted. H.L. reviewed and edited the manuscript and approved the final draft submitted. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Natural Science Foundation of Jiangsu Province (SBK2021021787); the Major Project of the Health Commission of Jiangsu Province (ZD2022025); and the Key Project of the Nanjing Health Commission (ZKX20048).
Conflicts of Interest
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
References
- Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Leemans, C.R.; Braakhuis, B.J.; Brakenhoff, R.H. The molecular biology of head and neck cancer. Nat. Rev. Cancer 2011, 11, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Yang, F.; Yin, Y.; Liu, S.; Li, P.; Zhang, X.; Chen, D.; Liu, Y.; Wang, J.; Wang, K.; et al. Prevalence of Human Papillomavirus Type-16 in Head and Neck Cancer Among the Chinese Population: A Meta-Analysis. Front. Oncol. 2018, 8, 619. [Google Scholar] [CrossRef] [PubMed]
- Ndiaye, C.; Mena, M.; Alemany, L.; Arbyn, M.; Castellsagué, X.; Laporte, L.; Bosch, F.X.; de Sanjosé, S.; Trottier, H. HPV DNA, E6/E7 mRNA, and p16INK4a detection in head and neck cancers: A systematic review and meta-analysis. Lancet Oncol. 2014, 15, 1319–1331. [Google Scholar] [CrossRef] [PubMed]
- Chow, L.Q.M. Head and Neck Cancer. N. Engl. J. Med. 2020, 382, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Kiyota, N.; Hasegawa, Y.; Takahashi, S.; Yokota, T.; Yen, C.J.; Iwae, S.; Shimizu, Y.; Hong, R.L.; Goto, M.; Kang, J.H.; et al. A randomized, open-label, Phase III clinical trial of nivolumab vs. therapy of investigator’s choice in recurrent squamous cell carcinoma of the head and neck: A subanalysis of Asian patients versus the global population in checkmate 141. Oral. Oncol. 2017, 73, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Larkins, E.; Blumenthal, G.M.; Yuan, W.; He, K.; Sridhara, R.; Subramaniam, S.; Zhao, H.; Liu, C.; Yu, J.; Goldberg, K.B.; et al. FDA Approval Summary: Pembrolizumab for the Treatment of Recurrent or Metastatic Head and Neck Squamous Cell Carcinoma with Disease Progression on or After Platinum-Containing Chemotherapy. Oncologist 2017, 22, 873–878. [Google Scholar] [CrossRef] [PubMed]
- Marquard, F.E.; Jücker, M. PI3K/AKT/mTOR signaling as a molecular target in head and neck cancer. Biochem. Pharmacol. 2020, 172, 113729. [Google Scholar] [CrossRef] [PubMed]
- Miroddi, M.; Sterrantino, C.; Simmonds, M.; Caridi, L.; Calapai, G.; Phillips, R.S.; Stewart, L.A. Systematic review and meta-analysis of the risk of severe and life-threatening thromboembolism in cancer patients receiving anti-EGFR monoclonal antibodies (cetuximab or panitumumab). Int. J. Cancer 2016, 139, 2370–2380. [Google Scholar] [CrossRef]
- Mesia, R.; Iglesias, L.; Lambea, J.; Martínez-Trufero, J.; Soria, A.; Taberna, M.; Trigo, J.; Chaves, M.; García-Castaño, A.; Cruz, J. SEOM clinical guidelines for the treatment of head and neck cancer (2020). Clin. Transl. Oncol. 2021, 23, 913–921. [Google Scholar] [CrossRef]
- Miserocchi, G.; Spadazzi, C.; Calpona, S.; De Rosa, F.; Usai, A.; De Vita, A.; Liverani, C.; Cocchi, C.; Vanni, S.; Calabrese, C.; et al. Precision Medicine in Head and Neck Cancers: Genomic and Preclinical Approaches. J. Pers. Med. 2022, 12, 854. [Google Scholar] [CrossRef]
- Wang, Z.; Valera, J.C.; Zhao, X.; Chen, Q.; Gutkind, J.S. mTOR co-targeting strategies for head and neck cancer therapy. Cancer Metastasis Rev. 2017, 36, 491–502. [Google Scholar] [CrossRef]
- Isaacsson Velho, P.H.; Castro, G., Jr.; Chung, C.H. Targeting the PI3K Pathway in Head and Neck Squamous Cell Carcinoma. Am. Soc. Clin. Oncol. Educ. Book. 2015, 35, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Janecka-Widła, A.; Majchrzyk, K.; Mucha-Małecka, A.; Biesaga, B. EGFR/PI3K/Akt/mTOR pathway in head and neck squamous cell carcinoma patients with different HPV status. Pol. J. Pathol. 2021, 72, 296–314. [Google Scholar] [CrossRef]
- Yuan, T.L.; Cantley, L.C. PI3K pathway alterations in cancer: Variations on a theme. Oncogene 2008, 27, 5497–5510. [Google Scholar] [CrossRef] [PubMed]
- Courtney, K.D.; Corcoran, R.B.; Engelman, J.A. The PI3K pathway as drug target in human cancer. J. Clin. Oncol. 2010, 28, 1075–1083. [Google Scholar] [CrossRef]
- Su, Y.C.; Lee, W.C.; Wang, C.C.; Yeh, S.A.; Chen, W.H.; Chen, P.J. Targeting PI3K/AKT/mTOR Signaling Pathway as a Radiosensitization in Head and Neck Squamous Cell Carcinomas. Int. J. Mol. Sci. 2022, 23, 15749. [Google Scholar] [CrossRef]
- Molinolo, A.A.; Hewitt, S.M.; Amornphimoltham, P.; Keelawat, S.; Rangdaeng, S.; García, A.M.; Raimondi, A.R.; Jufe, R.; Itoiz, M.; Gao, Y.; et al. Dissecting the Akt/mammalian target of rapamycin signaling network: Emerging results from the head and neck cancer tissue array initiative. Clin. Cancer Res. 2007, 13, 4964–4973. [Google Scholar] [CrossRef]
- Ediriweera, M.K.; Tennekoon, K.H.; Samarakoon, S.R. Role of the PI3K/AKT/mTOR signaling pathway in ovarian cancer: Biological and therapeutic significance. Semin. Cancer Biol. 2019, 59, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Luo, J.; Guo, J.; Yao, X.; Jing, X.; Guo, F. The PI3K/AKT/mTOR signaling pathway in osteoarthritis: A narrative review. Osteoarthr. Cartil. 2020, 28, 400–409. [Google Scholar] [CrossRef]
- Liu, P.; Cheng, H.; Roberts, T.M.; Zhao, J.J. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discov. 2009, 8, 627–644. [Google Scholar] [CrossRef]
- Barberis, L.; Hirsch, E. Targeting phosphoinositide 3-kinase gamma to fight inflammation and more. Thromb. Haemost. 2008, 99, 279–285. [Google Scholar] [CrossRef]
- Sun, P.; Meng, L.H. Emerging roles of class I PI3K inhibitors in modulating tumor microenvironment and immunity. Acta Pharmacol. Sin. 2020, 41, 1395–1402. [Google Scholar] [CrossRef] [PubMed]
- Yudushkin, I. Getting the Akt Together: Guiding Intracellular Akt Activity by PI3K. Biomolecules 2019, 9, 67. [Google Scholar] [CrossRef]
- Gyori, D.; Chessa, T.; Hawkins, P.T.; Stephens, L.R. Class (I) Phosphoinositide 3-Kinases in the Tumor Microenvironment. Cancers 2017, 9, 24. [Google Scholar] [CrossRef]
- He, Y.; Sun, M.M.; Zhang, G.G.; Yang, J.; Chen, K.S.; Xu, W.W.; Li, B. Targeting PI3K/Akt signal transduction for cancer therapy. Signal Transduct. Target. Ther. 2021, 6, 425. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhang, X. The role of PI3K/AKT/FOXO signaling in psoriasis. Arch. Dermatol. Res. 2019, 311, 83–91. [Google Scholar] [CrossRef]
- Hinz, N.; Jücker, M. Distinct functions of AKT isoforms in breast cancer: A comprehensive review. Cell Commun. Signal. 2019, 17, 154. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Liu, G.; Guo, J.; Su, Z. The PI3K/AKT pathway in obesity and type 2 diabetes. Int. J. Biol. Sci. 2018, 14, 1483–1496. [Google Scholar] [CrossRef]
- Revathidevi, S.; Munirajan, A.K. Akt in cancer: Mediator and more. Semin. Cancer Biol. 2019, 59, 80–91. [Google Scholar] [CrossRef]
- Basu, A.; Lambring, C.B. Akt Isoforms: A Family Affair in Breast Cancer. Cancers 2021, 13, 3445. [Google Scholar] [CrossRef] [PubMed]
- Weako, J.; Jang, H.; Keskin, O.; Nussinov, R.; Gursoy, A. The structural basis of Akt PH domain interaction with calmodulin. Biophys. J. 2021, 120, 1994–2008. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Shi, X.; Sheng, K.; Han, G.; Li, W.; Zhao, Q.; Jiang, B.; Feng, J.; Li, J.; Gu, Y. PI3K/Akt signaling transduction pathway, erythropoiesis and glycolysis in hypoxia (Review). Mol. Med. Rep. 2019, 19, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Wadhwa, B.; Makhdoomi, U.; Vishwakarma, R.; Malik, F. Protein kinase B: Emerging mechanisms of isoform-specific regulation of cellular signaling in cancer. Anticancer Drugs 2017, 28, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Risso, G.; Blaustein, M.; Pozzi, B.; Mammi, P.; Srebrow, A. Akt/PKB: One kinase, many modifications. Biochem. J. 2015, 468, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, N.; Gavin, M.; Loro, E.; Sostre-Colón, J.; Roberson, P.A.; Uehara, K.; Rivera-Fuentes, N.; Neinast, M.; Arany, Z.; Kimball, S.R.; et al. AKT controls protein synthesis and oxidative metabolism via combined mTORC1 and FOXO1 signalling to govern muscle physiology. J. Cachexia Sarcopenia Muscle 2022, 13, 495–514. [Google Scholar] [CrossRef] [PubMed]
- Walkowski, B.; Kleibert, M.; Majka, M.; Wojciechowska, M. Insight into the Role of the PI3K/Akt Pathway in Ischemic Injury and Post-Infarct Left Ventricular Remodeling in Normal and Diabetic Heart. Cells 2022, 11, 1553. [Google Scholar] [CrossRef]
- Glaviano, A.; Foo, A.S.C.; Lam, H.Y.; Yap, K.C.H.; Jacot, W.; Jones, R.H.; Eng, H.; Nair, M.G.; Makvandi, P.; Geoerger, B.; et al. PI3K/AKT/mTOR signaling transduction pathway and targeted therapies in cancer. Mol. Cancer 2023, 22, 138. [Google Scholar] [CrossRef]
- Zhou, H.; Huang, S. The complexes of mammalian target of rapamycin. Curr. Protein Pept. Sci. 2010, 11, 409–424. [Google Scholar] [CrossRef]
- Unni, N.; Arteaga, C.L. Is Dual mTORC1 and mTORC2 Therapeutic Blockade Clinically Feasible in Cancer? JAMA Oncol. 2019, 5, 1564–1565. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Jhanwar-Uniyal, M.; Wainwright, J.V.; Mohan, A.L.; Tobias, M.E.; Murali, R.; Gandhi, C.D.; Schmidt, M.H. Diverse signaling mechanisms of mTOR complexes: mTORC1 and mTORC2 in forming a formidable relationship. Adv. Biol. Regul. 2019, 72, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Dodd, K.M.; Yang, J.; Shen, M.H.; Sampson, J.R.; Tee, A.R. mTORC1 drives HIF-1α and VEGF-A signalling via multiple mechanisms involving 4E-BP1, S6K1 and STAT3. Oncogene 2015, 34, 2239–2250. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Langer, R.; Ferrara, N. Targeting angiogenesis in oncology, ophthalmology and beyond. Nat. Rev. Drug Discov. 2023, 22, 476–495. [Google Scholar] [CrossRef] [PubMed]
- Rapley, J.; Oshiro, N.; Ortiz-Vega, S.; Avruch, J. The mechanism of insulin-stimulated 4E-BP protein binding to mammalian target of rapamycin (mTOR) complex 1 and its contribution to mTOR complex 1 signaling. J. Biol. Chem. 2011, 286, 38043–38053. [Google Scholar] [CrossRef] [PubMed]
- Le Bacquer, O.; Combe, K.; Montaurier, C.; Salles, J.; Giraudet, C.; Patrac, V.; Domingues-Faria, C.; Guillet, C.; Louche, K.; Boirie, Y.; et al. Muscle metabolic alterations induced by genetic ablation of 4E-BP1 and 4E-BP2 in response to diet-induced obesity. Mol. Nutr. Food Res. 2017, 61, 1700128. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Lee, G.; Pickering, B.F.; Jang, C.; Park, J.H.; He, L.; Mathur, L.; Kim, S.S.; Jung, S.; Tang, H.W.; et al. mTORC1 promotes cell growth via m6A-dependent mRNA degradation. Mol. Cell 2021, 81, 2064–2075.e8. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Klein, P.S. Glycogen synthase kinase-3 and alternative splicing. Wiley Interdiscip. Rev. RNA 2018, 9, e1501. [Google Scholar] [CrossRef]
- Arioka, M.; Takahashi-Yanaga, F. Glycogen synthase kinase-3 inhibitor as a multi-targeting anti-rheumatoid drug. Biochem. Pharmacol. 2019, 165, 207–213. [Google Scholar] [CrossRef]
- Golpich, M.; Amini, E.; Hemmati, F.; Ibrahim, N.M.; Rahmani, B.; Mohamed, Z.; Raymond, A.A.; Dargahi, L.; Ghasemi, R.; Ahmadiani, A. Glycogen synthase kinase-3 beta (GSK-3β) signaling: Implications for Parkinson’s disease. Pharmacol. Res. 2015, 97, 16–26. [Google Scholar] [CrossRef]
- Mishra, R. Glycogen synthase kinase 3 beta: Can it be a target for oral cancer. Mol. Cancer 2010, 9, 144. [Google Scholar] [CrossRef] [PubMed]
- Ghorbani, A.; Jeddi-Tehrani, M.; Saidpour, A.; Safa, M.; Bayat, A.A.; Zand, H. PI3K/AKT and Mdm2 activation are associated with inhibitory effect of cAMP increasing agents on DNA damage-induced cell death in human pre-B NALM-6 cells. Arch. Biochem. Biophys. 2015, 566, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Chen, Y.; Liu, G.; Li, C.; Song, Y.; Cao, Z.; Li, W.; Hu, J.; Lu, C.; Liu, Y. PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death Dis. 2020, 11, 797. [Google Scholar] [CrossRef]
- Vazquez, F.; Devreotes, P. Regulation of PTEN function as a PIP3 gatekeeper through membrane interaction. Cell Cycle 2006, 5, 1523–1527. [Google Scholar] [CrossRef] [PubMed]
- Karar, J.; Maity, A. PI3K/AKT/mTOR Pathway in Angiogenesis. Front. Mol. Neurosci. 2011, 4, 51. [Google Scholar] [CrossRef] [PubMed]
- Du, G.; Sun, J.; Zhang, Y. Perfluorooctanoic acid impaired glucose homeostasis through affecting adipose AKT pathway. Cytotechnology 2018, 70, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Martini, M.; De Santis, M.C.; Braccini, L.; Gulluni, F.; Hirsch, E. PI3K/AKT signaling pathway and cancer: An updated review. Ann. Med. 2014, 46, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Wu, Y.; He, P.; Fan, Y.; Zhong, X.; Zheng, H.; Luo, T. PI3K/AKT/mTOR-Targeted Therapy for Breast Cancer. Cells 2022, 11, 2508. [Google Scholar] [CrossRef] [PubMed]
- Cochicho, D.; Esteves, S.; Rito, M.; Silva, F.; Martins, L.; Montalvão, P.; Cunha, M.; Magalhães, M.; da Costa, R.M.G.; Felix, A. PIK3CA Gene Mutations in HNSCC: Systematic Review and Correlations with HPV Status and Patient Survival. Cancers 2022, 14, 1286. [Google Scholar] [CrossRef]
- Fang, W.L.; Huang, K.H.; Lan, Y.T.; Lin, C.H.; Chang, S.C.; Chen, M.H.; Chao, Y.; Lin, W.C.; Lo, S.S.; Li, A.F.; et al. Mutations in PI3K/AKT pathway genes and amplifications of PIK3CA are associated with patterns of recurrence in gastric cancers. Oncotarget 2016, 7, 6201–6220. [Google Scholar] [CrossRef]
- Li, M.; Liu, F.; Zhang, F.; Zhou, W.; Jiang, X.; Yang, Y.; Qu, K.; Wang, Y.; Ma, Q.; Wang, T.; et al. Genomic ERBB2/ERBB3 mutations promote PD-L1-mediated immune escape in gallbladder cancer: A whole-exome sequencing analysis. Gut 2019, 68, 1024–1033. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.M.; Deuker, M.M.; Baguley, B.C.; McMahon, M. PIK3CA-mutated melanoma cells rely on cooperative signaling through mTORC1/2 for sustained proliferation. Pigment. Cell Melanoma Res. 2017, 30, 353–367. [Google Scholar] [CrossRef] [PubMed]
- Moura, A.C.; Assad, D.X.; Santos, J.A.D.; de Toledo, I.P.; Barra, G.B.; Castilho, R.M.; Squarize, C.H.; Guerra, E.N.S. Worldwide prevalence of PI3K-AKT-mTOR pathway mutations in head and neck cancer: A systematic review and meta-analysis. Crit. Rev. Oncol. Hematol. 2021, 160, 103284. [Google Scholar] [CrossRef] [PubMed]
- Gutkind, J.S.; Day, T.A.; Lippman, S.M.; Szabo, E. Targeting mTOR in Head and Neck Cancer-Response. Clin. Cancer Res. 2019, 25, 6555. [Google Scholar] [CrossRef] [PubMed]
- Sirico, M.; D’Angelo, A.; Gianni, C.; Casadei, C.; Merloni, F.; De Giorgi, U. Current State and Future Challenges for PI3K Inhibitors in Cancer Therapy. Cancers 2023, 15, 703. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Li, S.; Li, J.; Wang, D.; Li, Q. Effect of microRNA-135a on Cell Proliferation, Migration, Invasion, Apoptosis and Tumor Angiogenesis Through the IGF-1/PI3K/Akt Signaling Pathway in Non-Small Cell Lung Cancer. Cell Physiol. Biochem. 2017, 42, 1431–1446. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.Z.; Wu, S.C.; Chang, C.M.; Lin, C.L.; Kwan, A.L. Arctigenin, a Potent Ingredient of Arctium lappa L., Induces Endothelial Nitric Oxide Synthase and Attenuates Subarachnoid Hemorrhage-Induced Vasospasm through PI3K/Akt Pathway in a Rat Model. BioMed Res. Int. 2015, 2015, 490209. [Google Scholar] [CrossRef] [PubMed]
- Wicks, E.E.; Semenza, G.L. Hypoxia-inducible factors: Cancer progression and clinical translation. J. Clin. Investig. 2022, 132, e159839. [Google Scholar] [CrossRef] [PubMed]
- Nag, S.; Zhang, X.; Srivenugopal, K.S.; Wang, M.H.; Wang, W.; Zhang, R. Targeting MDM2-p53 interaction for cancer therapy: Are we there yet? Curr. Med. Chem. 2014, 21, 553–574. [Google Scholar] [CrossRef]
- Chin, Y.R.; Toker, A. The actin-bundling protein palladin is an Akt1-specific substrate that regulates breast cancer cell migration. Mol. Cell 2010, 38, 333–344. [Google Scholar] [CrossRef]
- Cheng, T.C.; Din, Z.H.; Su, J.H.; Wu, Y.J.; Liu, C.I. Sinulariolide Suppresses Cell Migration and Invasion by Inhibiting Matrix Metalloproteinase-2/-9 and Urokinase through the PI3K/AKT/mTOR Signaling Pathway in Human Bladder Cancer Cells. Mar. Drugs 2017, 15, 238. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.R.; Brunet, A.; Greenberg, M.E. Cellular survival: A play in three Akts. Genes. Dev. 1999, 13, 2905–2927. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ng, P.K.-S.; Kucherlapati, M.; Chen, F.; Liu, Y.; Tsang, Y.H.; de Velasco, G.; Jeong, K.J.; Akbani, R.; Hadjipanayis, A.; et al. A Pan-Cancer Proteogenomic Atlas of PI3K/AKT/mTOR Pathway Alterations. Cancer Cell 2017, 31, 820–832.e3. [Google Scholar] [CrossRef]
- Chow, L.M.; Baker, S.J. PTEN function in normal and neoplastic growth. Cancer Lett. 2006, 241, 184–196. [Google Scholar] [CrossRef] [PubMed]
- Song, M.S.; Salmena, L.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell Biol. 2012, 13, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Bergholz, J.S.; Wang, Q.; Wang, Q.; Ramseier, M.; Prakadan, S.; Wang, W.; Fang, R.; Kabraji, S.; Zhou, Q.; Gray, G.K.; et al. PI3Kβ controls immune evasion in PTEN-deficient breast tumours. Nature 2023, 617, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.W.; Pérez-Torres, M.; Narasanna, A.; Guix, M.; Stål, O.; Pérez-Tenorio, G.; Gonzalez-Angulo, A.M.; Hennessy, B.T.; Mills, G.B.; Kennedy, J.P.; et al. Loss of Phosphatase and Tensin homologue deleted on chromosome 10 engages ErbB3 and insulin-like growth factor-I receptor signaling to promote antiestrogen resistance in breast cancer. Cancer Res. 2009, 69, 4192–4201. [Google Scholar] [CrossRef]
- Fusco, N.; Sajjadi, E.; Venetis, K.; Gaudioso, G.; Lopez, G.; Corti, C.; Rocco, E.G.; Criscitiello, C.; Malapelle, U.; Invernizzi, M. PTEN Alterations and Their Role in Cancer Management: Are We Making Headway on Precision Medicine? Genes 2020, 11, 719. [Google Scholar] [CrossRef]
- Vidotto, T.; Melo, C.M.; Lautert-Dutra, W.; Chaves, L.P.; Reis, R.B.; Squire, J.A. Pan-cancer genomic analysis shows hemizygous PTEN loss tumors are associated with immune evasion and poor outcome. Sci. Rep. 2023, 13, 5049. [Google Scholar] [CrossRef]
- Hu, J.; Li, G.; Liu, Z.; Ma, H.; Yuan, W.; Lu, Z.; Zhang, D.; Ling, H.; Zhang, F.; Liu, Y.; et al. Bicarbonate transporter SLC4A7 promotes EMT and metastasis of HNSCC by activating the PI3K/AKT/mTOR signaling pathway. Mol. Carcinog. 2023, 62, 628–640. [Google Scholar] [CrossRef]
- Yu, M.; Chen, J.; Xu, Z.; Yang, B.; He, Q.; Luo, P.; Yan, H.; Yang, X. Development and safety of PI3K inhibitors in cancer. Arch. Toxicol. 2023, 97, 635–650. [Google Scholar] [CrossRef]
- Hillmann, P.; Fabbro, D. PI3K/mTOR Pathway Inhibition: Opportunities in Oncology and Rare Genetic Diseases. Int. J. Mol. Sci. 2019, 20, 5792. [Google Scholar] [CrossRef]
- Desilets, A.; Soulières, D. PI3K Inhibition for Squamous Cell Head and Neck Carcinoma. Cancer J. 2022, 28, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Maira, S.M.; Pecchi, S.; Huang, A.; Burger, M.; Knapp, M.; Sterker, D.; Schnell, C.; Guthy, D.; Nagel, T.; Wiesmann, M.; et al. Identification and characterization of NVP-BKM120, an orally available pan-class I PI3-kinase inhibitor. Mol. Cancer Ther. 2012, 11, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Bendell, J.C.; Rodon, J.; Burris, H.A.; de Jonge, M.; Verweij, J.; Birle, D.; Demanse, D.; De Buck, S.S.; Ru, Q.C.; Peters, M.; et al. Phase I, dose-escalation study of BKM120, an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. J. Clin. Oncol. 2012, 30, 282–290. [Google Scholar] [CrossRef]
- Brachmann, S.M.; Kleylein-Sohn, J.; Gaulis, S.; Kauffmann, A.; Blommers, M.J.; Kazic-Legueux, M.; Laborde, L.; Hattenberger, M.; Stauffer, F.; Vaxelaire, J.; et al. Characterization of the mechanism of action of the pan class I PI3K inhibitor NVP-BKM120 across a broad range of concentrations. Mol. Cancer Ther. 2012, 11, 1747–1757. [Google Scholar] [CrossRef]
- Kong, D.; Yamori, T.; Yamazaki, K.; Dan, S. In vitro multifaceted activities of a specific group of novel phosphatidylinositol 3-kinase inhibitors on hotspot mutant PIK3CA. Investig. New Drugs 2014, 32, 1134–1143. [Google Scholar] [CrossRef]
- Ando, Y.; Inada-Inoue, M.; Mitsuma, A.; Yoshino, T.; Ohtsu, A.; Suenaga, N.; Sato, M.; Kakizume, T.; Robson, M.; Quadt, C.; et al. Phase I dose-escalation study of buparlisib (BKM120), an oral pan-class I PI3K inhibitor, in Japanese patients with advanced solid tumors. Cancer Sci. 2014, 105, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Koul, D.; Shen, R.; Kim, Y.W.; Kondo, Y.; Lu, Y.; Bankson, J.; Ronen, S.M.; Kirkpatrick, D.L.; Powis, G.; Yung, W.K. Cellular and in vivo activity of a novel PI3K inhibitor, PX-866, against human glioblastoma. Neuro-Oncology 2010, 12, 559–569. [Google Scholar] [CrossRef]
- Keysar, S.B.; Astling, D.P.; Anderson, R.T.; Vogler, B.W.; Bowles, D.W.; Morton, J.J.; Paylor, J.J.; Glogowska, M.J.; Le, P.N.; Eagles-Soukup, J.R.; et al. A patient tumor transplant model of squamous cell cancer identifies PI3K inhibitors as candidate therapeutics in defined molecular bins. Mol. Oncol. 2013, 7, 776–790. [Google Scholar] [CrossRef]
- Jung, K.; Kang, H.; Mehra, R. Targeting phosphoinositide 3-kinase (PI3K) in head and neck squamous cell carcinoma (HNSCC). Cancers Head. Neck 2018, 3, 3. [Google Scholar] [CrossRef]
- Marret, G.; Isambert, N.; Rezai, K.; Gal, J.; Saada-Bouzid, E.; Rolland, F.; Chausson, M.; Borcoman, E.; Alt, M.; Klijanienko, J.; et al. Phase I trial of copanlisib, a selective PI3K inhibitor, in combination with cetuximab in patients with recurrent and/or metastatic head and neck squamous cell carcinoma. Investig. New Drugs 2021, 39, 1641–1648. [Google Scholar] [CrossRef] [PubMed]
- Furet, P.; Guagnano, V.; Fairhurst, R.A.; Imbach-Weese, P.; Bruce, I.; Knapp, M.; Fritsch, C.; Blasco, F.; Blanz, J.; Aichholz, R.; et al. Discovery of NVP-BYL719 a potent and selective phosphatidylinositol-3 kinase alpha inhibitor selected for clinical evaluation. Bioorg. Med. Chem. Lett. 2013, 23, 3741–3748. [Google Scholar] [CrossRef] [PubMed]
- Fritsch, C.; Huang, A.; Chatenay-Rivauday, C.; Schnell, C.; Reddy, A.; Liu, M.; Kauffmann, A.; Guthy, D.; Erdmann, D.; De Pover, A.; et al. Characterization of the novel and specific PI3Kα inhibitor NVP-BYL719 and development of the patient stratification strategy for clinical trials. Mol. Cancer Ther. 2014, 13, 1117–1129. [Google Scholar] [CrossRef] [PubMed]
- Chaves, P.; Garrido, M.; Oliver, J.; Pérez-Ruiz, E.; Barragan, I.; Rueda-Domínguez, A. Preclinical models in head and neck squamous cell carcinoma. Br. J. Cancer 2023, 128, 1819–1827. [Google Scholar] [CrossRef] [PubMed]
- Tinhofer, I.; Braunholz, D.; Klinghammer, K. Preclinical models of head and neck squamous cell carcinoma for a basic understanding of cancer biology and its translation into efficient therapies. Cancers Head Neck 2020, 5, 9. [Google Scholar] [CrossRef]
- Keam, B.; Kim, S.; Ahn, Y.O.; Kim, T.M.; Lee, S.H.; Kim, D.W.; Heo, D.S. In vitro anticancer activity of PI3K alpha selective inhibitor BYL719 in head and neck cancer. Anticancer Res. 2015, 35, 175–182. [Google Scholar]
- Zhao, W.; Qiu, Y.; Kong, D. Class I phosphatidylinositol 3-kinase inhibitors for cancer therapy. Acta Pharm. Sin. B 2017, 7, 27–37. [Google Scholar] [CrossRef]
- Hirai, H.; Sootome, H.; Nakatsuru, Y.; Miyama, K.; Taguchi, S.; Tsujioka, K.; Ueno, Y.; Hatch, H.; Majumder, P.K.; Pan, B.S.; et al. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol. Cancer Ther. 2010, 9, 1956–1967. [Google Scholar] [CrossRef]
- Knowles, J.A.; Golden, B.; Yan, L.; Carroll, W.R.; Helman, E.E.; Rosenthal, E.L. Disruption of the AKT pathway inhibits metastasis in an orthotopic model of head and neck squamous cell carcinoma. Laryngoscope 2011, 121, 2359–2365. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.B.; Goh, B.C.; Lim, W.T.; Hui, E.P.; Tan, E.H.; Gde, L.L.; Lo, K.W.; Li, L.; Loong, H.; Foster, N.R.; et al. Multicenter phase II study of the AKT inhibitor MK-2206 in recurrent or metastatic nasopharyngeal carcinoma from patients in the mayo phase II consortium and the cancer therapeutics research group (MC1079). Investig. New Drugs 2015, 33, 985–991. [Google Scholar] [CrossRef]
- Burkett, W.C.; Zhao, Z.; Newton, M.A.; Sun, W.; Deng, B.; Secord, A.A.; Zhou, C.; Bae-Jump, V. Ipatasertib, an oral AKT inhibitor, in combination with carboplatin exhibits anti-proliferative effects in uterine serous carcinoma. Ann. Med. 2023, 55, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Argiris, A.; Cohen, E.; Karrison, T.; Esparaz, B.; Mauer, A.; Ansari, R.; Wong, S.; Lu, Y.; Pins, M.; Dancey, J.; et al. A phase II trial of perifosine, an oral alkylphospholipid, in recurrent or metastatic head and neck cancer. Cancer Biol. Ther. 2006, 5, 766–770. [Google Scholar] [CrossRef][Green Version]
- Hilgard, P.; Klenner, T.; Stekar, J.; Nössner, G.; Kutscher, B.; Engel, J. D-21266, a new heterocyclic alkylphospholipid with antitumour activity. Eur. J. Cancer 1997, 33, 442–446. [Google Scholar] [CrossRef] [PubMed]
- Figg, W.D.; Monga, M.; Headlee, D.; Shah, A.; Chau, C.H.; Peer, C.; Messman, R.; Elsayed, Y.A.; Murgo, A.J.; Melillo, G.; et al. A phase I and pharmacokinetic study of oral perifosine with different loading schedules in patients with refractory neoplasms. Cancer Chemother. Pharmacol. 2014, 74, 955–967. [Google Scholar] [CrossRef] [PubMed]
- Day, T.A.; Shirai, K.; O’Brien, P.E.; Matheus, M.G.; Godwin, K.; Sood, A.J.; Kompelli, A.; Vick, J.A.; Martin, D.; Vitale-Cross, L.; et al. Inhibition of mTOR Signaling and Clinical Activity of Rapamycin in Head and Neck Cancer in a Window of Opportunity Trial. Clin. Cancer Res. 2019, 25, 1156–1164. [Google Scholar] [CrossRef] [PubMed]
- Cohen, E.E.; Sharma, M.R.; Janisch, L.; Llobrera, M.; House, L.; Wu, K.; Ramirez, J.; Fleming, G.F.; Stadler, W.M.; Ratain, M.J. A phase I study of sirolimus and bevacizumab in patients with advanced malignancies. Eur. J. Cancer 2011, 47, 1484–1489. [Google Scholar] [CrossRef]
- Harsha, C.; Banik, K.; Ang, H.L.; Girisa, S.; Vikkurthi, R.; Parama, D.; Rana, V.; Shabnam, B.; Khatoon, E.; Kumar, A.P.; et al. Targeting AKT/mTOR in Oral Cancer: Mechanisms and Advances in Clinical Trials. Int. J. Mol. Sci. 2020, 21, 3285. [Google Scholar] [CrossRef]
- Massarelli, E.; Lin, H.; Ginsberg, L.E.; Tran, H.T.; Lee, J.J.; Canales, J.R.; Williams, M.D.; Blumenschein, G.R., Jr.; Lu, C.; Heymach, J.V.; et al. Phase II trial of everolimus and erlotinib in patients with platinum-resistant recurrent and/or metastatic head and neck squamous cell carcinoma. Ann. Oncol. 2015, 26, 1476–1480. [Google Scholar] [CrossRef]
- Saba, N.F.; Hurwitz, S.J.; Magliocca, K.; Kim, S.; Owonikoko, T.K.; Harvey, D.; Ramalingam, S.S.; Chen, Z.; Rogerio, J.; Mendel, J.; et al. Phase 1 and pharmacokinetic study of everolimus in combination with cetuximab and carboplatin for recurrent/metastatic squamous cell carcinoma of the head and neck. Cancer 2014, 120, 3940–3951. [Google Scholar] [CrossRef]
- Geiger, J.L.; Bauman, J.E.; Gibson, M.K.; Gooding, W.E.; Varadarajan, P.; Kotsakis, A.; Martin, D.; Gutkind, J.S.; Hedberg, M.L.; Grandis, J.R.; et al. Phase II trial of everolimus in patients with previously treated recurrent or metastatic head and neck squamous cell carcinoma. Head Neck 2016, 38, 1759–1764. [Google Scholar] [CrossRef]
- Fury, M.G.; Sherman, E.; Ho, A.L.; Xiao, H.; Tsai, F.; Nwankwo, O.; Sima, C.; Heguy, A.; Katabi, N.; Haque, S.; et al. A phase 1 study of everolimus plus docetaxel plus cisplatin as induction chemotherapy for patients with locally and/or regionally advanced head and neck cancer. Cancer 2013, 119, 1823–1831. [Google Scholar] [CrossRef]
- Fury, M.G.; Lee, N.Y.; Sherman, E.; Ho, A.L.; Rao, S.; Heguy, A.; Shen, R.; Korte, S.; Lisa, D.; Ganly, I.; et al. A phase 1 study of everolimus + weekly cisplatin + intensity modulated radiation therapy in head-and-neck cancer. Int. J. Radiat. Oncol. Biol. Phys. 2013, 87, 479–486. [Google Scholar] [CrossRef]
- O’Reilly, K.E.; Rojo, F.; She, Q.B.; Solit, D.; Mills, G.B.; Smith, D.; Lane, H.; Hofmann, F.; Hicklin, D.J.; Ludwig, D.L.; et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006, 66, 1500–1508. [Google Scholar] [CrossRef]
- Rozengurt, E.; Soares, H.P.; Sinnet-Smith, J. Suppression of feedback loops mediated by PI3K/mTOR induces multiple overactivation of compensatory pathways: An unintended consequence leading to drug resistance. Mol. Cancer Ther. 2014, 13, 2477–2488. [Google Scholar] [CrossRef]
- Hall, C.P.; Reynolds, C.P.; Kang, M.H. Modulation of Glucocorticoid Resistance in Pediatric T-cell Acute Lymphoblastic Leukemia by Increasing BIM Expression with the PI3K/mTOR Inhibitor BEZ235. Clin. Cancer Res. 2016, 22, 621–632. [Google Scholar] [CrossRef]
- Gazi, M.; Moharram, S.A.; Marhäll, A.; Kazi, J.U. The dual specificity PI3K/mTOR inhibitor PKI-587 displays efficacy against T-cell acute lymphoblastic leukemia (T-ALL). Cancer Lett. 2017, 392, 9–16. [Google Scholar] [CrossRef]
- Bei, S.; Li, F.; Li, H.; Li, J.; Zhang, X.; Sun, Q.; Feng, L. Inhibition of gastric cancer cell growth by a PI3K-mTOR dual inhibitor GSK1059615. Biochem. Biophys. Res. Commun. 2019, 511, 13–20. [Google Scholar] [CrossRef]
- Xie, J.; Li, Q.; Ding, X.; Gao, Y. GSK1059615 kills head and neck squamous cell carcinoma cells possibly via activating mitochondrial programmed necrosis pathway. Oncotarget 2017, 8, 50814–50823. [Google Scholar] [CrossRef]
- Machl, A.; Wilker, E.W.; Tian, H.; Liu, X.; Schroeder, P.; Clark, A.; Huck, B.R. M2698 is a potent dual-inhibitor of p70S6K and Akt that affects tumor growth in mouse models of cancer and crosses the blood-brain barrier. Am. J. Cancer Res. 2016, 6, 806–818. [Google Scholar]
- Tsimberidou, A.M.; Shaw, J.V.; Juric, D.; Verschraegen, C.; Weise, A.M.; Sarantopoulos, J.; Lopes, G.; Nemunaitis, J.; Mita, M.; Park, H.; et al. Phase 1 study of M2698, a p70S6K/AKT dual inhibitor, in patients with advanced cancer. J. Hematol. Oncol. 2021, 14, 127. [Google Scholar] [CrossRef]
- Di Nicolantonio, F.; Arena, S.; Tabernero, J.; Grosso, S.; Molinari, F.; Macarulla, T.; Russo, M.; Cancelliere, C.; Zecchin, D.; Mazzucchelli, L.; et al. Deregulation of the PI3K and KRAS signaling pathways in human cancer cells determines their response to everolimus. J. Clin. Investig. 2010, 120, 2858–2866. [Google Scholar] [CrossRef]
- Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Kempf, R.C.; Long, J.; Laidler, P.; Mijatovic, S.; Maksimovic-Ivanic, D.; Stivala, F.; Mazzarino, M.C.; et al. Roles of the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways in controlling growth and sensitivity to therapy-implications for cancer and aging. Aging 2011, 3, 192–222. [Google Scholar] [CrossRef]
- Mohan, S.; Broek, R.V.; Shah, S.; Eytan, D.F.; Pierce, M.L.; Carlson, S.G.; Coupar, J.F.; Zhang, J.; Cheng, H.; Chen, Z.; et al. MEK Inhibitor PD-0325901 Overcomes Resistance to PI3K/mTOR Inhibitor PF-5212384 and Potentiates Antitumor Effects in Human Head and Neck Squamous Cell Carcinoma. Clin. Cancer Res. 2015, 21, 3946–3956. [Google Scholar] [CrossRef]
- Fujimoto, Y.; Morita, T.Y.; Ohashi, A.; Haeno, H.; Hakozaki, Y.; Fujii, M.; Kashima, Y.; Kobayashi, S.S.; Mukohara, T. Combination treatment with a PI3K/Akt/mTOR pathway inhibitor overcomes resistance to anti-HER2 therapy in PIK3CA-mutant HER2-positive breast cancer cells. Sci. Rep. 2020, 10, 21762. [Google Scholar] [CrossRef]
- Ranganna, K.; Selvam, C.; Shivachar, A.; Yousefipour, Z. Histone Deacetylase Inhibitors as Multitarget-Directed Epi-Drugs in Blocking PI3K Oncogenic Signaling: A Polypharmacology Approach. Int. J. Mol. Sci. 2020, 21, 8198. [Google Scholar] [CrossRef]
- Gupta, M.; Ansell, S.M.; Novak, A.J.; Kumar, S.; Kaufmann, S.H.; Witzig, T.E. Inhibition of histone deacetylase overcomes rapamycin-mediated resistance in diffuse large B-cell lymphoma by inhibiting Akt signaling through mTORC2. Blood 2009, 114, 2926–2935. [Google Scholar] [CrossRef]
- To, K.K.W.; Fu, L.W. CUDC-907, a dual HDAC and PI3K inhibitor, reverses platinum drug resistance. Investig. New Drugs 2018, 36, 10–19. [Google Scholar] [CrossRef]
- Erlich, R.B.; Kherrouche, Z.; Rickwood, D.; Endo-Munoz, L.; Cameron, S.; Dahler, A.; Hazar-Rethinam, M.; de Long, L.M.; Wooley, K.; Guminski, A.; et al. Preclinical evaluation of dual PI3K-mTOR inhibitors and histone deacetylase inhibitors in head and neck squamous cell carcinoma. Br. J. Cancer 2012, 106, 107–115. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).