

Intracellular Iron Deficiency and Abnormal Metabolism, Not Ferroptosis, Contributes to Homocysteine-Induced Vascular Endothelial Cell Death

 and
and
Abstract
:1. Introduction
2. Material and Methods
2.1. Chemicals and Reagents
2.2. Cell Viability and Cytotoxicity Assay
2.3. Real-Time Cell Proliferation and Cytotoxicity Analysis by xCELLigence
2.4. Live and Dead Cell Staining
2.5. Cell Cycle and Cell Apoptosis Analysis by Flow Cytometry
2.6. Intracellular MDA, SOD, GSH, ATP and Iron Assay
2.7. Detection of Intracellular ROS, Mitochondrial Content, and Membrane Potential
2.8. Real-Time PCR Analysis
2.9. Western Blotting Analysis
2.10. Statistical Analysis
3. Results
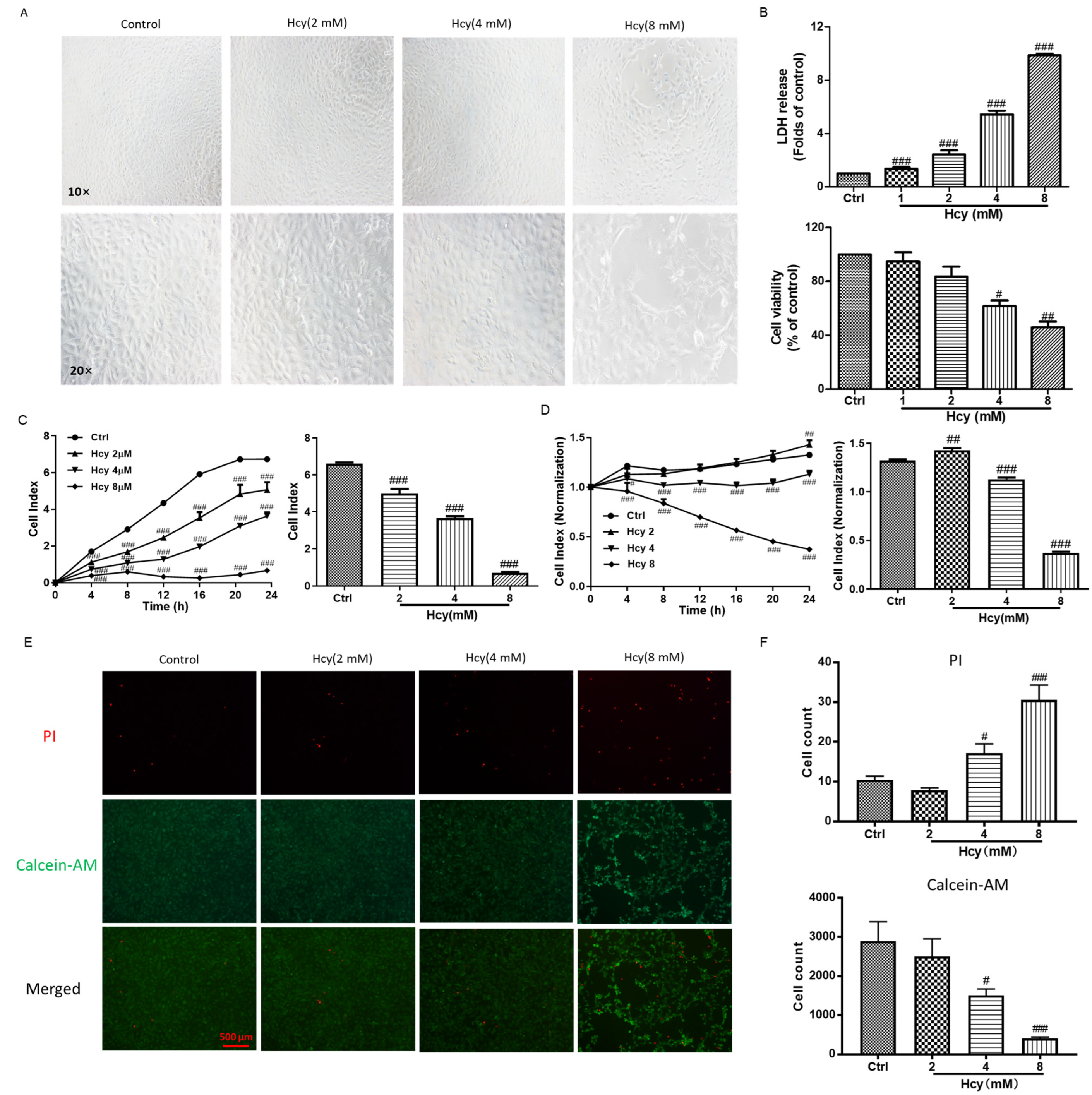
3.1. Homocysteine (Hcy)-Induced Vascular Endothelial Cell Death by Impairing Cell Viability and Cell Proliferation
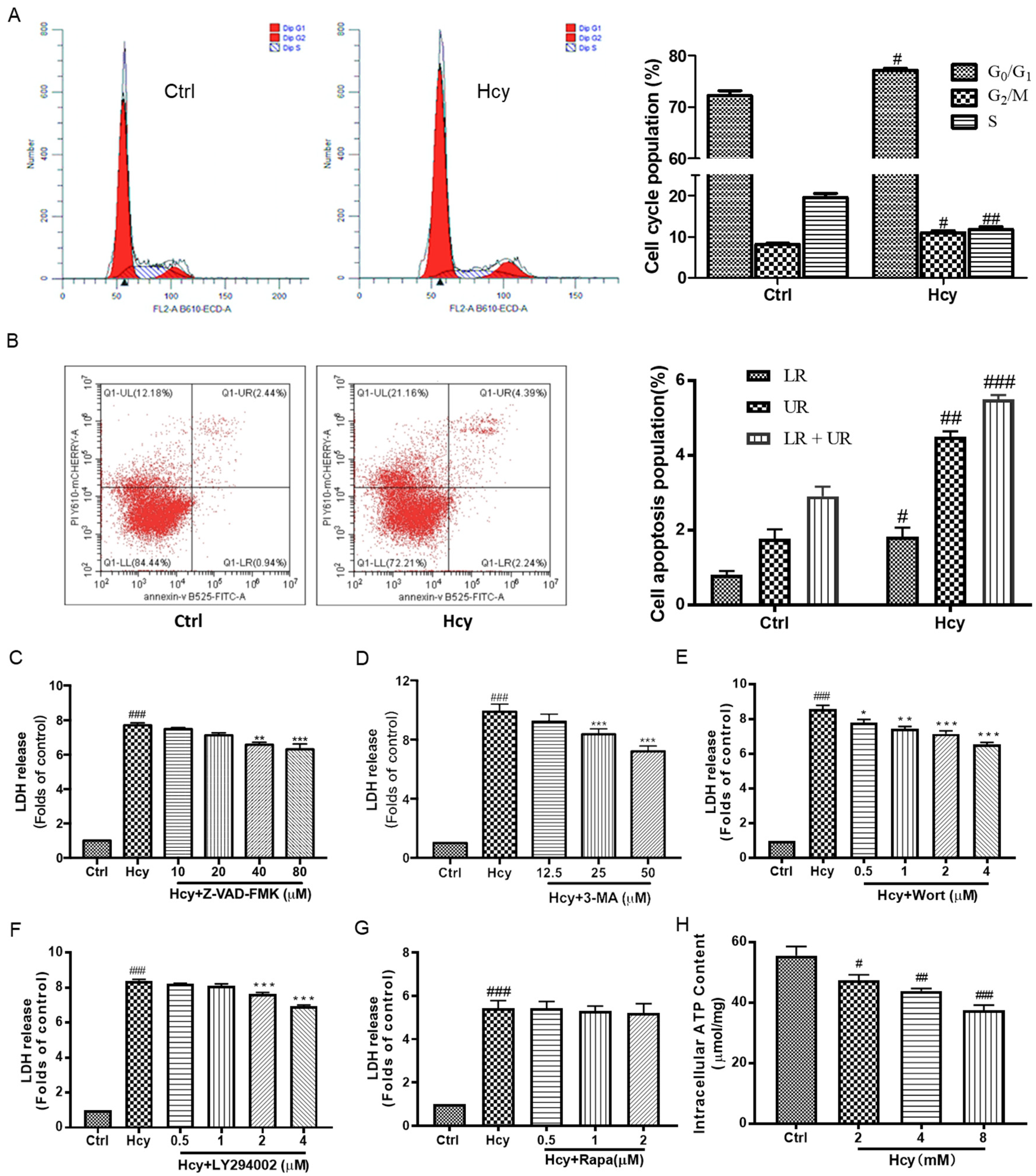
3.2. Hcy-Induced Vascular Endothelial Cell Death Involved in Cell Cycle Arrest, Apoptosis, and Autophagy as Well as Impairment of Energy Metabolism
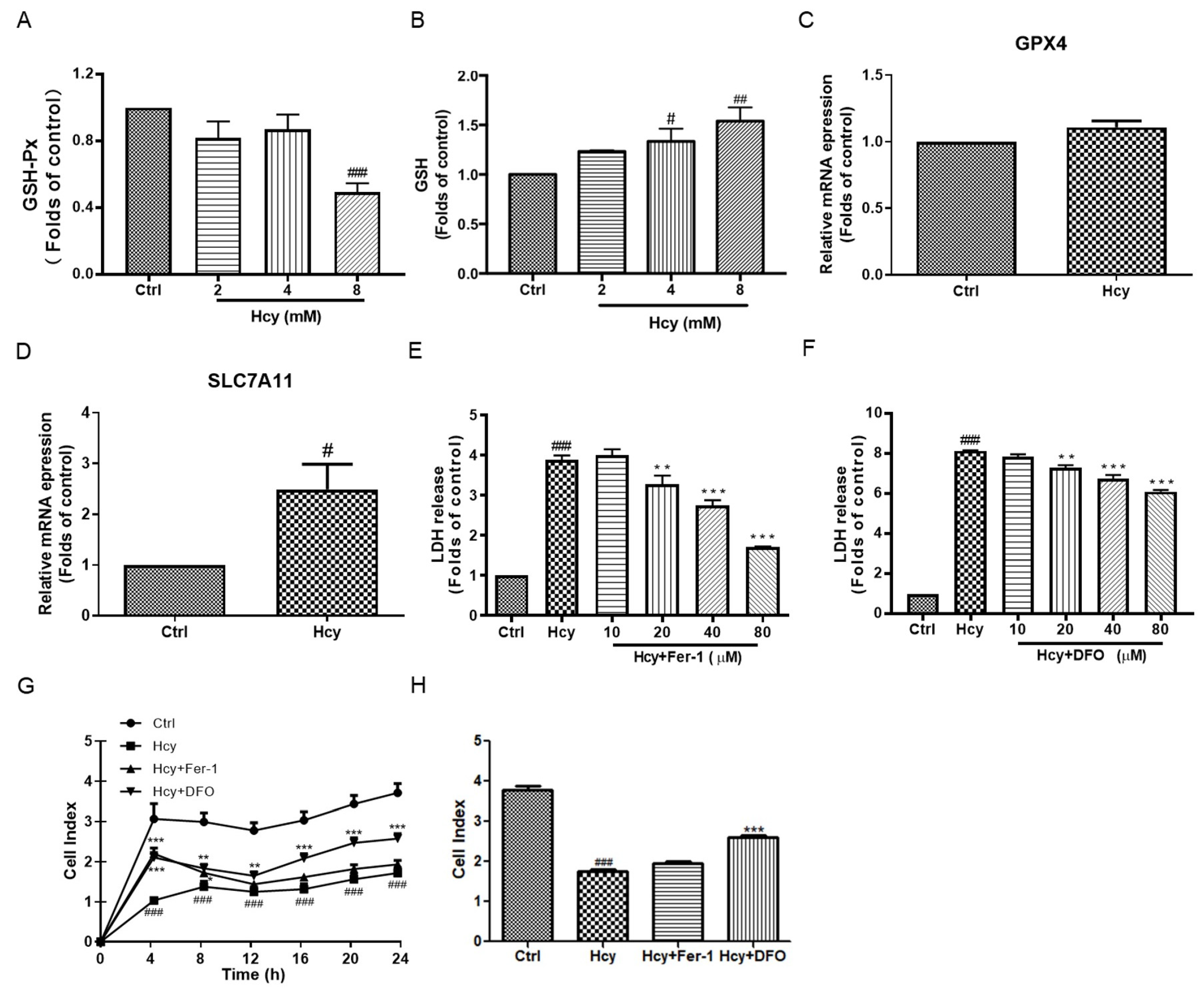
3.3. Hcy Disrupted the Intracellular Antioxidant System in Vascular Endothelial Cells
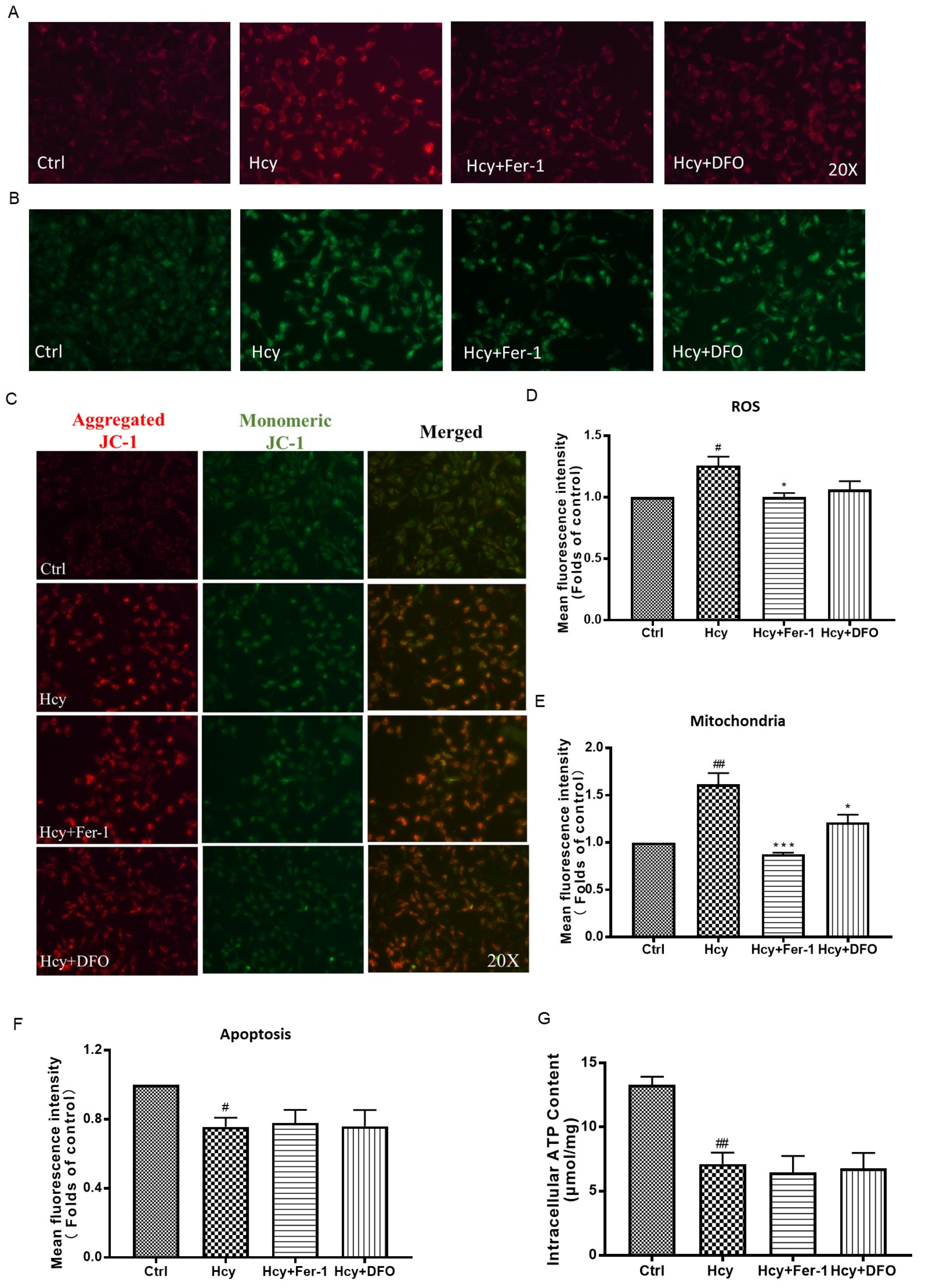
3.4. Ferroptosis Might Not Participate in Hcy-Induced Vascular Endothelial Death
3.5. Hcy Caused Intracellular Iron Deficiency and Abnormal Iron Metabolism in Vascular Endothelial Cells
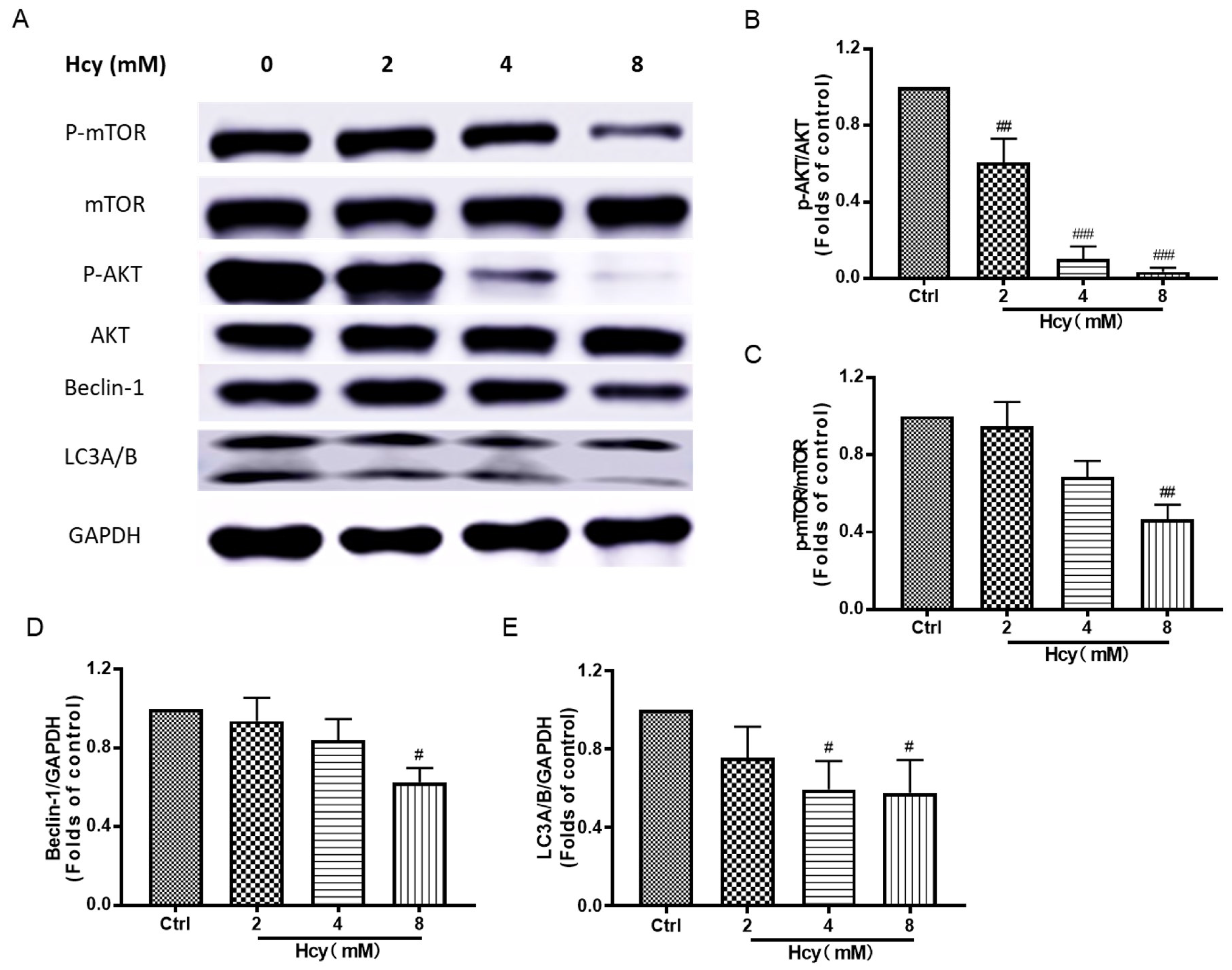
3.6. Hcy Suppressed the Akt/mTOR, MAPKs, and Nrf2/HO-1 Signaling in Vascular Endothelial Cells
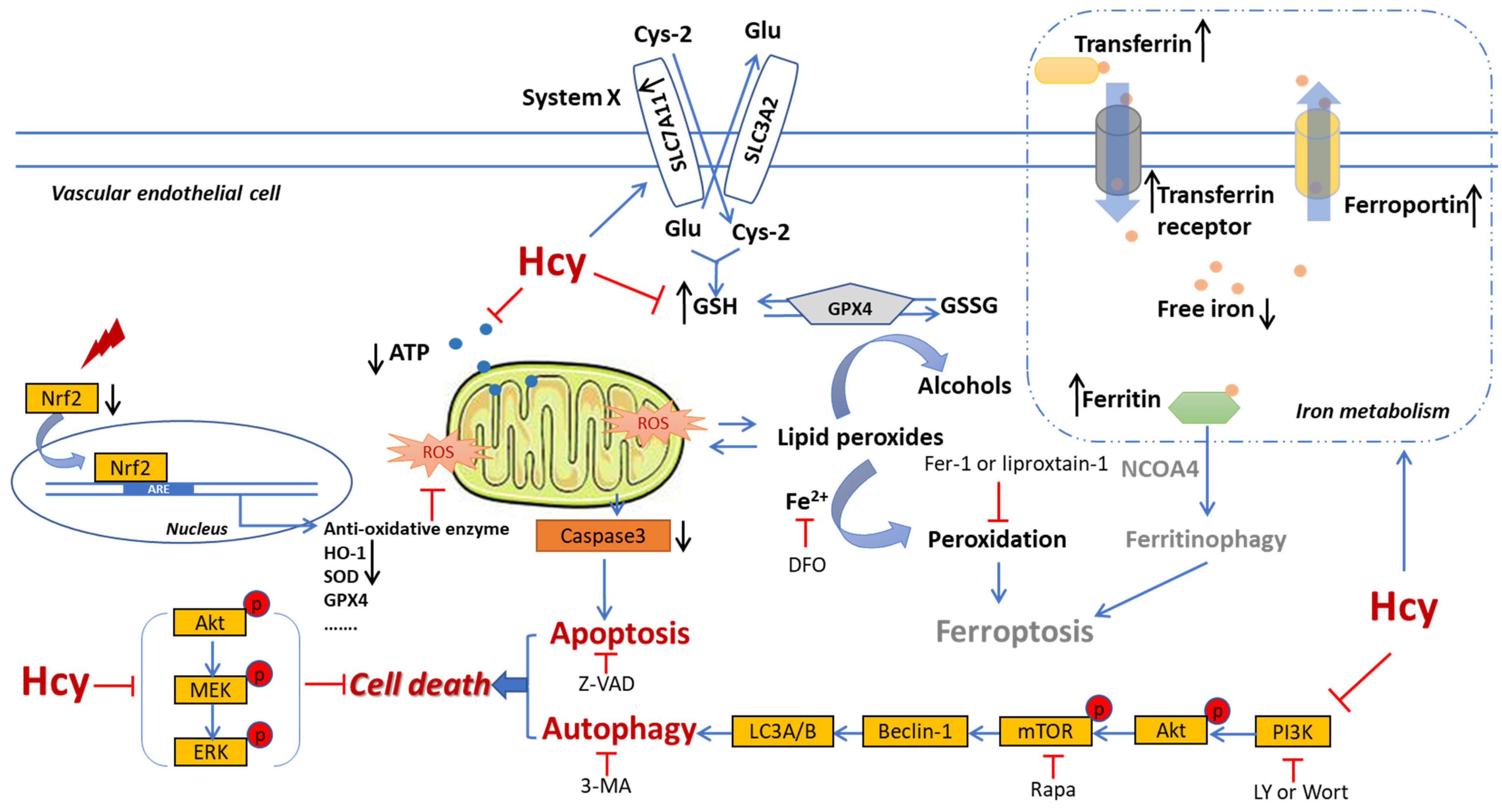
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Son, P.; Lewis, L. Hyperhomocysteinemia. In StatPearls; StatPearls Publishing LLC: Treasure Island, FL, USA, 2022. [Google Scholar]
- Guieu, R.; Ruf, J.; Mottola, G. Hyperhomocysteinemia and cardiovascular diseases. Ann. Biol. Clin. 2022, 80, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Al Mutairi, F. Hyperhomocysteinemia: Clinical Insights. J. Cent. Nerv. Syst. Dis. 2020, 12, 1179573520962230. [Google Scholar] [CrossRef] [PubMed]
- McCully, K.S. Homocysteine and the pathogenesis of atherosclerosis. Expert Rev. Clin. Pharmacol. 2015, 8, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Li, F.F.; Yuan, S.Q.; Tang, H.K.; Zhou, J.H.; He, Q.Y.; Baker, J.S.; Dong, Y.H.; Yang, Y.D. Prevalence of Hyperhomocysteinemia in China: An Updated Meta-Analysis. Biology 2021, 10, 959. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Yang, Y.; Peng, D.; Chen, L.; Luo, J. Hyperhomocysteinemia as a metabolic disorder parameter is independently associated with the severity of coronary heart disease. Saudi Med. J. 2015, 36, 839–846. [Google Scholar] [CrossRef]
- You, A.; Li, Y.; Shen, C.; Fan, H.; He, J.; Liu, Z.; Xue, Q.; Zhang, Y.; Zheng, L. Associations of non-traditional cardiovascular risk factors and body mass index with metabolic syndrome in the Chinese elderly population. Diabetol. Metab. Syndr. 2023, 15, 129. [Google Scholar] [CrossRef]
- Martí-Carvajal, A.J.; Solà, I.; Lathyris, D.; Dayer, M. Homocysteine-lowering interventions for preventing cardiovascular events. Cochrane Database Syst. Rev. 2017, 8, Cd006612. [Google Scholar] [CrossRef]
- Paganelli, F.; Mottola, G.; Fromonot, J.; Marlinge, M.; Deharo, P.; Guieu, R.; Ruf, J. Hyperhomocysteinemia and Cardiovascular Disease: Is the Adenosinergic System the Missing Link? Int. J. Mol. Sci. 2021, 22, 1690. [Google Scholar] [CrossRef]
- Esse, R.; Barroso, M.; Tavares de Almeida, I.; Castro, R. The Contribution of Homocysteine Metabolism Disruption to Endothelial Dysfunction: State-of-the-Art. Int. J. Mol. Sci. 2019, 20, 867. [Google Scholar] [CrossRef]
- Ganguly, P.; Alam, S.F. Role of homocysteine in the development of cardiovascular disease. Nutr. J. 2015, 14, 6. [Google Scholar] [CrossRef]
- Ahmad, A.; Corban, M.T.; Toya, T.; Sara, J.D.; Lerman, B.; Park, J.Y.; Lerman, L.O.; Lerman, A. Coronary Microvascular Endothelial Dysfunction in Patients With Angina and Nonobstructive Coronary Artery Disease Is Associated With Elevated Serum Homocysteine Levels. J. Am. Heart Assoc. 2020, 9, e017746. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Wang, Q.S.; Yang, X.; Zhu, D.; Sun, Y.; Niu, N.; Yao, J.; Dong, B.H.; Jiang, S.; Tang, L.L.; et al. Homocysteine Causes Endothelial Dysfunction via Inflammatory Factor-Mediated Activation of Epithelial Sodium Channel (ENaC). Front. Cell Dev. Biol. 2021, 9, 672335. [Google Scholar] [CrossRef] [PubMed]
- Aykutoglu, G.; Tartik, M.; Darendelioglu, E.; Ayna, A.; Baydas, G. Melatonin and vitamin E alleviate homocysteine-induced oxidative injury and apoptosis in endothelial cells. Mol. Biol. Rep. 2020, 47, 5285–5293. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.S.; Trinath, J.; Reddy, G.B. Implication of homocysteine in protein quality control processes. Biochimie 2019, 165, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, Y.; Tang, J.; Zhao, S.; Li, C.; Huang, Y.P.; Yi, M. Oxymatrine Inhibits Homocysteine-Mediated Autophagy via MIF/mTOR Signaling in Human Umbilical Vein Endothelial Cells. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2018, 45, 1893–1903. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, P.; Tatarkova, Z.; Sivonova, M.K.; Racay, P.; Lehotsky, J. Homocysteine and Mitochondria in Cardiovascular and Cerebrovascular Systems. Int. J. Mol. Sci. 2020, 21, 7698. [Google Scholar] [CrossRef]
- Sato, K.; Nishii, T.; Sato, A.; Tatsunami, R. Autophagy activation is required for homocysteine-induced apoptosis in bovine aorta endothelial cells. Heliyon 2020, 6, e03315. [Google Scholar] [CrossRef]
- Tsikas, D. Homocysteine and copper ions: Is their interaction responsible for cardiovascular-related damage? Amino Acids 2021, 53, 1297–1298. [Google Scholar] [CrossRef]
- Gupta, M.; Meehan-Atrash, J.; Strongin, R.M. Identifying a role for the interaction of homocysteine and copper in promoting cardiovascular-related damage. Amino Acids 2021, 53, 739–744. [Google Scholar] [CrossRef]
- Chen, X.; Li, J.; Kang, R.; Klionsky, D.J.; Tang, D. Ferroptosis: Machinery and regulation. Autophagy 2021, 17, 2054–2081. [Google Scholar] [CrossRef]
- Baggott, J.E.; Tamura, T. Homocysteine, iron and cardiovascular disease: A hypothesis. Nutrients 2015, 7, 1108–1118. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Huang, X.; Zhang, Q.; Katz, S.D. Iron sucrose augments homocysteine-induced endothelial dysfunction in normal subjects. Kidney Int. 2006, 69, 679–684. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Dimayuga, C.; Hudaihed, A.; Katz, S.D. Effect of dexrazoxane on homocysteine-induced endothelial dysfunction in normal subjects. Arterioscler. Thromb. Vasc. Biol. 2002, 22, E15–E18. [Google Scholar] [CrossRef] [PubMed]
- Borkowska, A.; Ziolkowski, W.; Kaczor, K.; Herman-Antosiewicz, A.; Knap, N.; Wronska, A.; Antosiewicz, J. Homocysteine-induced decrease in HUVEC cells’ resistance to oxidative stress is mediated by Akt-dependent changes in iron metabolism. Eur. J. Nutr. 2021, 60, 1619–1631. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.Y.; Zhao, W.R.; Xiao, Y.; Zhou, X.M.; Huang, C.; Shi, W.T.; Zhang, J.; Ye, Q.; Chen, X.L.; Tang, J.Y. Antiangiogenesis effect of timosaponin AIII on HUVECs in vitro and zebrafish embryos in vivo. Acta Pharmacol. Sin. 2019, 12, 260–269. [Google Scholar] [CrossRef]
- Zhou, Z.Y.; Huan, L.Y.; Zhao, W.R.; Tang, N.; Jin, Y.; Tang, J.Y. Spatholobi Caulis extracts promote angiogenesis in HUVECs in vitro and in zebrafish embryos in vivo via up-regulation of VEGFRs. J. Ethnopharmacol. 2017, 200, 74–83. [Google Scholar] [CrossRef]
- Huang, B.; Zhou, Z.Y.; Li, S.; Huang, X.H.; Tang, J.Y.; Hoi, M.P.M.; Lee, S.M.Y. Tanshinone I prevents atorvastatin-induced cerebral hemorrhage in zebrafish and stabilizes endothelial cell-cell adhesion by inhibiting VE-cadherin internalization and actin-myosin contractility. Pharmacol. Res. 2018, 128, 389–398. [Google Scholar] [CrossRef]
- Zhou, Z.-Y.; Zhao, W.-R.; Xiao, Y.; Zhang, J.; Tang, J.-Y.; Lee, S.M.-Y. Mechanism Study of the Protective Effects of Sodium Tanshinone IIA Sulfonate against Atorvastatin-Induced Cerebral Hemorrhage in Zebrafish: Transcriptome Analysis. Front. Pharmacol. 2020, 11, 551745. [Google Scholar] [CrossRef]
- Zhao, W.-R.; Shi, W.-T.; Zhang, J.; Zhang, K.-Y.; Qing, Y.; Tang, J.-Y.; Chen, X.-L.; Zhou, Z.-Y. Tribulus terrestris L. Extract Protects against Lipopolysaccharide-Induced Inflammation in RAW 264.7 Macrophage and Zebrafish via Inhibition of Akt/MAPKs and NF-κB/iNOS-NO Signaling Pathways. Evid. Based Complement. Altern. Med. 2021, 2021, 6628561. [Google Scholar] [CrossRef]
- Zhou, Z.Y.; Xiao, Y.; Zhao, W.R.; Zhang, J.; Shi, W.T.; Ma, Z.L.; Ye, Q.; Chen, X.L.; Tang, N.; Tang, J.Y. Pro-angiogenesis effect and transcriptome profile of Shuxinyin formula in zebrafish. Phytomedicine Int. J. Phytother. Phytopharm. 2019, 65, 153083. [Google Scholar] [CrossRef]
- Zhou, Z.Y.; Xu, J.Q.; Zhao, W.R.; Chen, X.L.; Jin, Y.; Tang, N.; Tang, J.Y. Ferulic acid relaxed rat aortic, small mesenteric and coronary arteries by blocking voltage-gated calcium channel and calcium desensitization via dephosphorylation of ERK1/2 and MYPT1. Eur. J. Pharmacol. 2017, 815, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Nagarajan, A.; Uchil, P.D. Analysis of Cell Viability by the Lactate Dehydrogenase Assay. Cold Spring Harb. Protoc. 2018, 2018, 29858337. [Google Scholar] [CrossRef] [PubMed]
- Cacciapuoti, G.; Manna, C.; Napoli, D.; Zappia, V.; Porcelli, M. Homocysteine-induced endothelial cell adhesion is related to adenosine lowering and is not mediated by S-adenosylhomocysteine. FEBS Lett. 2007, 581, 4567–4570. [Google Scholar] [CrossRef] [PubMed]
- D’Arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.Y.; Shi, W.T.; Zhang, J.; Zhao, W.R.; Xiao, Y.; Zhang, K.Y.; Ma, J.; Tang, J.Y.; Wang, Y. Sodium tanshinone IIA sulfonate protects against hyperhomocysteine-induced vascular endothelial injury via activation of NNMT/SIRT1-mediated NRF2/HO-1 and AKT/MAPKs signaling in human umbilical vascular endothelial cells. Biomed. Pharmacother. Biomed. Pharmacother. 2023, 158, 114137. [Google Scholar] [CrossRef]
- Hannibal, L.; Blom, H.J. Homocysteine and disease: Causal associations or epiphenomenons? Mol. Aspects Med. 2017, 53, 36–42. [Google Scholar] [CrossRef]
- Zhang, Q.; Li, Q.; Chen, Y.; Huang, X.; Yang, I.H.; Cao, L.; Wu, W.K.; Tan, H.M. Homocysteine-impaired angiogenesis is associated with VEGF/VEGFR inhibition. Front. Biosci. 2012, 4, 2525–2535. [Google Scholar] [CrossRef]
- Pan, L.; Yu, G.; Huang, J.; Zheng, X.; Xu, Y. Homocysteine inhibits angiogenesis through cytoskeleton remodeling. Biosci. Rep. 2017, 37, BSR20170860. [Google Scholar] [CrossRef]
- Sim, W.C.; Han, I.; Lee, W.; Choi, Y.J.; Lee, K.Y.; Kim, D.G.; Jung, S.H.; Oh, S.H.; Lee, B.H. Inhibition of homocysteine-induced endoplasmic reticulum stress and endothelial cell damage by l-serine and glycine. Toxicol. Vitr. 2016, 34, 138–145. [Google Scholar] [CrossRef]
- Wang, H.; Yoshizumi, M.; Lai, K.; Tsai, J.C.; Perrella, M.A.; Haber, E.; Lee, M.E. Inhibition of growth and p21ras methylation in vascular endothelial cells by homocysteine but not cysteine. J. Biol. Chem. 1997, 272, 25380–25385. [Google Scholar] [CrossRef]
- Zhang, Y.; Ouyang, J.; Zhan, L.; Li, Y.; Li, S.; He, Y.; Wang, H.; Zhang, X. Autophagy in homocysteine-induced HUVEC senescence. Exp. Ther. Med. 2023, 26, 354. [Google Scholar] [CrossRef] [PubMed]
- Aminzadeh, A.; Mehrzadi, S. Melatonin attenuates homocysteine-induced injury in human umbilical vein endothelial cells. Fundam. Clin. Pharmacol. 2018, 32, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Liu, L.; Zhou, K.; Ding, L.; Zeng, J.; Zhang, W. Anti-Oxidant and Anti-Endothelial Dysfunctional Properties of Nano-Selenium in vitro and in vivo of Hyperhomocysteinemic Rats. Int. J. Nanomed. 2020, 15, 4501–4521. [Google Scholar] [CrossRef] [PubMed]
- Angelova, P.R.; Esteras, N.; Abramov, A.Y. Mitochondria and lipid peroxidation in the mechanism of neurodegeneration: Finding ways for prevention. Med. Res. Rev. 2021, 41, 770–784. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Ma, X.; An, Y.; Sun, Y.; Dou, W.; Li, M.; Bao, H.; Zhang, C. Green Tea Polyphenols Alleviate Hydrogen Peroxide-Induced Oxidative Stress, Inflammation, and Apoptosis in Bovine Mammary Epithelial Cells by Activating ERK1/2-NFE2L2-HMOX1 Pathways. Front. Vet. Sci. 2021, 8, 804241. [Google Scholar] [CrossRef]
- Meng, F.; Tan, L.; Cai, P.; Wang, J. Effects of polystyrene nanoplastics on growth and hemolysin production of microalgae Karlodinium veneficum. Aquat. Toxicol. 2024, 266, 106810. [Google Scholar] [CrossRef]
- Yan, Y.; Teng, H.; Hang, Q.; Kondiparthi, L.; Lei, G.; Horbath, A.; Liu, X.; Mao, C.; Wu, S.; Zhuang, L.; et al. SLC7A11 expression level dictates differential responses to oxidative stress in cancer cells. Nat. Commun. 2023, 14, 3673. [Google Scholar] [CrossRef]
- Jiang, Y.; Sun, M. SLC7A11: The Achilles heel of tumor? Front. Immunol. 2024, 15, 1438807. [Google Scholar] [CrossRef]
- Koppula, P.; Zhuang, L.; Gan, B. Cystine transporter SLC7A11/xCT in cancer: Ferroptosis, nutrient dependency, and cancer therapy. Protein Cell 2021, 12, 599–620. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Chu, C.; Wang, X.; Yang, C.; Chen, F.; Shi, L.; Xu, W.; Wang, K.; Liu, B.; Wang, C.; Sun, D.; et al. Neutrophil extracellular traps drive intestinal microvascular endothelial ferroptosis by impairing Fundc1-dependent mitophagy. Redox Biol. 2023, 67, 102906. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.J.; Frazer, D.M. Current understanding of iron homeostasis. Am. J. Clin. Nutr. 2017, 106, 1559s–1566s. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.X.; Zhang, B.; Xia, R.; Jia, Q.Y. Inflammation, apoptosis and autophagy as critical players in vascular dementia. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 9601–9614. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhu, R.; Zhang, L. Triclosan stimulates human vascular endothelial cell injury via repression of the PI3K/Akt/mTOR axis. Chemosphere 2020, 241, 125077. [Google Scholar] [CrossRef]
- Wei, B.; Sun, C.; Wan, H.; Shou, Q.; Han, B.; Sheng, M.; Li, L.; Kai, G. Bioactive components and molecular mechanisms of Salvia miltiorrhiza Bunge in promoting blood circulation to remove blood stasis. J. Ethnopharmacol. 2023, 317, 116697. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| transferrin | 5′-AGCCTGCACTTTCCGTAGAC-3′ | 5′-AACCACTTGGGCCAGTGAAA-3′ |
| transferrin receptor | 5′-GTTAGGGGCCGCCATCC-3′ | 5′-AAGAACACTAGCGCGTCCTC-3′ |
| ferritin light chain | 5′-CACCTGACCAACCTCCACAG-3′ | 5′-CGTGCTTGAGAGTGAGCCTT-3′ |
| ferritin heavy chain | 5′-ACTTTGACCGCGATGATGTG-3′ | 5′-CCTGAAGGAAGATTCGGCCA-3′ |
| ferroportin | 5′-TCAGTTTGCAACATGTCTGTACC-3′ | 5′-GCAACGTATTGCAGTCTCCAT-3′ |
| SLC7A11 | 5′-TGACTGGAGTCCCTGCGTAT-3′ | 5′-TCTTCTTCTGGTACAACTTCCAGT-3′ |
| GPX4 | 5’-CAGTGAGGCAAGACCGAAGT-3’ | 5’-CCGAACTGGTTACACGGGAA-3’ |
| β-actin | 5′-GGGCATGGGTCAGAAGGATT-3′ | 5′-TCGATGGGGTACTTCAGGGT-3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, W.; Zhang, J.; Zhao, W.; Yue, M.; Ma, J.; Zeng, S.; Tang, J.; Wang, Y.; Zhou, Z. Intracellular Iron Deficiency and Abnormal Metabolism, Not Ferroptosis, Contributes to Homocysteine-Induced Vascular Endothelial Cell Death. Biomedicines 2024, 12, 2301. https://doi.org/10.3390/biomedicines12102301
Shi W, Zhang J, Zhao W, Yue M, Ma J, Zeng S, Tang J, Wang Y, Zhou Z. Intracellular Iron Deficiency and Abnormal Metabolism, Not Ferroptosis, Contributes to Homocysteine-Induced Vascular Endothelial Cell Death. Biomedicines. 2024; 12(10):2301. https://doi.org/10.3390/biomedicines12102301
Chicago/Turabian StyleShi, Wenting, Jing Zhang, Wairong Zhao, Meiyan Yue, Jie Ma, Silu Zeng, Jingyi Tang, Yu Wang, and Zhongyan Zhou. 2024. "Intracellular Iron Deficiency and Abnormal Metabolism, Not Ferroptosis, Contributes to Homocysteine-Induced Vascular Endothelial Cell Death" Biomedicines 12, no. 10: 2301. https://doi.org/10.3390/biomedicines12102301
APA StyleShi, W., Zhang, J., Zhao, W., Yue, M., Ma, J., Zeng, S., Tang, J., Wang, Y., & Zhou, Z. (2024). Intracellular Iron Deficiency and Abnormal Metabolism, Not Ferroptosis, Contributes to Homocysteine-Induced Vascular Endothelial Cell Death. Biomedicines, 12(10), 2301. https://doi.org/10.3390/biomedicines12102301