
Macrophages as Potential Therapeutic Targets in Acute Myeloid Leukemia
 , , , and
, , , and
Abstract
1. Introduction
2. The Functions of Macrophages
3. Activation of Macrophages
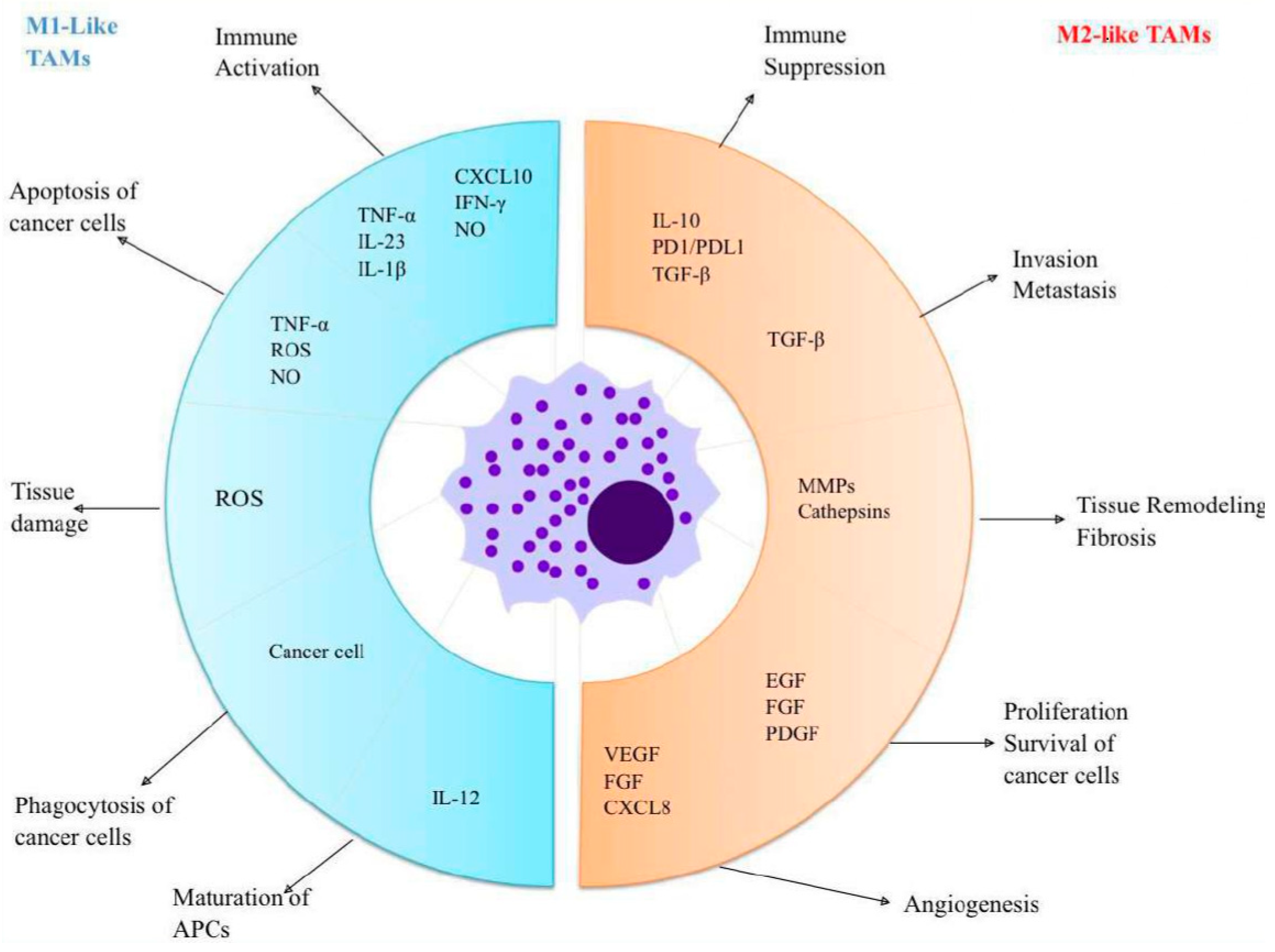
4. Tumor-Associated Macrophages
5. M2 Macrophages in AML
6. Therapeutic Targets of Macrophages
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Saultz, J.N.; Garzon, R. Acute Myeloid Leukemia: A Concise Review. J. Clin. Med. 2016, 5, 33. [Google Scholar] [CrossRef] [PubMed]
- Franklin, R.A.; Liao, W.; Sarkar, A.; Kim, M.V.; Bivona, M.R.; Liu, K.; Pamer, E.G.; Li, M.O. The Cellular and Molecular Origin of Tumor-Associated Macrophages. Science 2014, 344, 921–925. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zheng, G. Macrophages in Leukemia Microenvironment. Blood Sci. 2019, 1, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Mosser, D.M.; Edwards, J.P. Exploring the Full Spectrum of Macrophage Activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Li, Y.; You, M.J.; Yang, Y.; Hu, D.; Tian, C. The Role of Tumor-Associated Macrophages in Leukemia. Acta Haematol. 2020, 143, 112–117. [Google Scholar] [CrossRef]
- Cooper, M.D.; Alder, M.N. The Evolution of Adaptive Immune Systems. Cell 2006, 124, 815–822. [Google Scholar] [CrossRef]
- Tauber, A.I. Metchnikoff and the Phagocytosis Theory. Nat. Rev. Mol. Cell Biol. 2003, 4, 897–901. [Google Scholar] [CrossRef]
- van Furth, R. Origin and Kinetics of Monocytes and Macrophages. Semin. Hematol. 1970, 7, 125–141. [Google Scholar]
- Volkman, A.; Chang, N.C.; Strausbauch, P.H.; Morahan, P.S. Differential Effects of Chronic Monocyte Depletion on Macrophage Populations. Lab. Investig. 1983, 49, 291–298. [Google Scholar]
- Sawyer, R.T.; Strausbauch, P.H.; Volkman, A. Resident Macrophage Proliferation in Mice Depleted of Blood Monocytes by Strontium-89. Lab. Investig. 1982, 46, 165–170. [Google Scholar]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate Mapping Analysis Reveals That Adult Microglia Derive from Primitive Macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [PubMed]
- Schulz, C.; Perdiguero, E.G.; Chorro, L.; Szabo-Rogers, H.; Cagnard, N.; Kierdorf, K.; Prinz, M.; Wu, B.; Jacobsen, S.E.; Pollard, J.W.; et al. A Lineage of Myeloid Cells Independent of Myb and Hematopoietic Stem Cells. Science 2012, 336, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Yona, S.; Kim, K.W.; Wolf, Y.; Mildner, A.; Varol, D.; Breker, M.; Strauss-Ayali, D.; Viukov, S.; Guilliams, M.; Misharin, A.; et al. Fate Mapping Reveals Origins and Dynamics of Monocytes and Tissue Macrophages under Homeostasis. Immunity 2013, 38, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Epelman, S.; Lavine, K.J.; Beaudin, A.E.; Sojka, D.K.; Carrero, J.A.; Calderon, B.; Brija, T.; Gautier, E.L.; Ivanov, S.; Satpathy, A.T.; et al. Embryonic and Adult-Derived Resident Cardiac Macrophages Are Maintained through Distinct Mechanisms at Steady State and During Inflammation. Immunity 2014, 40, 91–104. [Google Scholar] [CrossRef]
- Hashimoto, D.; Chow, A.; Noizat, C.; Teo, P.; Beasley, M.B.; Leboeuf, M.; Becker, C.D.; See, P.; Price, J.; Lucas, D.; et al. Tissue-Resident Macrophages Self-Maintain Locally Throughout Adult Life with Minimal Contribution from Circulating Monocytes. Immunity 2013, 38, 792–804. [Google Scholar] [CrossRef]
- Guilliams, M.; De Kleer, I.; Henri, S.; Post, S.; Vanhoutte, L.; De Prijck, S.; Deswarte, K.; Malissen, B.; Hammad, H.; Lambrecht, B.N. Alveolar Macrophages Develop from Fetal Monocytes That Differentiate into Long-Lived Cells in the First Week of Life Via Gm-Csf. J. Exp. Med. 2013, 210, 1977–1992. [Google Scholar] [CrossRef]
- Jakubzick, C.; Gautier, E.L.; Gibbings, S.L.; Sojka, D.K.; Schlitzer, A.; Johnson, T.E.; Ivanov, S.; Duan, Q.; Bala, S.; Condon, T.; et al. Minimal Differentiation of Classical Monocytes as They Survey Steady-State Tissues and Transport Antigen to Lymph Nodes. Immunity 2013, 39, 599–610. [Google Scholar] [CrossRef]
- Gordon, S.; Martinez, F.O. Alternative Activation of Macrophages: Mechanism and Functions. Immunity 2010, 32, 593–604. [Google Scholar] [CrossRef]
- Yao, Y.; Xu, X.H.; Jin, L. Macrophage Polarization in Physiological and Pathological Pregnancy. Front. Immunol. 2019, 10, 792. [Google Scholar] [CrossRef]
- Janeway, C.A., Jr. How the Immune System Works to Protect the Host from Infection: A Personal View. Proc. Natl. Acad. Sci. USA 2001, 98, 7461–7468. [Google Scholar] [CrossRef]
- Arango Duque, G.; Descoteaux, A. Macrophage Cytokines: Involvement in Immunity and Infectious Diseases. Front. Immunol. 2014, 5, 491. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, D.; Iida, T.; Nakase, H. The Phagocytic Function of Macrophage-Enforcing Innate Immunity and Tissue Homeostasis. Int. J. Mol. Sci. 2017, 19, 92. [Google Scholar] [CrossRef] [PubMed]
- Unanue, E.R.; Beller, D.I.; Calderon, J.; Kiely, J.M.; Stadecker, M.J. Regulation of Immunity and Inflammation by Mediators from Macrophages. Am. J. Pathol. 1976, 85, 465–478. [Google Scholar] [PubMed]
- Jackaman, C.; Tomay, F.; Duong, L.; Razak, N.B.A.; Pixley, F.J.; Metharom, P.; Nelson, D.J. Aging and Cancer: The Role of Macrophages and Neutrophils. Ageing Res. Rev. 2017, 36, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Kraus, R.F.; Gruber, M.A. Neutrophils-from Bone Marrow to First-Line Defense of the Innate Immune System. Front. Immunol. 2021, 12, 767175. [Google Scholar] [CrossRef]
- Beutler, B.A. The Role of Tumor Necrosis Factor in Health and Disease. J. Rheumatol. Suppl. 1999, 57, 16–21. [Google Scholar]
- Pace, J.L.; Russell, S.W.; Schreiber, R.D.; Altman, A.; Katz, D.H. Macrophage Activation: Priming Activity from a T-Cell Hybridoma Is Attributable to Interferon-Gamma. Proc. Natl. Acad. Sci. USA 1983, 80, 3782–3786. [Google Scholar] [CrossRef]
- Italiani, P.; Boraschi, D. From Monocytes to M1/M2 Macrophages: Phenotypical Vs. Functional Differentiation. Front. Immunol. 2014, 5, 514. [Google Scholar] [CrossRef]
- Gordon, S. Phagocytosis: An Immunobiologic Process. Immunity 2016, 44, 463–475. [Google Scholar] [CrossRef]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 Macrophages and the Th1/Th2 Paradigm. J. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef]
- Patel, U.; Rajasingh, S.; Samanta, S.; Cao, T.; Dawn, B.; Rajasingh, J. Macrophage Polarization in Response to Epigenetic Modifiers During Infection and Inflammation. Drug Discov. Today 2017, 22, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef] [PubMed]
- Porta, C.; Riboldi, E.; Ippolito, A.; Sica, A. Molecular and Epigenetic Basis of Macrophage Polarized Activation. Semin. Immunol. 2015, 27, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Mulder, R.; Banete, A.; Basta, S. Spleen-Derived Macrophages Are Readily Polarized into Classically Activated (M1) or Alternatively Activated (M2) States. Immunobiology 2014, 219, 737–745. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J.J.; Paul, W.E. Mechanisms Underlying Lineage Commitment and Plasticity of Helper Cd4+ T Cells. Science 2010, 327, 1098–1102. [Google Scholar] [CrossRef]
- Wang, N.; Liang, H.; Zen, K. Molecular Mechanisms That Influence the Macrophage M1-M2 Polarization Balance. Front. Immunol. 2014, 5, 614. [Google Scholar] [CrossRef]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Macrophage Polarization: Different Gene Signatures in M1(Lps+) Vs. Classically and M2(Lps-) Vs. Alternatively Activated Macrophages. Front. Immunol. 2019, 10, 1084. [Google Scholar] [CrossRef]
- Murray, P.J.; Smale, S.T. Restraint of Inflammatory Signaling by Interdependent Strata of Negative Regulatory Pathways. Nat. Immunol. 2012, 13, 916–924. [Google Scholar] [CrossRef]
- Graff, J.W.; Dickson, A.M.; Clay, G.; McCaffrey, A.P.; Wilson, M.E. Identifying Functional Micrornas in Macrophages with Polarized Phenotypes. J. Biol. Chem. 2012, 287, 21816–21825. [Google Scholar] [CrossRef]
- Yang, X.; Feng, W.; Wang, R.; Yang, F.; Wang, L.; Chen, S.; Ru, Y.; Cheng, T.; Zheng, G. Repolarizing Heterogeneous Leukemia-Associated Macrophages with More M1 Characteristics Eliminates Their Pro-Leukemic Effects. Oncoimmunology 2018, 7, e1412910. [Google Scholar] [CrossRef]
- Wang, L.X.; Zhang, S.X.; Wu, H.J.; Rong, X.L.; Guo, J. M2b Macrophage Polarization and Its Roles in Diseases. J. Leukoc. Biol. 2019, 106, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Mesaros, O.; Jimbu, L.; Neaga, A.; Popescu, C.; Berceanu, I.; Tomuleasa, C.; Fetica, B.; Zdrenghea, M. Macrophage Polarization in Chronic Lymphocytic Leukemia: Nurse-Like Cells Are the Caretakers of Leukemic Cells. Biomedicines 2020, 8, 516. [Google Scholar] [CrossRef] [PubMed]
- Atri, C.; Guerfali, F.Z.; Laouini, D. Role of Human Macrophage Polarization in Inflammation During Infectious Diseases. Int. J. Mol. Sci. 2018, 19, 1801. [Google Scholar] [CrossRef] [PubMed]
- Ferrante, C.J.; Pinhal-Enfield, G.; Elson, G.; Cronstein, B.N.; Hasko, G.; Outram, S.; Leibovich, S.J. The Adenosine-Dependent Angiogenic Switch of Macrophages to an M2-Like Phenotype Is Independent of Interleukin-4 Receptor Alpha (Il-4ralpha) Signaling. Inflammation 2013, 36, 921–931. [Google Scholar] [CrossRef] [PubMed]
- Zizzo, G.; Hilliard, B.A.; Monestier, M.; Cohen, P.L. Efficient Clearance of Early Apoptotic Cells by Human Macrophages Requires M2c Polarization and Mertk Induction. J. Immunol. 2012, 189, 3508–3520. [Google Scholar] [CrossRef]
- Akinduro, O.; Weber, T.S.; Ang, H.; Haltalli, M.L.R.; Ruivo, N.; Duarte, D.; Rashidi, N.M.; Hawkins, E.D.; Duffy, K.R.; Celso, C.L. Proliferation Dynamics of Acute Myeloid Leukaemia and Haematopoietic Progenitors Competing for Bone Marrow Space. Nat. Commun. 2018, 9, 519. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, Y. Tumor-Associated Macrophages: From Basic Research to Clinical Application. J. Hematol. Oncol. 2017, 10, 58. [Google Scholar] [CrossRef]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-Associated Macrophages as Treatment Targets in Oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Riabov, V.; Gudima, A.; Wang, N.; Mickley, A.; Orekhov, A.; Kzhyshkowska, J. Role of Tumor Associated Macrophages in Tumor Angiogenesis and Lymphangiogenesis. Front. Physiol. 2014, 5, 75. [Google Scholar] [CrossRef]
- He, Z.; Zhang, S. Tumor-Associated Macrophages and Their Functional Transformation in the Hypoxic Tumor Microenvironment. Front. Immunol. 2021, 12, 741305. [Google Scholar] [CrossRef]
- Zhukova, O.V.; Kovaleva, T.F.; Arkhipova, E.V.; Ryabov, S.A.; Mukhina, I.V. Tumor-Associated Macrophages: Role in the Pathological Process of Tumorigenesis and Prospective Therapeutic Use (Review). Biomed. Rep. 2020, 13, 47. [Google Scholar] [CrossRef] [PubMed]
- Larionova, I.; Tuguzbaeva, G.; Ponomaryova, A.; Stakheyeva, M.; Cherdyntseva, N.; Pavlov, V.; Choinzonov, E.; Kzhyshkowska, J. Tumor-Associated Macrophages in Human Breast, Colorectal, Lung, Ovarian and Prostate Cancers. Front. Oncol. 2020, 10, 566511. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Yang, C.; Wang, S.; Shi, D.; Zhang, C.; Lin, X.; Xiong, B. M2 Macrophages Confer Resistance to 5-Fluorouracil in Colorectal Cancer through the Activation of Ccl22/Pi3k/Akt Signaling. Onco Targets Ther. 2019, 12, 3051–3063. [Google Scholar] [CrossRef] [PubMed]
- Pei, B.X.; BSun, S.; Zhang, Z.F.; Wang, A.L.; Ren, P. Interstitial Tumor-Associated Macrophages Combined with Tumor-Derived Colony-Stimulating Factor-1 and Interleukin-6, a Novel Prognostic Biomarker in Non-Small Cell Lung Cancer. J. Thorac. Cardiovasc. Surg. 2014, 148, 1208–1216 e2. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Chen, B.; Yang, Z. The Role of Tumor-Associated Macrophages in Colorectal Carcinoma Progression. Cell Physiol. Biochem. 2014, 45, 356–365. [Google Scholar] [CrossRef]
- Jafarzadeh, N.; Safari, Z.; Pornour, M.; Amirizadeh, N.; Moghadam, M.F.; Sadeghizadeh, M. Alteration of Cellular and Immune-Related Properties of Bone Marrow Mesenchymal Stem Cells and Macrophages by K562 Chronic Myeloid Leukemia Cell Derived Exosomes. J. Cell Physiol. 2019, 234, 3697–3710. [Google Scholar] [CrossRef]
- Petty, A.J.; Yang, Y. Tumor-Associated Macrophages: Implications in Cancer Immunotherapy. Immunotherapy 2017, 9, 289–302. [Google Scholar] [CrossRef]
- Qian, B.Z.; Pollard, J.W. Macrophage Diversity Enhances Tumor Progression and Metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef]
- Sumitomo, R.; Hirai, T.; Fujita, M.; Murakami, H.; Otake, Y.; Huang, C.L. M2 Tumor-Associated Macrophages Promote Tumor Progression in Non-Small-Cell Lung Cancer. Exp. Ther. Med. 2019, 18, 4490–4498. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage Plasticity and Polarization: In Vivo Veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Shimizu, K.; Iyoda, T.; Okada, M.; Yamasaki, S.; Fujii, S.I. Immune Suppression and Reversal of the Suppressive Tumor Microenvironment. Int. Immunol. 2018, 30, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Solinas, G.; Germano, G.; Mantovani, A.; Allavena, P. Tumor-Associated Macrophages (Tam) as Major Players of the Cancer-Related Inflammation. J. Leukoc. Biol. 2009, 86, 1065–1073. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Schioppa, T.; Mantovani, A.; Allavena, P. Tumour-Associated Macrophages Are a Distinct M2 Polarised Population Promoting Tumour Progression: Potential Targets of Anti-Cancer Therapy. Eur. J. Cancer 2006, 42, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Halder, K.; Bose, A.; Bhattacharya, P.; Gupta, G.; Karmahapatra, S.; Das, S.; Chaudhuri, S.; Majumdar, S.B.; Majumdar, S. Tlr Signaling-Mediated Differential Histone Modification at Il-10 and Il-12 Promoter Region Leads to Functional Impairments in Tumor-Associated Macrophages. Carcinogenesis 2011, 32, 1789–1797. [Google Scholar] [CrossRef]
- Chen, Y.; Song, Y.; Du, W.; Gong, L.; Chang, H.; Zou, Z. Tumor-Associated Macrophages: An Accomplice in Solid Tumor Progression. J. Biomed. Sci. 2019, 26, 78. [Google Scholar] [CrossRef]
- Lampiasi, N.; Russo, R.; Zito, F. The Alternative Faces of Macrophage Generate Osteoclasts. Biomed Res. Int. 2016, 2016, 9089610. [Google Scholar] [CrossRef]
- Shirabe, K.; Mano, Y.; Muto, J.; Matono, R.; Motomura, T.; Toshima, T.; Takeishi, K.; Uchiyama, H.; Yoshizumi, T.; Taketomi, A.; et al. Role of Tumor-Associated Macrophages in the Progression of Hepatocellular Carcinoma. Surg. Today 2012, 42, 1–7. [Google Scholar] [CrossRef]
- Addison, C.L.; Arenberg, D.A.; Morris, S.B.; Xue, Y.Y.; Burdick, M.D.; Mulligan, M.S.; Iannettoni, M.D.; Strieter, R.M. The Cxc Chemokine, Monokine Induced by Interferon-Gamma, Inhibits Non-Small Cell Lung Carcinoma Tumor Growth and Metastasis. Hum. Gene Ther. 2000, 11, 247–261. [Google Scholar] [CrossRef]
- Ruytinx, P.; Proost, P.; Van Damme, J.; Struyf, S. Chemokine-Induced Macrophage Polarization in Inflammatory Conditions. Front. Immunol. 2018, 9, 1930. [Google Scholar] [CrossRef]
- Sica, A.; Saccani, A.; Bottazzi, B.; Bernasconi, S.; Allavena, P.; Gaetano, B.; Fei, F.; LaRosa, G.; Scotton, C.; Balkwill, F.; et al. Defective Expression of the Monocyte Chemotactic Protein-1 Receptor Ccr2 in Macrophages Associated with Human Ovarian Carcinoma. J. Immunol. 2000, 164, 733–738. [Google Scholar] [CrossRef]
- Li, Y.; Zheng, Y.; Li, T.; Wang, Q.; Qian, J.; Lu, Y.; Zhang, M.; Bi, E.; Yang, M.; Reu, F.; et al. Chemokines Ccl2, 3, 14 Stimulate Macrophage Bone Marrow Homing, Proliferation; Polarization in Multiple Myeloma. Oncotarget 2015, 6, 24218–24229. [Google Scholar] [CrossRef] [PubMed]
- Pimenta, D.B.; Varela, V.A.; Datoguia, T.S.; Caraciolo, V.B.; Lopes, G.H.; Pereira, W.O. The Bone Marrow Microenvironment Mechanisms in Acute Myeloid Leukemia. Front. Cell Dev. Biol. 2021, 9, 764698. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, J.; Chen, Z.; Luo, J.; Guo, W.; Sun, L.; Lin, L. Targeting M2-Like Tumor-Associated Macrophages Is a Potential Therapeutic Approach to Overcome Antitumor Drug Resistance. NPJ Precis. Oncol. 2024, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Gao, W.; Su, M.; Nice, E.C.; Zhang, W.; Lin, J.; Xie, N. Adaptive Mechanisms of Tumor Therapy Resistance Driven by Tumor Microenvironment. Front. Cell Dev. Biol. 2021, 9, 641469. [Google Scholar] [CrossRef]
- Li, M.; He, L.; Zhu, J.; Zhang, P.; Liang, S. Targeting Tumor-Associated Macrophages for Cancer Treatment. Cell Biosci. 2022, 12, 85. [Google Scholar] [CrossRef]
- Wang, H.; Wang, X.; Zhang, X.; Xu, W. The Promising Role of Tumor-Associated Macrophages in the Treatment of Cancer. Drug Resist. Updates 2024, 73, 101041. [Google Scholar] [CrossRef]
- Tang, X. Tumor-Associated Macrophages as Potential Diagnostic and Prognostic Biomarkers in Breast Cancer. Cancer Lett. 2013, 332, 3–10. [Google Scholar] [CrossRef]
- Chen, Y.L. Prognostic Significance of Tumor-Associated Macrophages in Patients with Nasopharyngeal Carcinoma: A Meta-Analysis. Medicine 2020, 99, e21999. [Google Scholar] [CrossRef]
- Hadiloo, K.; Taremi, S.; Heidari, M.; Esmaeilzadeh, A. The Car Macrophage Cells, a Novel Generation of Chimeric Antigen-Based Approach against Solid Tumors. Biomark. Res. 2023, 11, 103. [Google Scholar] [CrossRef]
- Li, W.; Wang, F.; Guo, R.; Bian, Z.; Song, Y. Targeting Macrophages in Hematological Malignancies: Recent Advances and Future Directions. J. Hematol. Oncol. 2022, 15, 110. [Google Scholar] [CrossRef]
- Chulpanova, D.S.; Kitaeva, K.V.; Green, A.R.; Rizvanov, A.A.; Solovyeva, V.V. Molecular Aspects and Future Perspectives of Cytokine-Based Anti-Cancer Immunotherapy. Front. Cell Dev. Biol. 2020, 8, 402. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.J.; Gu, Y.; Wang, C.Z.; Jin, Y.; Wen, X.M.; Ma, J.C.; Tang, L.J.; Mao, Z.W.; Qian, J.; Lin, J. The M2 Macrophage Marker Cd206: A Novel Prognostic Indicator for Acute Myeloid Leukemia. Oncoimmunology 2020, 9, 1683347. [Google Scholar] [CrossRef] [PubMed]
- Barnes, N.G. Targeting Tams. Nat. Rev. Chem. 2022, 6, 678. [Google Scholar] [CrossRef] [PubMed]
- Hino, C.; Pham, B.; Park, D.; Yang, C.; Nguyen, M.H.K.; Kaur, S.; Reeves, M.E.; Xu, Y.; Nishino, K.; Pu, L.; et al. Targeting the Tumor Microenvironment in Acute Myeloid Leukemia: The Future of Immunotherapy and Natural Products. Biomedicines 2022, 10, 1410. [Google Scholar] [CrossRef]
- Jalte, M.; Abbassi, M.; El Mouhi, H.; Belghiti, H.D.; Ahakoud, M.; Bekkari, H. Flt3 Mutations in Acute Myeloid Leukemia: Unraveling the Molecular Mechanisms and Implications for Targeted Therapies. Cureus 2023, 15, e45765. [Google Scholar] [CrossRef]
- Nepstad, I.; Hatfield, K.J.; Gronningsaeter, I.S.; Reikvam, H. The Pi3k-Akt-Mtor Signaling Pathway in Human Acute Myeloid Leukemia (Aml) Cells. Int. J. Mol. Sci. 2020, 21, 2907. [Google Scholar] [CrossRef]
- Nepstad, I.; Hatfield, K.J.; Gronningsaeter, I.S.; Aasebo, E.; Hernandez-Valladares, M.; Hagen, K.M.; Rye, K.P.; Berven, F.S.; Selheim, F.; Reikvam, H.; et al. Effects of Insulin and Pathway Inhibitors on the Pi3k-Akt-Mtor Phosphorylation Profile in Acute Myeloid Leukemia Cells. Signal Transduct. Target. Ther. 2019, 4, 20. [Google Scholar] [CrossRef]
- Pillinger, G.; Loughran, N.V.; Piddock, R.E.; Shafat, M.S.; Zaitseva, L.; Abdul-Aziz, A.; Lawes, M.J.; Bowles, K.M.; Rushworth, S.A. Targeting Pi3kdelta and Pi3kgamma Signalling Disrupts Human Aml Survival and Bone Marrow Stromal Cell Mediated Protection. Oncotarget 2016, 7, 39784–39795. [Google Scholar] [CrossRef]
- He, Y.; Sun, M.M.; Zhang, G.G.; Yang, J.; Chen, K.S.; Xu, W.W.; Li, B. Targeting Pi3k/Akt Signal Transduction for Cancer Therapy. Signal Transduct. Target. Ther. 2021, 6, 425. [Google Scholar] [CrossRef]
- Hallowell, R.W.; Collins, S.L.; Craig, J.M.; Zhang, Y.; Oh, M.; Illei, P.B.; Chan-Li, Y.; Vigeland, C.L.; Mitzner, W.; Scott, A.L.; et al. Mtorc2 Signalling Regulates M2 Macrophage Differentiation in Response to Helminth Infection and Adaptive Thermogenesis. Nat. Commun. 2017, 8, 14208. [Google Scholar] [CrossRef]
- Collins, S.L.; Oh, M.H.; Sun, I.H.; Chan-Li, Y.; Zhao, L.; Powell, J.D.; Horton, M.R. Mtorc1 Signaling Regulates Proinflammatory Macrophage Function and Metabolism. J. Immunol. 2021, 207, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Oki, T.; Mercier, F.; Kato, H.; Jung, Y.; McDonald, T.O.; Spencer, J.A.; Mazzola, M.C.; van Gastel, N.; Lin, C.P.; Michor, F.; et al. Imaging Dynamic Mtorc1 Pathway Activity in Vivo Reveals Marked Shifts That Support Time-Specific Inhibitor Therapy in Aml. Nat. Commun. 2021, 12, 245. [Google Scholar] [CrossRef] [PubMed]
- Mussai, F.; De Santo, C.; Abu-Dayyeh, I.; Booth, S.; Quek, L.; McEwen-Smith, R.M.; Qureshi, A.; Dazzi, F.; Vyas, P.; Cerundolo, V. Acute Myeloid Leukemia Creates an Arginase-Dependent Immunosuppressive Microenvironment. Blood 2013, 122, 749–758. [Google Scholar] [CrossRef]
- Herschbein, L.; Liesveld, J.L. Dueling for Dual Inhibition: Means to Enhance Effectiveness of Pi3k/Akt/Mtor Inhibitors in Aml. Blood Rev. 2018, 32, 235–248. [Google Scholar] [CrossRef]
- Al-Matary, Y.S.; Botezatu, L.; Opalka, B.; Hones, J.M.; Lams, R.F.; Thivakaran, A.; Schutte, J.; Koster, R.; Lennartz, K.; Schroeder, T.; et al. Acute Myeloid Leukemia Cells Polarize Macrophages Towards a Leukemia Supporting State in a Growth Factor Independence 1 Dependent Manner. Haematologica 2016, 101, 1216–1227. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Zhang, J.; Qian, L.; Miao, Y.; Song, W.; Liu, H.; Li, R. Moz Forms an Autoregulatory Feedback Loop with Mir-223 in Aml and Monocyte/Macrophage Development. iScience 2019, 11, 189–204. [Google Scholar] [CrossRef] [PubMed]
- Thomas, L.W.; Lam, C.; Edwards, S.W. Mcl-1; the Molecular Regulation of Protein Function. FEBS Lett. 2010, 584, 2981–2989. [Google Scholar] [CrossRef]
- Miari, K.E.; Guzman, M.L.; Wheadon, H.; Williams, M.T.S. Macrophages in Acute Myeloid Leukaemia: Significant Players in Therapy Resistance and Patient Outcomes. Front. Cell Dev. Biol. 2021, 9, 692800. [Google Scholar] [CrossRef]
- Mazur, G.; Wrobel, T.; Butrym, A.; Kapelko-Slowik, K.; Poreba, R.; Kuliczkowski, K. Increased Monocyte Chemoattractant Protein 1 (Mcp-1/Ccl-2) Serum Level in Acute Myeloid Leukemia. Neoplasma 2007, 54, 285–289. [Google Scholar]
- Sierra-Filardi, E.; Nieto, C.; Dominguez-Soto, A.; Barroso, R.; Sanchez-Mateos, P.; Puig-Kroger, A.; Lopez-Bravo, M.; Joven, J.; Ardavin, C.; Rodriguez-Fernandez, J.L.; et al. Ccl2 Shapes Macrophage Polarization by Gm-Csf and M-Csf: Identification of Ccl2/Ccr2-Dependent Gene Expression Profile. J. Immunol. 2014, 192, 3858–3867. [Google Scholar] [CrossRef]
- Merle, M.; Fischbacher, D.; Liepert, A.; Grabrucker, C.; Kroell, T.; Kremser, A.; Dreyssig, J.; Freudenreich, M.; Schuster, F.; Borkhardt, A.; et al. Serum Chemokine-Release Profiles in Aml-Patients Might Contribute to Predict the Clinical Course of the Disease. Immunol. Investig. 2020, 49, 365–385. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, O.J.; Johansen, J.S.; Klausen, T.W.; Mylin, A.K.; Kristensen, J.S.; Kjeldsen, E.; Johnsen, H.E. High Serum Concentration of Ykl-40 Is Associated with Short Survival in Patients with Acute Myeloid Leukemia. Clin. Cancer Res. 2005, 11 Pt 1, 8644–8652. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Gu, Z.; Xu, Y.; Jiang, L.; Zhu, W.; Wang, W. Chi3l1 (Chitinase 3 Like 1) Upregulation Is Associated with Macrophage Signatures in Esophageal Cancer. Bioengineered 2021, 12, 7882–7892. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.W.; Chiang, Y.C.; Sun, N.Y.; Chen, Y.L.; Chang, C.F.; Tai, Y.J.; Chen, C.A.; Cheng, W.F. Chi3l1 Results in Poor Outcome of Ovarian Cancer by Promoting Properties of Stem-Like Cells. Endocr. Relat. Cancer 2019, 26, 73–88. [Google Scholar] [CrossRef]
- Zhao, H.; Huang, M.; Jiang, L. Potential Roles and Future Perspectives of Chitinase 3-Like 1 in Macrophage Polarization and the Development of Diseases. Int. J. Mol. Sci. 2023, 24, 16149. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; Kadia, T.M.; DiNardo, C.D.; Welch, M.A.; Ravandi, F. Acute Myeloid Leukemia: Treatment and Research Outlook for 2021 and the Md Anderson Approach. Cancer 2021, 127, 1186–1207. [Google Scholar] [CrossRef]
- Hamdan, S.O.; Sughayer, M.; Khader, M.; Tbakhi, A.; Khudirat, S.; Hejazi, A.; AlRyalat, S.; Bustami, N.; Aladily, T.N. Programmed Death Ligand-1 Is Frequently Expressed in Primary Acute Myeloid Leukemia and B-Acute Lymphoblastic Leukemia. Clin. Lab. 2022, 68, 748–754. [Google Scholar] [CrossRef]
- Marra, A.; Akarca, A.U.; Martino, G.; Ramsay, A.; Ascani, S.; Perriello, V.M.; O’Nions, J.; Wilson, A.J.; Gupta, R.; Childerhouse, A.; et al. Cd47 Expression in Acute Myeloid Leukemia Varies According to Genotype. Haematologica 2023, 108, 3491–3495. [Google Scholar] [CrossRef]
- Marques-Piubelli, M.L.; Kumar, B.; Basar, R.; Panowski, S.; Srinivasan, S.; Norwood, K.; Prashad, S.; Szenes, V.; Balakumaran, A.; Arandhya, A.; et al. Increased Expression of Cd70 in Relapsed Acute Myeloid Leukemia after Hypomethylating Agents. Virchows Arch. 2024. [Google Scholar] [CrossRef]
- Taylor, J.G.; Truelove, E.; Clear, A.; Calaminici, M.; Gribben, J.G. Pdl1 Shapes the Classical Hodgkin Lymphoma Microenvironment without Inducing T-Cell Exhaustion. Haematologica 2023, 108, 1068–1082. [Google Scholar] [CrossRef]
- Jimbu, L.; Mesaros, O.; Neaga, A.; Nanut, A.M.; Tomuleasa, C.; Dima, D.; Bocsan, C.; Zdrenghea, M. The Potential Advantage of Targeting Both Pd-L1/Pd-L2/Pd-1 and Il-10-Il-10r Pathways in Acute Myeloid Leukemia. Pharmaceuticals 2021, 14, 1105. [Google Scholar] [CrossRef] [PubMed]
- Pu, Y.; Ji, Q. Tumor-Associated Macrophages Regulate Pd-1/Pd-L1 Immunosuppression. Front. Immunol. 2022, 13, 874589. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Lu, M.; Cao, F.; Wu, G.; Gao, F.; Pang, H.; Li, Y.; Zhang, Y.; Xing, H.; Liang, C.; et al. Single-Cell Map of Diverse Immune Phenotypes in the Acute Myeloid Leukemia Microenvironment. Biomark. Res. 2021, 9, 15. [Google Scholar] [CrossRef] [PubMed]
- Jayasingam, S.D.; Citartan, M.; Thang, T.H.; Zin, A.A.M.; Ang, K.C.; Ch’ng, E.S. Evaluating the Polarization of Tumor-Associated Macrophages into M1 and M2 Phenotypes in Human Cancer Tissue: Technicalities and Challenges in Routine Clinical Practice. Front. Oncol. 2019, 9, 1512. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.L.; Yang, F.; Huang, Z.Q.; Li, Y.Y.; Shi, H.Y.; Sun, Q.; Ma, Y.; Wang, Y.; Zhang, Y.; Yang, S.; et al. T Cells, Nk Cells; Tumor-Associated Macrophages in Cancer Immunotherapy and the Current State of the Art of Drug Delivery Systems. Front. Immunol. 2023, 14, 1199173. [Google Scholar] [CrossRef]
- Qian, B.Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. Ccl2 Recruits Inflammatory Monocytes to Facilitate Breast-Tumour Metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef]
- Cannarile, M.A.; Weisser, M.; Jacob, W.; Jegg, A.M.; Ries, C.H.; Ruttinger, D. Colony-Stimulating Factor 1 Receptor (Csf1r) Inhibitors in Cancer Therapy. J. Immunother. Cancer 2017, 5, 53. [Google Scholar] [CrossRef]
- Denny, W.A.; Flanagan, J.U. Small-Molecule Csf1r Kinase Inhibitors; Review of Patents 2015-Present. Expert Opin. Ther. Pat. 2021, 31, 107–117. [Google Scholar] [CrossRef]
- Aharinejad, S.; Abraham, D.; Paulus, P.; Abri, H.; Hofmann, M.; Grossschmidt, K.; Schafer, R.; Stanley, E.R.; Hofbauer, R. Colony-Stimulating Factor-1 Antisense Treatment Suppresses Growth of Human Tumor Xenografts in Mice. Cancer Res. 2002, 62, 5317–5324. [Google Scholar]
- Lin, C.C. Clinical Development of Colony-Stimulating Factor 1 Receptor (Csf1r) Inhibitors. J. Immunother. Precis. Oncol. 2021, 4, 105–114. [Google Scholar] [CrossRef]
- Benner, B.; Good, L.; Quiroga, D.; Schultz, T.E.; Kassem, M.; Carson, W.E.; Cherian, M.A.; Sardesai, S.; Wesolowski, R. Pexidartinib, a Novel Small Molecule Csf-1r Inhibitor in Use for Tenosynovial Giant Cell Tumor: A Systematic Review of Pre-Clinical and Clinical Development. Drug Des. Dev. Ther. 2020, 14, 1693–1704. [Google Scholar] [CrossRef] [PubMed]
- Wesolowski, R.; Sharma, N.; Reebel, L.; Rodal, M.B.; Peck, A.; West, B.L.; Marimuthu, A.; Severson, P.; Karlin, D.A.; Dowlati, A.; et al. Phase Ib Study of the Combination of Pexidartinib (Plx3397), a Csf-1r Inhibitor, and Paclitaxel in Patients with Advanced Solid Tumors. Ther. Adv. Med. Oncol. 2019, 11, 1758835919854238. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, E.; Kelly, C.; D’Angelo, S.P.; Dickson, M.A.; Gounder, M.; Keohan, M.L.; Movva, S.; Condy, M.; Adamson, T.; McFadyen, C.R.; et al. A Phase I Study of Binimetinib (Mek162) Combined with Pexidartinib (Plx3397) in Patients with Advanced Gastrointestinal Stromal Tumor. Oncologist 2019, 24, 1309-e983. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, B.A.; Chapin, B.F.; Jindal, S.; Duan, F.; Basu, S.; Yadav, S.S.; Gu, A.D.; Espejo, A.B.; Kinder, M.; Pettaway, C.A.; et al. Immune and Pathologic Responses in Patients with Localized Prostate Cancer Who Received Daratumumab (Anti-Cd38) or Edicotinib (Csf-1r Inhibitor). J. Immunother. Cancer 2023, 11, e006262. [Google Scholar] [CrossRef]
- clinicaltrials.gov. Available online: https://clinicaltrials.gov/study/NCT03557970?cond=acute%20myeloid%20leukemia&intr=CSF1R&viewType=Card&rank=1 (accessed on 31 August 2024).
- D’Incalci, M.; Galmarini, C.M. A Review of Trabectedin (Et-743): A Unique Mechanism of Action. Mol. Cancer Ther. 2010, 9, 2157–2163. [Google Scholar] [CrossRef]
- Herrero, A.B.; Martin-Castellanos, C.; Marco, E.; Gago, F.; Moreno, S. Cross-Talk between Nucleotide Excision and Homologous Recombination DNA Repair Pathways in the Mechanism of Action of Antitumor Trabectedin. Cancer Res. 2006, 66, 8155–8162. [Google Scholar] [CrossRef]
- Liguori, M.; Buracchi, C.; Pasqualini, F.; Bergomas, F.; Pesce, S.; Sironi, M.; Grizzi, F.; Mantovani, A.; Belgiovine, C.; Allavena, P. Functional Trail Receptors in Monocytes and Tumor-Associated Macrophages: A Possible Targeting Pathway in the Tumor Microenvironment. Oncotarget 2016, 7, 41662–41676. [Google Scholar] [CrossRef]
- Germano, G.; Frapolli, R.; Belgiovine, C.; Anselmo, A.; Pesce, S.; Liguori, M.; Erba, E.; Uboldi, S.; Zucchetti, M.; Pasqualini, F.; et al. Role of Macrophage Targeting in the Antitumor Activity of Trabectedin. Cancer Cell 2013, 23, 249–262. [Google Scholar] [CrossRef]
- Rogers, T.L.; Holen, I. Tumour Macrophages as Potential Targets of Bisphosphonates. J. Transl. Med. 2011, 9, 177. [Google Scholar] [CrossRef]
- Van Acker, H.H.; Anguille, S.; Willemen, Y.; Smits, E.L.; Van Tendeloo, V.F. Bisphosphonates for Cancer Treatment: Mechanisms of Action and Lessons from Clinical Trials. Pharmacol. Ther. 2016, 158, 24–40. [Google Scholar] [CrossRef]
- Frith, J.C.; Rogers, M.J. Antagonistic Effects of Different Classes of Bisphosphonates in Osteoclasts and Macrophages in vitro. J. Bone Miner. Res. 2003, 18, 204–212. [Google Scholar] [CrossRef] [PubMed]
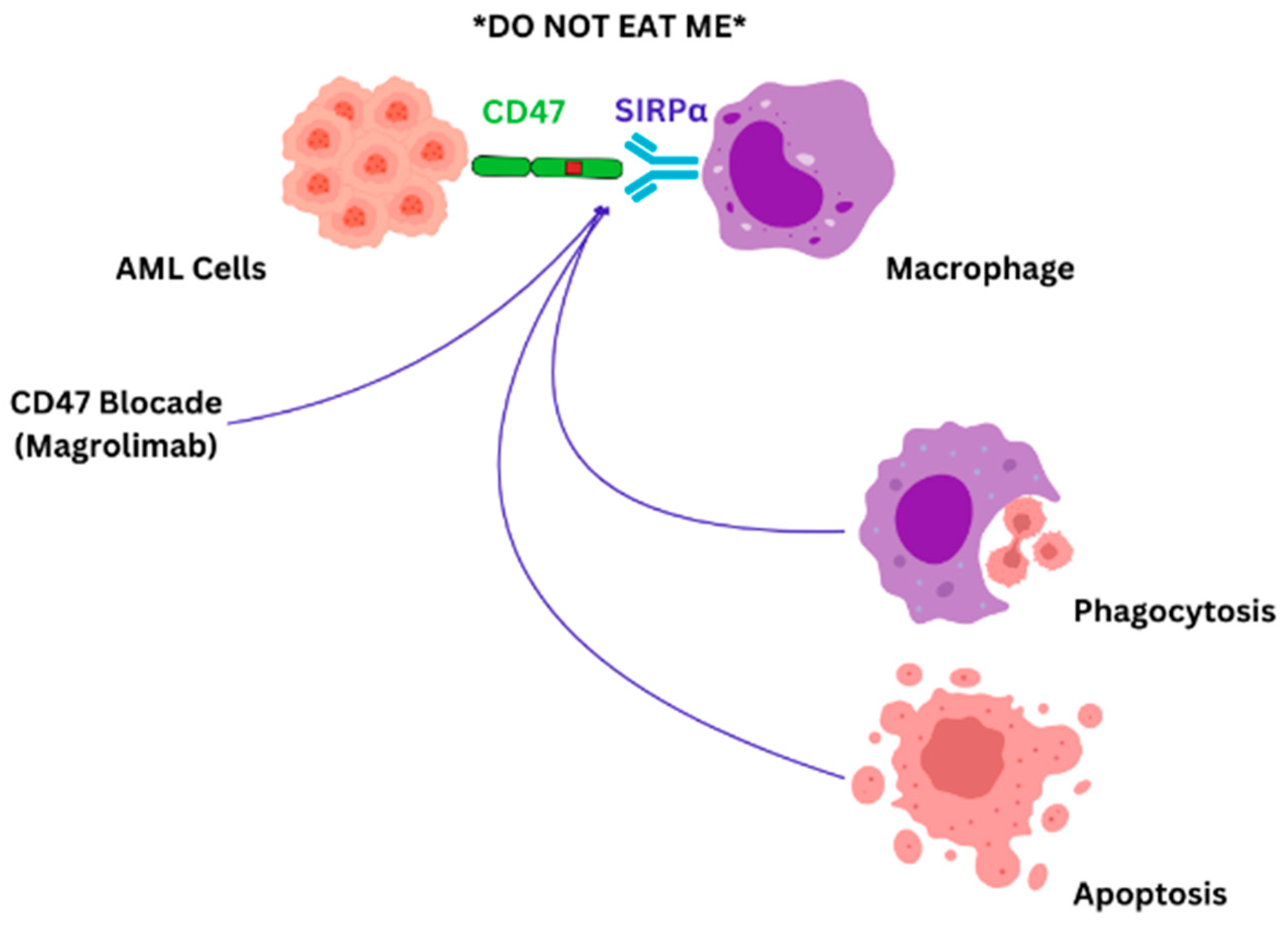
- Weiskopf, K. Cancer Immunotherapy Targeting the Cd47/Sirpalpha Axis. Eur. J. Cancer 2017, 76, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Huang, Q.; Xiao, W.; Zhao, Y.; Pi, J.; Xu, H.; Zhao, H.; Xu, J.; Evans, C.E.; Jin, H. Advances in Anti-Tumor Treatments Targeting the Cd47/Sirpalpha Axis. Front. Immunol. 2020, 11, 18. [Google Scholar]
- Takimoto, C.H.; Chao, M.P.; Gibbs, C.; McCamish, M.A.; Liu, J.; Chen, J.Y.; Majeti, R.; Weissman, I.L. The Macrophage ‘Do Not Eat Me’ Signal, Cd47, Is a Clinically Validated Cancer Immunotherapy Target. Ann. Oncol. 2019, 30, 486–489. [Google Scholar] [CrossRef]
- Yamada-Hunter, S.A.; Theruvath, J.; McIntosh, B.J.; Freitas, K.A.; Lin, F.; Radosevich, M.T.; Leruste, A.; Dhingra, S.; Martinez-Velez, N.; Xu, P.; et al. Engineered Cd47 Protects T Cells for Enhanced Antitumour Immunity. Nature 2024, 630, 457–465. [Google Scholar] [CrossRef]
- Xu, L.; Wang, X.; Zhang, T.; Meng, X.; Zhao, W.; Pi, C.; Yang, Y.G. Expression of a Mutant Cd47 Protects against Phagocytosis without Inducing Cell Death or Inhibiting Angiogenesis. Cell Rep. Med. 2024, 5, 101450. [Google Scholar] [CrossRef]
- Haddad, F.; Daver, N. Targeting Cd47/Sirpalpha in Acute Myeloid Leukemia and Myelodysplastic Syndrome: Preclinical and Clinical Developments of Magrolimab. J. Immunother. Precis. Oncol. 2021, 4, 67–71. [Google Scholar] [CrossRef]
- Shin, D.Y. Tp53 Mutation in Acute Myeloid Leukemia: An Old Foe Revisited. Cancers 2023, 15, 4816. [Google Scholar] [CrossRef]
- Daver, N.G.; Vyas, P.; Kambhampati, S.; Al Malki, M.M.; Larson, R.A.; Asch, A.S.; Mannis, G.; Chai-Ho, W.; Tanaka, T.N.; Bradley, T.J.; et al. Tolerability and Efficacy of the Anticluster of Differentiation 47 Antibody Magrolimab Combined with Azacitidine in Patients with Previously Untreated Aml: Phase Ib Results. J. Clin. Oncol. 2023, 41, 4893–4904. [Google Scholar] [CrossRef]
- Chao, M.P.; Takimoto, C.H.; Feng, D.D.; McKenna, K.; Gip, P.; Liu, J.; Volkmer, J.P.; Weissman, I.L.; Majeti, R. Therapeutic Targeting of the Macrophage Immune Checkpoint Cd47 in Myeloid Malignancies. Front. Oncol. 2019, 9, 1380. [Google Scholar] [CrossRef]
- Sallman, D.A.; Al Malki, M.M.; Asch, A.S.; Wang, E.S.; Jurcic, J.G.; Bradley, T.J.; Flinn, I.W.; Pollyea, D.A.; Kambhampati, S.; Tanaka, T.N.; et al. Magrolimab in Combination with Azacitidine in Patients with Higher-Risk Myelodysplastic Syndromes: Final Results of a Phase Ib Study. J. Clin. Oncol. 2023, 41, 2815–2826. [Google Scholar] [CrossRef]
- Daver, N.; Senapati, J.; Maiti, A.; Loghavi, S.; Kadia, T.M.; DiNardo, C.D.; Pemmaraju, N.; Jabbour, E.; Montalban-Bravo, G.; Tang, G.; et al. Phase I/Ii Study of Azacitidine (Aza) with Venetoclax (Ven) and Magrolimab (Magro) in Patients (Pts) with Newly Diagnosed (Nd) Older/Unfit or High-Risk Acute Myeloid Leukemia (Aml) and Relapsed/Refractory (R/R) Aml. Blood 2022, 140 (Suppl. 1), 141–144. [Google Scholar] [CrossRef]
- Daver, N.G.; Liu, K.; Kuwahara, S.B.; Caldwell, K.; Vyas, P. Aml-577 a Phase Iii, Randomized Trial of Magrolimab in Combination with Venetoclax and Azacitidine in Previously Untreated Patients with Acute Myeloid Leukemia Who Are Ineligible for Intensive Chemotherapy (Enhance-3). Clin. Lymphoma Myeloma Leuk. 2023, 23, S313–S314. [Google Scholar] [CrossRef]
- Gilead Sciences, Inc. Gilead Statement on Discontinuation of Phase 3 Enhance-3 Study in Aml. Available online: https://www.gilead.com/company/company-statements/2024/gilead-statement-on-discontinuation-of-phase-3-enhance-3-study-in-aml (accessed on 31 August 2024).
- Garcia-Manero, G.; Przespolewski, A.; Abaza, Y.; Byrne, M.; Fong, A.P.; Jin, F.; Forgie, A.J.; Tsiatis, A.C.; Guan, S.; Erba, H.P. Evorpacept (Alx148), a Cd47-Blocking Myeloid Checkpoint Inhibitor, in Combination with Azacitidine and Venetoclax in Patients with Acute Myeloid Leukemia (Aspen-05): Results from Phase 1a Dose Escalation Part. Blood 2022, 140, 9046–9047. [Google Scholar] [CrossRef]
- Zeidan, A.M.; DeAngelo, D.J.; Palmer, J.; Seet, C.S.; Tallman, M.S.; Wei, X.; Raymon, H.; Sriraman, P.; Kopytek, S.; Bewersdorf, J.P.; et al. Phase 1 Study of Anti-Cd47 Monoclonal Antibody Cc-90002 in Patients with Relapsed/Refractory Acute Myeloid Leukemia and High-Risk Myelodysplastic Syndromes. Ann. Hematol. 2022, 101, 557–569. [Google Scholar] [CrossRef] [PubMed]
- Sallman, D.A.; Donnellan, W.B.; Asch, A.S.; Lee, D.J.; Al Malki, M.; Marcucci, G.; Pollyea, D.A.; Kambhampati, S.; Komrokji, R.S.; Van Elk, J.; et al. The First-in-Class Anti-Cd47 Antibody Hu5f9-G4 Is Active and Well Tolerated Alone or with Azacitidine in Aml and Mds Patients: Initial Phase 1b Results. J. Clin. Oncol. 2019, 37, 7009. [Google Scholar] [CrossRef]
- Clinicaltrials.gov. Available online: https://clinicaltrials.gov/study/NCT05607199?cond=acute%20myeloid%20leukemia&intr=cd47%20&page=2&rank=11#collaborators-and-investigators (accessed on 31 August 2024).
- Clinicaltrials.gov. Available online: https://clinicaltrials.gov/study/NCT06008405?cond=acute%20myeloid%20leukemia&intr=cd47%20&rank=9 (accessed on 31 August 2024).
- Clinicaltrials.gov. Available online: https://clinicaltrials.gov/study/NCT06387420?cond=acute%20myeloid%20leukemia&intr=cd47%20&page=1&rank=5 (accessed on 31 August 2024).
- Rannikko, J.H.; Bono, P.; Hynninen, J.; Hollmen, M. Bexmarilimab Activates Human Tumor-Associated Macrophages to Support Adaptive Immune Responses in Interferon-Poor Immune Microenvironments. Cancer Immunol. Res. 2024, 12, 48–59. [Google Scholar] [CrossRef]
- Kontro, M.; Stein, A.S.; Pyörälä, M.; Rimpiläinen, J.; Siitonen, T.; Hollmén, M.; Fjaellskog, M.-L.; Pawlitzky, I.; Zeidan, A.M.; Daver, N. Encouraging Efficacy Observed in Bexmab Study: A Phase 1/2 Study to Assess Safety and Efficacy of Bexmarilimab in Combination with Standard of Care in Myeloid Malignancies. Blood 2023, 142 (Suppl. 1), 2915. [Google Scholar] [CrossRef]
- Weinhauser, I.; Pereira-Martins, D.A.; Almeida, L.Y.; Hilberink, J.R.; Silveira, D.R.A.; Quek, L.; Ortiz, C.; Araujo, C.L.; Bianco, T.M.; Lucena-Araujo, A.; et al. M2 Macrophages Drive Leukemic Transformation by Imposing Resistance to Phagocytosis and Improving Mitochondrial Metabolism. Sci. Adv. 2023, 9, eadf8522. [Google Scholar] [CrossRef]
- Trasanidis, N.; Katsarou, A.; Ponnusamy, K.; Shen, Y.A.; Kostopoulos, I.V.; Bergonia, B.; Keren, K.; Reema, P.; Xiao, X.; Szydlo, R.M.; et al. Systems Medicine Dissection of Chr1q-Amp Reveals a Novel Pbx1-Foxm1 Axis for Targeted Therapy in Multiple Myeloma. Blood 2022, 139, 1939–1953. [Google Scholar] [CrossRef]
- Liu, S.X.; Zhou, Y.; Zhao, L.; Zhou, L.S.; Sun, J.; Liu, G.J.; Du, Y.S.; Zhou, Y.N. Thiostrepton Confers Protection against Reactive Oxygen Species-Related Apoptosis by Restraining Foxm1-Triggerred Development of Gastric Cancer. Free Radic. Biol. Med. 2022, 193 Pt 1, 385–404. [Google Scholar] [CrossRef] [PubMed]
- Weinhauser, I.; Pereira-Martins, D.A.; Hilberink, J.R.; Brouwers-Vos, A.; Rego, E.M.; Huls, G.; Schuringa, J.J. Thiostrepton Induces Cell Death of Acute Myeloid Leukemia Blasts and the Associated Macrophage Population. Haematologica 2024, 109, 639–645. [Google Scholar] [CrossRef] [PubMed]
- La Fleur, L.; Botling, J.; He, F.; Pelicano, C.; Zhou, C.; He, C.; Palano, G.; Mezheyeuski, A.; Micke, P.; Ravetch, J.V.; et al. Targeting Marco and Il37r on Immunosuppressive Macrophages in Lung Cancer Blocks Regulatory T Cells and Supports Cytotoxic Lymphocyte Function. Cancer Res. 2021, 81, 956–967. [Google Scholar] [CrossRef] [PubMed]
- Eisinger, S.; Sarhan, D.; Boura, V.F.; Ibarlucea-Benitez, I.; Tyystjarvi, S.; Oliynyk, G.; Arsenian-Henriksson, M.; Lane, D.; Wikstrom, S.L.; Kiessling, R.; et al. Targeting a Scavenger Receptor on Tumor-Associated Macrophages Activates Tumor Cell Killing by Natural Killer Cells. Proc. Natl. Acad. Sci. USA 2020, 117, 32005–32016. [Google Scholar] [CrossRef]
- Weigel, B.J.; Cooley, S.; DeFor, T.; Weisdorf, D.J.; Panoskaltsis-Mortari, A.; Chen, W.; Blazar, B.R.; Miller, J.S. Prolonged Subcutaneous Administration of 852a, a Novel Systemic Toll-Like Receptor 7 Agonist, to Activate Innate Immune Responses in Patients with Advanced Hematologic Malignancies. Am. J. Hematol. 2012, 87, 953–956. [Google Scholar] [CrossRef]
- Sumaiya, K.; Langford, D.; Natarajaseenivasan, K.; Shanmughapriya, S. Macrophage Migration Inhibitory Factor (Mif): A Multifaceted Cytokine Regulated by Genetic and Physiological Strategies. Pharmacol. Ther. 2022, 233, 108024. [Google Scholar] [CrossRef]
- Jankauskas, S.S.; Wong, D.W.L.; Bucala, R.; Djudjaj, S.; Boor, P. Evolving Complexity of Mif Signaling. Cell Signal 2019, 57, 76–88. [Google Scholar] [CrossRef]
- Abdul-Aziz, A.M.; Shafat, M.S.; Mehta, T.K.; Di Palma, F.; Lawes, M.J.; Rushworth, S.A.; Bowles, K.M. Mif-Induced Stromal Pkcbeta/Il8 Is Essential in Human Acute Myeloid Leukemia. Cancer Res. 2017, 77, 303–311. [Google Scholar] [CrossRef]
- Islam, M.; Mohamed, E.H.; Esa, E.; Kamaluddin, N.R.; Zain, S.M.; Yusoff, Y.M.; Assenov, Y.; Mohamed, Z.; Zakaria, Z. Circulating Cytokines and Small Molecules Follow Distinct Expression Patterns in Acute Myeloid Leukaemia. Br. J. Cancer 2017, 117, 1551–1556. [Google Scholar] [CrossRef]
- Smirnova, T.; Spertini, C.; Spertini, O. Csf1r Inhibition Combined with Gm-Csf Reprograms Macrophages and Disrupts Protumoral Interplays with Aml Cells. Cancers 2021, 13, 5289. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Activated | Phenotype | The Main Secreted Cytokines | Function |
|---|---|---|---|
| LPS, IFN-γ | M1 macrophages | IL-1, IL-6, IL-8, IL-12, TNF-α, iNOS | Pro-inflammatory, Cytotoxic Antitumorigenic |
| IL-4, IL-13, IL-10, M-CSF | M2 macrophages | ||
| M2a | Il-10, CCL13, CCL17, CCL22 | ||
| M2b | IL-10, CCL1, IL-12 | Immunosuppressive Pro-tumorigenic Anti-inflammatory | |
| M2c | CCL16, CCL18 | ||
| M2d | IL-10, IL-12, TGF-β, VEGF, CCL5, CXCL12, CXCL16 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mesaros, O.; Onciul, M.; Matei, E.; Joldes, C.; Jimbu, L.; Neaga, A.; Serban, O.; Zdrenghea, M.; Nanut, A.M. Macrophages as Potential Therapeutic Targets in Acute Myeloid Leukemia. Biomedicines 2024, 12, 2306. https://doi.org/10.3390/biomedicines12102306
Mesaros O, Onciul M, Matei E, Joldes C, Jimbu L, Neaga A, Serban O, Zdrenghea M, Nanut AM. Macrophages as Potential Therapeutic Targets in Acute Myeloid Leukemia. Biomedicines. 2024; 12(10):2306. https://doi.org/10.3390/biomedicines12102306
Chicago/Turabian StyleMesaros, Oana, Madalina Onciul, Emilia Matei, Corina Joldes, Laura Jimbu, Alexandra Neaga, Oana Serban, Mihnea Zdrenghea, and Ana Maria Nanut. 2024. "Macrophages as Potential Therapeutic Targets in Acute Myeloid Leukemia" Biomedicines 12, no. 10: 2306. https://doi.org/10.3390/biomedicines12102306
APA StyleMesaros, O., Onciul, M., Matei, E., Joldes, C., Jimbu, L., Neaga, A., Serban, O., Zdrenghea, M., & Nanut, A. M. (2024). Macrophages as Potential Therapeutic Targets in Acute Myeloid Leukemia. Biomedicines, 12(10), 2306. https://doi.org/10.3390/biomedicines12102306