Abstract
RASopathies, a group of neurodevelopmental congenital disorders stemming from mutations in the RAS/MAPK pathway, present a unique opportunity to delve into the intricacies of complex neurological disorders. Afflicting approximately one in a thousand newborns, RASopathies manifest as abnormalities across multiple organ systems, with a pronounced impact on the central and peripheral nervous system. In the pursuit of understanding RASopathies’ neurobiology and establishing phenotype–genotype relationships, in vivo non-mammalian models have emerged as indispensable tools. Species such as Danio rerio, Drosophila melanogaster, Caenorhabditis elegans, Xenopus species and Gallus gallus embryos have proven to be invaluable in shedding light on the intricate pathways implicated in RASopathies. Despite some inherent weaknesses, these genetic models offer distinct advantages over traditional rodent models, providing a holistic perspective on complex genetics, multi-organ involvement, and the interplay among various pathway components, offering insights into the pathophysiological aspects of mutations-driven symptoms. This review underscores the value of investigating the genetic basis of RASopathies for unraveling the underlying mechanisms contributing to broader neurological complexities. It also emphasizes the pivotal role of non-mammalian models in serving as a crucial preliminary step for the development of innovative therapeutic strategies.
1. Introduction
RASopathies are a group of syndromic disorders primarily caused by germline mutations in genes that encode any of the proteins along the rat sarcoma/mitogen-activated protein kinase (RAS/MAPK) signaling network. The core of this cascade comprises the membrane-bound RAS-GTPase, the protein kinases RAF and MEK, and the transcription factor ERK (extracellular signal-related kinase), which control numerous cellular and physiological processes, including organism development, cell cycle control, cell proliferation and differentiation, cell survival, and death [1,2]. These disorders manifest as multisystem syndromes, including, but not limited to, neurofibromatosis type 1 (NF1), Noonan syndrome (NS), Legius syndrome (LS), Costello syndrome (CS), cardio–facio–cutaneous (CFC) syndrome and SYNGAP1 encephalopathy (SE) [3]. Most RASopathies are associated with an autosomal dominant mode of inheritance, one exception being Noonan syndrome, where recessive phenotypes caused by mutations in LZTR1 and SPRED2 have been described [4,5]. Although each RASopathy syndrome is individually rare, collectively, this family of congenital disorders is one of the largest in the world, affecting approximately 1 in 1000 individuals [6]. Patients affected by these disorders have common clinical features such as short stature, dysmorphic facial features, cardiac structural and functional defects, and lymphatic dysfunction [7]. The central and peripheral nervous systems of patients are also frequently and severely affected, as evidenced by structural malformations of the brain, such as macrocephaly [8], neurocognitive deficits [9], and intellectual disability, as well as an increased risk of tumorigenesis [10]. The symptoms of RASopathies can either present at birth or become apparent later in life. Regrettably, finding effective targeted therapies remains a challenge, and available treatment options are currently scarce or non-existent [11]. It is therefore essential to better define the genetic, molecular and cellular etiologies of these complex congenital diseases to identify effective treatments. The investigation of RASopathies also offers profound insights into the intricate landscape of complex neurological disorders, enriching our comprehension of the molecular mechanisms dictating neurological health. This scientific endeavor contributes fundamentally to the evolving landscape of precision medicine, forging new pathways for the diagnosis and treatment of a spectrum of neurodevelopmental challenges.
Previous works based on in vivo modelling of RASopathies highlight the relevance of developing animal models for the study of these complex diseases [7,12]. Additional studies addressing neurological disorders linked to RASopathies emphasize the involvement of the RAS/MAPK pathway in human neurodevelopmental processes. These investigations explore the impacts of mutations within pathway components on both human subjects and mice [9,13]. In this context, our emphasis lies in elucidating recent advancements in our understanding of the molecular and cellular underpinnings of neurological symptoms in RASopathies, with a special focus on genetically modified non-mammalian animal models. The zebrafish Danio rerio, the fly Drosophila melanogaster, the worm Caenorhabditis elegans, the frogs Xenopus laevis and Xenopus tropicalis and the chick Gallus gallus stand as valuable models for studying the effects of genetic factors on the RAS/MAPK pathway. These models provide unique advantages for studying genetic modifiers and dissecting interactions within the RAS/MAPK pathway while adhering to the “Three Rs” principles—reduction, refinement and replacement—which are aimed at minimizing the number of animals used, refining experimental procedures for animal welfare, and replacing animals with alternative methods whenever possible. Additionally, these models can be effective in capturing some of the complexity and variability seen in the clinical presentations of these conditions. Nevertheless, it is essential to recognize the constraints inherent in these models and to back up observations by using validation in mammalian and human cellular models.
2. Neurological Manifestations of RASopathies
The RAS/MAPK pathway is involved in the control of processes of embryonic and postnatal development, such as cell specification and axon growth, and also in the control of adult plasticity of the central and peripheral nervous systems [14,15]. Consequently, genetic mutations associated with RASopathies give rise to multiple neurological impairments [9]: (i) structural intracranial anomalies like Chiari I malformation [16], syringomyelia, cerebral vascular anomalies, benign external hydrocephalus, craniosynostosis and posterior fossa anomalies [17]; (ii) neuropathies or headaches; (iii) seizures [18]; and (iv) diverse cognitive deficits, such as psychomotor delay [19] and cognitive abnormalities [20]. As in other neurodevelopmental disorders, these cognitive deficits consist in language delays [21], deficits in learning ability and memory [20], and social impairments that are partly reminiscent of autism spectrum disorders [22]. All these neurological disorders tend to be lifelong and compromise the well-being of patients and their families. Furthermore, individuals with RASopathies face a heightened susceptibility to both benign and malignant nervous system tumors, such as low-grade gliomas in NS and Chiari 1 malformations in CS [8], an increased rhabdomyosarcoma risk [23], as well as other risks [2,24,25]. RASopathies that manifest with neurological disorders include NF1, LS, NS, CS, CFC syndrome and SE (Table 1). Further details of the neurological manifestations present in each disorder are shown in the following.

Table 1.
Neurological phenotypes in patients with RASopathies; the most common neurological features observed in individuals affected by RASopathies. The ✔ symbol denotes the presence of a specific phenotype within the respective RASopathy, while the X symbol indicates its absence. The category “Increased risk of tumors” encompasses both benign and malignant tumors. NF1 stands for neurofibromatosis type 1; LS stands for Legius syndrome; NS stands for Noonan syndrome; CS stands for Costello syndrome; CFC stands for cardio–facio–cutaneous syndrome; SE stands for SYNGAP1 encephalopathy; ADHD stands for attention-deficit/hyperactivity disorder; ASD stands for autism spectrum disorder.
Neurofibromatosis type 1 (NF1) is a dominant autosomal disorder caused by mutations in the NF1 gene [26]. These mutations render NF1-derived neurofibromin protein unable to promote the intrinsic GTPase activity of RAS to hydrolyze RAS-bound GTP to GDP, ultimately activating the RAS signaling pathway [27]. NF1 occurs in approximately one out of every 3200 people, and symptoms normally begin in newborns and infants [28]. Neurological symptoms may be different from person to person, may differ in number and can range from mild to severe (Table 1). Neurological abnormality is very frequently found in NF1 patients, including neurological symptoms and disorders such as hydrocephalus, macrocephaly, cerebrovascular events, neuropathy, seizures, epilepsy, and headache [18]. Patients may have cognitive defects, including mild intellectual disability, which is characterized by an intelligence quotient (IQ) ranging from 50 to 69, memory impairment, neurological speech impairment and specific learning disabilities, such as reading or writing, coordination, self-control or attention, that interfere with the ability to learn [29,30,31]. Behavioral problems may include attention deficits, hyperactivity and impulsivity, thus fulfilling the diagnostic criteria for attention-deficit/hyperactivity disorder (ADHD), as well as an increased incidence of autism spectrum disorder (ASD) [32,33]. NF1 individuals may develop nervous system defects including cerebellar ataxia, glaucoma, meningioma and neurofibromas. Synaptic plasticity alterations have also been detected in adult NF1 patients [34,35]. Finally, they have heightened risk of malignant tumors of the central nervous system (optic-pathway glioma, astrocytoma, and tumors in the brain, on the cranial nerves, or involving the spinal cord) and malignant peripheral nerve sheath tumors (MPNST) [25,36,37,38].
Legius syndrome (LS) is a dominant autosomal disorder, caused by heterozygous inactivating mutations in SPRED1 [39], which negatively regulates the RAS-mediated activation of BRAF, CRAF and neurofibromin [40,41]. The estimated birth prevalence of this disorder is not known, but it is supposed to be very rare, and the age of onset can vary, ranging from newborn to childhood, with a milder phenotype, compared with NF1 patients. Symptoms may include, in rare cases, learning impairments, attention problems, ADHD, abnormal brain imaging, Chiari type 1 malformation, intellectual impairment, speech difficulties and seizures [42] (Table 1). Occasional symptoms include short attention span, whereas frequent neurological symptoms include various affective, behavioral, cognitive and perceptual abnormalities, as well as specific learning disabilities (reading or writing, coordination, or attention, not related to a global deficiency of intelligence), hyperactivity and macrocephaly [43]. While development of tumors is common in NF1, they are rare in LS, with only a few cases reported in the literature [39,44,45]. These findings suggest that SPRED1 mutations may not confer the same tumorigenic risk as NF1 mutations, although further research is needed to fully understand the relationship between SPRED1 and tumorigenesis.
Noonan syndrome (NS) is a dominant autosomal disorder caused by heterozygous mutations in one of several RAS/MAPK pathway genes, although rare autosomal recessive forms have been described [4,46]. In all, 50% of NS cases are caused by mutation in PTPN11, 10% by mutation in SOS1, 10% by mutation in RIT1, 5% by mutation in RAF1, and an additional 5% of cases are caused by mutations in KRAS (Table S1). The remaining 20% of cases are caused by rarer mutations in up to 10 different genes (Table S1). NS occurs in approximately one in one thousand to one in twenty-five hundred individuals, with onset ranging from prenatal stages up to 11 years, and symptoms vary in number and severity among patients (Table 1). More than one-third of patients have neurocognitive delay, including lower IQ, ADHD, language problems (dysarthric speech, neurological speech impairment, and sensorineural hearing impairment), delayed verbal recall, and visual recognition memory deficits [47,48]. Social and emotional problems are also seen [47], as well as ASDs [49]. LTP-like synaptic plasticity induced by transcranial magnetic stimulation (TMS) is impaired in Noonan syndrome patients, suggesting underlying synaptic plasticity defects [34]. Other neurological features include high forehead, an intractable neuropathic pain linked to generalized or proximal nerve hypertrophy in the peripheral nerve system in adult patients [50], and an increased risk of developing neurological malignancies, such as low-grade glioma [51].
Costello syndrome (CS) is a RASopathy caused by heterozygous gain-of-function germline mutations in HRAS (Table S2), typically in the G12 position of HRAS (p.G12S variant) [52]. Age of onset can vary, ranging from prenatal to newborns. Individuals with CS typically present craniofacial abnormalities such as cerebral cortical atrophy, ventriculomegaly or hydrocephalus in the brain, macrocephaly, and Chiari malformation or demyelination of the basal ganglia [53]. They may also experience seizures and have an increased risk of developing benign or malignant tumors [54]. Cognitive impairment, including mild-to-moderate grade intellectual disability, and affected learning and memory processes are also common features [55] (Table 1). Studies using TMS protocols have found enhanced synaptic plasticity in CS patients, although these studies are from very small cohorts [34,56,57].
Cardio–facio–cutaneous syndrome (CFC) syndrome is a dominant genetic disorder caused by alterations in one of four genes: BRAF (~75%), MEK1/2 (~25%) and KRAS, which occurs in few individuals, although some other genes might be also associated with CFC syndrome [58] (Table S2). CFC syndrome is a very rare condition whose incidence is unknown, and its age of onset can vary, ranging from prenatal to newborns. Hypotonia, motor delay, speech delay and intellectual disability, and learning disability can be considered the main neurologic features of this syndrome, although it is also characterized by the presence of macrocephaly and brain structural malformations, among other symptoms [58] (Table 1). The presence of ASD is also high in CFC syndrome patients, supporting the importance of the RAS/MAPK pathway in the etiology of ASD [49]. Findings on magnetic resonance imaging have included prominent Virchow–Robin spaces, abnormal myelination and structural anomalies [53].
SYNGAP1 encephalopathy (SE) is caused by mutations in the SYNGAP1 gene. The SYNGAP1 gene, identified as a causative factor for autosomal dominant intellectual disability type 5 [59] and classified as a RASopathy protein, primarily impacts the central nervous system, functioning as a neuronal RAS GTPase-activating protein (GAP) with roles in the regulation of excitatory plasticity [60,61]. SYNGAP1 loss-of-function mutations have been reported in patients with intellectual disability, schizophrenia, seizures and epilepsy, with patients exhibiting ASD as well [61,62] (Table 1). In particular, large-scale genetic studies utilizing exome sequencing have now related mutations in SYNGAP1 to increased risk of both ASD [63] and schizophrenia [64]. Moreover, there is evidence of how decreased SYNGAP1 expression in human neurons precipitates alterations in dendritic and synaptic maturation, resulting in enlarged neurons, heightened excitatory synapse density and earlier onset of synaptic activity [65]. This evidence collectively highlights the role of SYNGAP1 in neurological processes.
3. Neuro-Phenotypic Complexity in RASopathies with Insights from Non-Mammalian Models
Establishing a genotype–phenotype relationship in RASopathies is challenging, given the substantial degree of clinical variability and incomplete penetrance of the associated phenotypes. For instance, mutations occurring in neural progenitor cells during neurogenesis can result in diverse tissue effects within the same person [66,67], and recent studies have demonstrated that particular mutations associated with each disorder disrupt central nervous system (CNS) development in a mutation-specific manner [13]. Non-mammalian in vivo models have proven particularly valuable for validating the impact of RASopathy genes on neurodevelopmental processes reliant on the RAS/MAPK signaling pathway [12,68,69]. Techniques to modulate RAS-MAPK signaling activity include clustered regularly interspaced short palindromic repeats (CRISPR), transcription activator-like effector nucleases (TALENs) or transposon-based methods in Zebrafish and Drosophila [12], RNA interference (RNAi) [70], CRISPR-Cas9 gene editing [68] and behavioral assays [71] in C. elegans, CRISPR and TALEN in Xenopus [72], and in ovo electroporation of expression vectors in chick embryos [73]. The employment of each technique depends on the final goal of the study. For example, overexpression techniques such as mRNA injection and transposon-based methods are desired to mimic a specific phenotype observed in humans or to assay for the activity of the mutant protein in vivo. However, if the aim is to accurately model a disease in heterozygous individuals, it is more effective to introduce mutations at the endogenous locus by using techniques such as TALENs or CRISPR [12]. Bearing this in mind, these models are adept at conducting phenotypic analyses across various biological scales, spanning from the molecular to the tissue level.
Danio rerio, the zebrafish, has emerged as a robust non-mammalian model for research due to its high fecundity and time- and cost-efficient genetic manipulation and real-time high-resolution imaging. Zebrafish embryos develop rapidly and have transparent bodies, making it easy to observe developmental defects and organ dysfunction. Several techniques can be employed to validate the consequences of RASopathy genes in neurodevelopmental processes reliant on MAPK signaling in zebrafish, including behavioral assays [74], high-content screening [75], and determining the major/minor axis ratio of embryos [5]. Also, various functional assays have been used for gene discovery, including in vitro luciferase assays and in vivo zebrafish modelling [76,77,78,79]. These techniques have greatly increased the ability to investigate the impact of certain mutations and how these lesions impact disease phenotype, positioning the zebrafish as a powerful in vivo tool for modeling neurological syndromes [80]. In addition, the zebrafish platform supports medium- to high-throughput preclinical drug screening to identify compounds that may represent novel treatment paradigms or even prevent cancer evolution [81]. In fact, zebrafish is emerging as a time- and cost-effective cancer avatar model to assess tumor phenotype and drug responses. For instance, zebrafish patient-derived xenografts (zPDX) may be suitable for providing precision cancer-medicine pipelines [82].
Nonetheless zebrafish has been used for the study of NF1, NS, CS and CFC syndromes (Figure 1). Zebrafish has proven to be an effective in vivo model for investigating NF1, with successful knockout of nf1a and nf1b paralogues displaying macrocephaly and increased oligodendrocyte progenitor cell (OPC) migration within the spinal cord, and defects in myelin structure formation lead to decreased myelination in these mutants [83] (Table S2). Additionally, nf1 mutants crossed with a p53 null background develop high-grade gliomas and MPNSTs, similar to what is observed in NF1 patients [83]. Also, NF1 variants associated with cognitive dysfunction have been linked to aberrant cAMP signaling [84]. Memory formation and recall have been tested in nf1 mutants, and deficits were improved by treatment with chemical stimulants for the cAMP pathway [85]. The over-expression of platelet-derived growth factor receptor alpha (PDGFRA) has been linked to malignant transformation of MPNSTs [86], and over-expression of PDGFRA in a nf1a+/−; nf1b−/−; p53m/m background accelerated the onset of MPNSTs [87]. Other studied neurological defects include macro- and microcephaly [88], various developmental abnormalities, [76] and tumors [84].

Figure 1.
RASopathies explored in non-mammalian models. Non-mammalian models employed in neuroscience research, highlighting the studied syndromes exclusively based on the relevant RASopathy genes. NF1 stands for neurofibromatosis type 1; NS stands for Noonan syndrome; CS stands for Costello syndrome; CFC stands for cardio–facio–cutaneous syndrome. Created with BioRender.com.
As with NF1, the zebrafish genome contains two PTPN11 genes (ptpn11a and ptpn11b), encoding Shp2a and Shp2b (Table S1), two proteins with a similar catalytic activity, although only Shp2a is indispensable during zebrafish development [79]. NS-associated neurological features, including shorter body axis length [79,89], craniofacial defects [90], and impaired long-term memory [91], can be attributed to the dynamic regulation of cell movement during zebrafish gastrulation and epiboly [92,93]. E-cadherin turnover, as well as ERK signaling, contribute to these processes. Noonan-associated PTPN11 and NRAS mutants can alter coordinated convergent-extension cell movements and result in oblong embryos with abnormal axis ratios [12,94]. Additionally, introducing ptpn11 variants associated to NS with multiple lentigines (NSML) in zebrafish embryos can increase the major/minor axis ratio [89] and cause craniofacial dysmorphia in adult fish [79]. Shp2D61G mutant zebrafish can display neurological NS traits, including craniofacial defects, which vary in severity among individuals [95]. The introduction of gain-of-function mutations in RIT1 into zebrafish embryos also causes NS and demonstrates a biological effect similar to mutations in other RASopathy-related genes [76]. The biochemical relationship between the RASopathy proteins KRAS, RIT1 and LZTR1 was tested in zebrafish and fruit flies; all LZTR1 orthologs preferentially interacted with RIT1 orthologs, indicating that this interaction is also conserved in less-complex model organisms [96]. Homozygosity of three different SPRED2 variants can cause developmental delay, intellectual disability, cardiac defects, short stature, skeletal anomalies, and a typical facial gestalt as major features linked to a recessive phenotype evocative of NS [5] (Table S2). Furthermore, activating RRAS2 mutations can cause NS, and several zebrafish models were generated to study this; larvae overexpressing RRAS2G24_G26dup or RRAS2Q72H variants, but not wild-type RRAS2, showed craniofacial defects and macrocephaly [97], and RRAS2Q72L caused severe developmental impairments and craniofacial defects, whereas RRAS2F75C resulted in no aberrant in vitro or in vivo phenotypes [97] (Table S2).
Zebrafish embryos have been employed to probe craniofacial anomalies associated with NS, a condition linked to A2ML1 variants detected in NS patients (Table S2). In this context, morphometric analysis of A2ML1 mutant-expressing embryos revealed substantial head broadening and facial blunting [77]. On the other hand, A2ML1 mutations showed no influence on zebrafish development [98]. Notably, since RAC1 activates PAK, which assists in the MEK/ERK pathway via phosphorylation of c-RAF and MEK1 [99], RAC1 variants implicated in developmental delay during neuronal maturation exhibited dominant-negative properties causing microcephaly, diminished neuronal proliferation, and cerebellar anomalies when overexpressed in zebrafish [88]. Intriguingly, the RAC1P29S variant induced RASopathy-like manifestations in zebrafish akin to activated BRAF and KRAS, mitigated by PAK and MEK inhibitors [100]. Furthermore, a RABL3S36* mutant linked to hereditary pancreatic cancer led to RASopathy features in a zebrafish model, with adult homozygous mutants displaying indicative swimming defects [101]. Expression of activated BRAF, KRAS, or RAC1 prompted pronounced body axis disruption and heightened ERK and PAK activity, contrasting with wild-type BRAF’s inert impact [100]. These findings collectively underscore the potency of zebrafish models in elucidating molecular and developmental mechanisms underlying diverse genetic disorders.
Transgenic zebrafish that ubiquitously expresses the constituently active HRASG12V in the germline display phenotypes consistent with CS: shortened body length, craniofacial dysmorphia and oncogene-induced senescence in the brain [102]. Overexpression of HRASG12V led to a 22% mortality rate at 2 dpf and a 60% mortality rate at 5 dpf in zebrafish embryos, with 6% of surviving embryos exhibiting brain hemorrhage at 5 dpf [103].
The biological consequences of CFC syndrome that have been associated with BRAF mutations have been studied in several non-mammalian models, including zebrafish. Expression of the common kinase-activating variant in CFC syndrome BRAFQ257R in zebrafish embryos increased the major/minor axis ratio [104]. Also, overexpression of BRAFV600E before gastrulation in zebrafish embryos caused severe embryonic malignancy with truncated posterior structure and compromised forebrain, while later induction led to craniofacial deformities resembling CFC syndrome [105] (Table S2). Activating mutations in MEK1 have different strengths, which are correlated with the severity of the phenotypes observed in human patients affected by these mutations [106]. Further, spatiotemporal resolution was used in zebrafish to discover that intrinsically active variants of MEK can both increase and reduce the levels of pathway activation in vivo [107].
Drosophila melanogaster has been a powerful genetic system with which to decipher fundamental cell and developmental biology questions, including the discovery of components of the RAS/MAPK pathway [108]. Drosophila presents many interesting attributes such as its short life-cycle, small size, high fecundity and a relatively compact genome [109]. Furthermore, more than half of Drosophila genes have orthologs in the human [110] and its complex brain and behavioral repertoire make it a particularly useful model for understanding neurobiology [111,112]. Results from this model organism may provide valuable insights into how pathogenic variants promote specific traits in humans, as well as enable tailored therapeutic approaches to treat them. Drosophila has been used for the study of NF1 and NS (Figure 1). Early work in Drosophila identified the ortholog of ERK as the terminal kinases of the RAS/ERK signaling cascade [113]. Later on, Drosophila RASopathy models were generated, generally emulating RAS gain-of-function phenotypes found in NS patients, by mutating the Drosophila ortholog of human PTPN11, corkscrew (csw) [90,91], and SOS2 [114]. Drosophila wing vein formation and eye development have been settled as excellent in vivo readouts for RAS signaling [115,116]. For example, expression of PTPN11 variants associated in NS-like in Drosophila results in ectopic wing vein and rough eye phenotypes [117], and NS-associated mutations in RIT1 also result in ectopic wing veins [76]. Studies have shown that the loss of neurofibromin affects the synaptic function of neurons, which is associated with a decrease in neurotransmitter release, changes in synaptic plasticity and abnormal activation of signaling pathways involved in neuronal development and function [118,119]. Although the NF1 mutations affect both types of memory, the GAP domain that regulates RAS affects only long-term memory [118,119], while the C-terminal domain reduces the immediate memory through regulation of adenyl cyclase [120]. Interestingly, this memory impairment could be rescued by expression of a human NF1 transgene [118,119]. Other studies also support the proposition that Drosophila lacking the NF1 homolog exhibit specific learning and memory deficits similar to those of many children with NF1 [121,122]. Expression of Dsor1Y130C, the MEK1 ortholog in Drosophila, leads to larval cuticle deficits and ectopic wing veins [106] (Table S2), and intrinsically active variants of MEK can produce either an increase or a decrease in pathway activation levels in vivo, as previously reported for zebrafish [107]. Aberrant RAS/MAPK signaling in the eye leads to small, rough eyes; aberrant signaling in the wing leads to ectopic wing development [7]. Drosophila Lztr1 preferentially regulates Ric rather than Ras levels, as seen with the corresponding mammalian orthologs [96]. However, a previous study showed that flies expressing RNA interference constructs against Lztr1 displayed minor defects in wing vein patterning [123]. Divergent outcomes between these two studies may arise from the incomplete knockdown and off-target effects in the RNA interference study by Bigenzahn et al. (2018), compared to the complete loss of function achieved in the null Lztr1 flies of the Cuevas-Navarro et al. (2022) study, which was potentially compounded by differences in experimental conditions.
Studies in Drosophila also identified novel modifiers (Dap160 and CCKLR-17D1) that implicate synaptic defects in the dNf1 growth deficiency [124]. This and other studies also have implicated the neuronal-specific RTK Anaplastic Lymphoma Kinase (ALK) as an upstream activator of RAS signaling in neurons [124,125]. Altering ALK activity in these animal models partially ameliorates NF1 cognitive deficits, suggesting that it represents a potential therapeutic target. More recently, 13 Drosophila RASopathy models expressing commonly observed RASopathy variants were generated and used to explore fly phenotypes, altered cellular signaling networks, and responses to drugs [126] (Table S2). Using up to 33 Drosophila models, it was planned to provide a broad overview of how RASopathy variants alter the signaling network in different tissues [11].
Caenorhabditis elegans stands as a potent model to unveil the pathogenic implications of RASopathy mutations. C. elegans is a small nematode worm that has become a promising model organism for studying neurological diseases due to its simple and well-characterized nervous system. It has a transparent body, enabling researchers to study the effects of genetic and environmental factors on its development, physiology, and behavior. With only 302 neurons and ~7000 synapses, it has the ability to recapitulate key aspects of neurological function, and to unravel complex signaling pathways involved in human disease systems [127,128]. The genome of C. elegans contains homologs of two-thirds of all human disease genes, including many related to neurological disorders [129]. In recent years, C. elegans has also been used for high-throughput drug screening, further highlighting its potential as a model organism for studying neurological diseases [130]. C. elegans has been used for the study of NS (Figure 1). Mutations in the MAPK1 gene cause the multivulva (Muv) phenotype, which is consistent with aberrant RAS/MAPK pathway activity. Up to five (p.Ile74Asn, p.His80Tyr, p.Ala174Val, p.Asp318Gly and p.Pro323Arg) de novo MAPK1 variants found in patients with a neurodevelopmental disease within the RASopathy phenotypic spectrum were expressed in C. elegans, and all of them were found to cause the Muv phenotype [68] (Table S2). Similarly, the SHOC2S2G variant, which is involved in neurodevelopmental disorders including NS [131], was found to introduce a N-myristoylation site, resulting in an impaired translocation to the nucleus upon growth factor stimulation, and engendered protruding vulva, a phenotype also associated with an aberrant signaling in the RAS/MAPK pathway [132] (Table S2). In addition, expression of the RRASG39dup, identified in a panel of NS patients, ortholog in C. elegans enhanced RAS signaling and engendered protruding vulva, as well as decreased egg-laying efficiency and accumulation of larvae inside the mother, supporting the proposition of the gain-of-function role of the mutation in RAS-1 function [133] (Table S2).
Xenopus has been used as a model organism in the study of embryology, cell biology, genetics, physiology, toxicology and disease, among other fields [134]. This amphibian has several advantages as a model system, including its external development, easy genetic manipulation, and availability of a variety of techniques for the study of neural development and function, along with the balance between mesoderm formation and the levels of MAPK [135]. Furthermore, one of the main advantages of its use is that the disruptions of development caused by RASopathy-associated mutations can be effectively modeled in Xenopus, presenting phenotypes that are highly reminiscent of the symptoms of patients and thus allowing the shedding of light into the mechanisms by which these diseases develop [69]. Xenopus has been used for the study of NS and CFC syndrome (Figure 1). Induction of a dominant-negative SHP2 in Xenopus blocks mesoderm formation by impairing ERK signaling, leading to arrest of gastrulation [136,137]. Also, Xenopus has been used to study the effects of mutations in the RAS/MAPK pathway on neural crest (NC) cell migration, a key process in the development of the peripheral nervous system [138]. Xenopus embryos expressing a dominant negative FGFR4 showed reduced NC gene expression at mid-neurula stages [14]. When a CFC syndrome germline variant in 14-3-3ζ (YWHAZ) was expressed in Xenopus tropicalis, it caused an increase in BRAF and RAF1 binding, ERK phosphorylation, and a decrease in body length [139]. Furthermore, recent studies in Xenopus have revealed the role of the RAS/MAPK pathway in the development of the visual system [140]. Overall, the use of Xenopus as a non-mammalian model system has proven to be a valuable tool in the study of the neurobiology of RASopathies.
4. Other Potential Models: Chick Embryos
Leveraging chick embryos as a model organism provides a valuable avenue for unraveling intricate molecular mechanisms underpinning neurological disorders. Aberrant RAS signaling activation in chick embryos notably induces shifts in axon guidance [141]. Additionally, mutations in RAS/MAPK components prompt alterations in cortical neuron migration and morphological changes [142]. Intriguingly, investigations have illuminated the pivotal role of PI3K as an anti-apoptotic transducer in neuroblasts and neurons, while also governing cellular migration within the neuroepithelium through Rho GTPases. Recent advancements showcase that an overactivation of ERK1/2 in the trunk neural tube, mediated by the constitutive active form of MEK1 (MEK1ca), drives shifts in the transcriptional profile of developing spinal cord cells. MEK1ca-transfected cells relinquish neuronal identity, expressing potential oncogenes like AQP1, highlighting MEK1’s promise as an in vivo model in uncovering mechanisms fostering neoplasia and malignancy in neural-origin ERK-induced tumorigenesis [73]. This model is notably advantageous for examining early embryonic development, a pivotal phase for brain maturation [143].
The chicken embryo, with a resemblance to the human embryo’s frontonasal mass surpassing that of the mouse, offers an invaluable platform for craniofacial research, bolstered by the capacity for replicative accumulation [144]. Techniques, including RNAi, morpholinos, promoter-driven DNA constructs for gain of function, ex ovo early embryo electroporation, and in ovo electroporation, stand at our disposal to investigate neurodevelopmental gene involvement in chick embryos [145]. A distinctive advantage lies in the chicken embryo’s capacity for comprehensive observation, enabling the association of genes or signals with phenotypes. These studies, uncovering the effects of RAS signaling modulation on chick embryo development, underscore the species’ potential in delving into the etiology of neurological disorders associated with RASopathies [144].
5. Limitations of Using Non-Mammalian Models
While non-mammalian models offer significant advantages for studying various diseases, including RASopathies, they are not without limitations that warrant consideration. In general terms, there are limitations when studying some of the most fundamental aspects of development, genetics, pathology, and disease mechanisms unique to humans in animal models. These drawbacks are even higher if the study is focused on disorders that affect the brain, where the most significant differences between humans and animal models have been found. Neuronal subtype complexity and human-specific aspects of gene expression and regulation are some of the key differences that limit the ability of animal models to recapitulate human brain development and to be used in identifying the underlying cellular and molecular mechanisms [146]. Another limitation to take in account is the difference between human and animal models in response to drugs, as pharmacokinetics and pharmacodynamics vary not only between species but also between individuals from the same species [147]. Also, it is well known that mammalian models are evolutionary closer to humans and thus are more similar to them than are non-mammalian models [148].
Zebrafish has a comparatively lower brain complexity when compared to humans and mammals, and this, coupled with the field’s relatively early stage, presents challenges. Also the variability in responses due to a low inbreeding rate can complicate data analysis [149]. Drosophila models mainly rely on human disease-causing gene overexpression in fly eyes, which lack the complexity and significant anatomical differences seen in the human brain [150]. In the case of C. elegans, its simplicity in terms of replicating human neural connections and cell interactions, which is critical for understanding the pathogenesis of neurodegenerative diseases, represents a limitation [129]. The yolk content within Xenopus embryos can hinder signal detection, and deep live imaging within early embryos is constrained in conventional imaging methods. Additionally, the longer generation time and reproduction cycle can impact experimental frequency [151]. Susceptibility to high doses of potentially lethal chemicals and the influences of poorly understood factors like breeder genetics, age, egg size, and incubation conditions are limitations in using chick embryos [152,153].
6. Effective Neurological Treatments and RASopathy Inhibitors Used in Non-Mammalian Models
Currently, there are no definitive cures for RASopathies. However, ongoing research focuses on the exploration of the RAS/MAPK signaling pathway, aiming to develop pharmaceutical interventions that may effectively alleviate neurological symptoms. However, the complexity of the signaling pathway and the diversity of the symptoms associated with RASopathies make drug development challenging. Still, there are several treatments and therapies available, based on the medical issues of each patient [7,11,19,154]. Neuropharmacology is a promising approach for the treatment of neurological symptoms in RASopathies, and studies using non-mammalian model organisms may help to identify potential therapeutic targets (Figure 2).
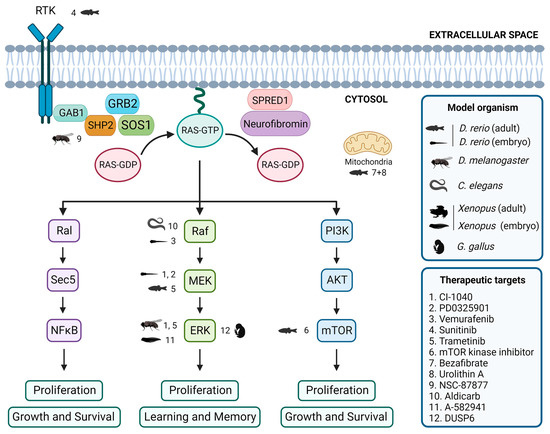
Figure 2.
Evaluation of inhibitors and drugs for RASopathies in non-mammalian models. Overview of inhibitors and drugs tested in non-mammalian models, showing the associations between tested drugs, target proteins, and the corresponding non-mammalian models. Created with BioRender.com.
In zebrafish embryos, studies have shown that expression of BRAFQ257R increased the major/minor axis ratio, a phenotype that was prevented by treatment with the MEK inhibitors CI-1040 and PD0325901 [104,155]. Similarly, the CFC-syndrome-like phenotypes associated with ectopic expression of BRAFV600E were profoundly ameliorated by simultaneous treatment with vemurafenib [105]. Furthermore, in the zebrafish model of NF1, treatment with sunitinib, a RTK inhibitor, effectively arrested the progression of transplanted MPNSTs, and combination treatment with both sunitinib and trametinib, a MEK inhibitor, enhanced this therapeutic effect [87]. Additionally, the study of the effects of various drugs on the zebrafish models of RASopathies has allowed the identification of targets of the RAS/MAPK pathway and the investigation of the mechanisms underlying the pathogeneses of RASopathies [69]. DNA Topo I and mTOR kinase inhibitors were identified as the most effective single agents in eliminating MPNST cells while avoiding excessive toxicity [156]. Treatment with a combination of bezafibrate and urolithin A provided a synergistic effect at a mitochondrial level, rescuing the genetic developmental defects in the CS zebrafish model [103].
The Drosophila model has been instrumental in identifying potential therapeutic targets for RASopathy-associated symptoms [126,157]. For example, preclinical studies using Drosophila have shown that inhibitors of the Ras/MAPK pathway, such as SHP2 inhibitor NSC-87877 [91] and MEK inhibitors CI-1040 [104] and trametinib [87], have potential therapeutic value. Studies in flies have also shown that activating the cAMP pathway can rescue neurofibromin deficiency [158]. In addition, the gene nemy has been identified as a potential therapeutic target for the treatment of cognitive impairment [159].
In C. elegans, expression of activated LIN-45/Raf in head acetylcholine neurons is sufficient to cause a waveform phenotype and hypersensitivity to the acetylcholinesterase inhibitor aldicarb, similar to an activated heterotrimeric G protein (Gq) mutant, suggesting that the ERK/MAPK pathway modulates the output of Gq-Rho signaling to control locomotion [160]. Furthermore, although not directly studied in RASopathy models, the GABAergic signaling pathway [161] and acetylcholine system [162] have been identified as potential therapeutic targets for treating cognitive impairment.
Several studies using Xenopus have identified potential therapeutic targets for treating neurological symptoms that may be present in patients affected by RASopathies. One study found that the use of A-582941, an agonist of the α7 nicotinic acetylcholine receptor (α7 nAChR), can lead to broad-spectrum efficacy at doses that enhance ERK1/2 and cAMP response element-binding protein (CREB) activation, and may represent a mechanism that offers potential for the improvement of cognitive deficits [163]. In another study, the effects of a 14-3-3ζ variant found in CFC syndrome patients were investigated, and the results suggested that the inhibition of the RAS/MAPK pathway could potentially be a therapeutic approach used to ameliorate CFC syndrome symptoms [139].
In chick embryos, only a limited number of studies have focused on the identification of key signaling pathways and targets that may be relevant in this context. One study investigated the effects of arsenic on NC cells in chick embryos and found that it induced gross abnormalities in craniofacial development and neural tube defects [164]. Inhibition of ERK1/2 activity by overexpressing the ERK1/2 phosphatase DUSP6 reduced the expression of the NC genes, although the way by which ERK1/2 signaling promotes NC induction is still unclear [14].
7. Discussion
The spectrum of neurological symptoms associated with RASopathies is underpinned by a myriad of factors, including the progressive nature of the disease, genetic modifiers, the intricate interplay of RASopathy-related gene mutations, and the relationships between tumor suppressors and neural tumors. Acknowledging the evolving nature of their symptoms over an individual’s lifespan and comprehending the genetic nuances that drive symptom diversity are paramount to a holistic understanding of the disease’s clinical landscape. Furthermore, a more comprehensive exploration of neural tumors within the context of RASopathies would provide a well-rounded depiction of the neurological intricacies that patients encounter. Importantly, delving into the intricacies of neural symptoms within the context of RASopathies not only contributes to a more thorough understanding of the associated neurological complexities, but also has broader implications, potentially shedding light on fundamental aspects of neurological diseases that are more complex.
The use of non-mammalian model organisms, such as zebrafish, Drosophila, C. elegans, Xenopus and chick embryos, has been instrumental in advancing our understanding of RASopathies and developing new neuropharmacological approaches. No single model can fully replicate human diseases, and further research is required to bridge the gap between these models and clinical settings. Non-mammalian model organisms provide a simplified system for studying the RAS/MAPK signaling pathway and the underlying mechanisms of neuroprotection and neurotoxicity, which can be challenging to study in mammalian models. Based on the current literature, it seems that some of the less common RASopathies, such as NSML and CFC syndrome, have not been studied as extensively as some of the more common RASopathies like NF1 and NS. There are still several common RASopathy variants identified in humans that have yet to be modeled in non-mammalian models. For example, the PTPN11 mutation A72T is common in NS but has not been studied in any non-mammalian model. Similarly, the RAF1 mutation G266E, which is also commonly found in NS, has not been studied in any non-mammalian models. In addition, some mutations that have been studied in non-mammalian models have not been identified as common variants in humans. For example, the RAF1 mutation S259A has been studied in zebrafish but is not a common variant in humans [165]. It is important to note that the study of RASopathies is an ongoing and constantly evolving field, and new variants are constantly being identified and studied. So, while some variants may not have been modeled yet, they may be studied in the future as new research is conducted.
Non-mammalian models have significantly contributed to the quest for effective treatments in the realm of RASopathies. Studies in C. elegans have underscored the intricate relationship between the ERK/MAPK pathway and locomotion, indirectly hinting at potential intervention points. Zebrafish models have shown promise in assessing RASopathy inhibitors, with MEK inhibitors and RTK inhibitors demonstrating potential therapeutic value. Drosophila has played a pivotal role in identifying therapeutic targets for RASopathy-associated cognitive deficits, including inhibitors of the RAS/MAPK pathway and the activation of the cAMP pathway. Xenopus models have unveiled potential targets for the amelioration of cognitive deficits in RASopathies, and although chick embryos are relatively less explored in this context, they have begun shedding light on the impact of environmental factors and ERK1/2 signaling. The chick embryo model emerges as an indispensable asset used for uncovering the underlying mechanisms of neurological disorders within the spectrum of RASopathies. Its utility extends to the identification of new neuroprotective compounds and a heightened comprehension of the RAS-MAPK pathway’s role in neurodevelopment. Significantly, the potential of chick embryos in drug discovery for RASopathies is underscored, demonstrating their transformative capacity in advancing therapeutic avenues. These non-mammalian models offer diverse advantages, from cost-effectiveness to the ability to study genetic modifiers and pathway interactions, collectively aiding in the quest for effective treatments in the complex landscape of neurobiology.
Current research suggests a dynamic interplay between non-mammalian, mouse and iPSC models in RASopathy research. Zebrafish and C. elegans models have identified key signaling pathways and potential therapeutic targets, which have then been validated and further characterized in mice. Subsequently, iPSC models can be used to refine these findings in a human context, considering individual patients’ variations. This multi-model approach strengthens the translational potential of research findings and increases the likelihood of successful therapeutic development. While non-mammalian models offer valuable starting points, mice and iPSCs play critical roles in validating and translating their findings towards clinical application. The future lies in a collaborative approach, leveraging the strengths of each model system to bridge the gap between basic research and effective therapies for RASopathies and other complex diseases. Additionally, advanced computational tools are emerging to integrate data from diverse models, providing a more comprehensive understanding of RASopathies.
While non-mammalian models have been invaluable in understanding RASopathies and developing therapeutic approaches, the future may hold even more revolutionary solutions. The emergence of powerful technologies like artificial intelligence (AI) has the potential to redefine the way we think about and develop therapies, potentially even questioning the need for traditional model systems altogether. These systems could identify intricate patterns and connections that might be missed by traditional research methods, leading to the discovery of novel therapeutic targets and personalized treatment strategies tailored to individual patients’ unique genetic makeup. AI could also be used to simulate complex biological processes, including disease progression and drug interactions, with a level of detail and accuracy that surpasses current model systems. However, it is crucial to remember that it is a tool, not a replacement for biological understanding, and it still needs further development to accurately predict or stimulate the outcome of a disease and drug interaction. Model systems, especially those incorporating human-derived cells or organoids, will still play a vital role in validating AI-generated predictions and testing the safety and efficacy of potential therapies in a more biological context. The convergence of AI and other emerging technologies will undoubtedly reshape the landscape of therapeutic discovery, offering exciting possibilities for the future of medicine.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/biomedicines12040841/s1, Table S1: Conservation of the RASopathy genes in non-mammalian model organisms. This table illustrates the percentage of identity between genes associated with RASopathies and their orthologs in non-mammalian models, focusing exclusively on those genes and models mentioned in the text. In genes associated with Noonan syndrome (NS), asterisks are used to indicate an additional connection with Noonan Syndrome with Multiple Lentigines (NSML, denoted by *) or syndromes resembling Noonan syndrome (**). Data have been obtained from DIOPT Ortholog Finder (accessed on 31 August 2023, https://www.flyrnai.org/cgi-bin/DRSC_orthologs.pl) and Ensembl Bio Mart (accessed on 31 August 2023, https://www.ensembl.org/info/data/biomart/index.html); Table S2: RASopathy-associated mutations studied in non-mammalian model organisms. Collation of investigated RASopathy-associated mutations in non-mammalian models emphasized in this review, along with observed related features. In genes related to Noonan syndrome (NS), double asterisks (**) signify an additional correlation between the mutations and Noonan-like syndrome.
Author Contributions
Conceptualization, J.L., M.R.-M., J.B.-F. and V.R.; investigation, J.L., M.R.-M. and V.R.; writing—original draft preparation, J.L., M.R.-M. and J.B.-F.; writing—review and editing, J.L., M.R.-M., J.B.-F., V.R., M.I.-G. and P.P.-M.; supervision, J.L.; funding acquisition, P.P.-M. All authors have read and agreed to the published version of the manuscript.
Funding
This study was made possible through the support of the Alicia Koplowitz Foundation, provided via the Research Grants program (Code: FAK21/001). Additionally, funding from the European Union—NextGenerationEU, under the “Programa Investigo,” played a crucial role in facilitating this study. We also extend our appreciation to Banco Santander and the University of Salamanca (USAL) for their financial backing through the “Becas para realizar estudios de Doctorado en la Universidad de Salamanca destinadas a estudiantes latinoamericanos” program. Specifically, the salary support for Mario Rodríguez Martín was sustained by the Programa Investigo, while Juan Báez Flores received a predoctoral fellowship from Banco Santander. We express our sincere gratitude to the Alicia Koplowitz Foundation, SEPE, the European Union, Banco Santander, and USAL for their instrumental contributions, which were vital for the successful completion of this study. The APC was waived by Biomedicines.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bahar, M.E.; Kim, H.J.; Kim, D.R. Targeting the RAS/RAF/MAPK Pathway for Cancer Therapy: From Mechanism to Clinical Studies. Signal Transduct. Target. Ther. 2023, 8, 455. [Google Scholar] [CrossRef] [PubMed]
- Cizmarova, M.; Kostalova, L.; Pribilincova, Z.; Lasabova, Z.; Hlavata, A.; Kovacs, L.; Ilencikova, D. Rasopathies—Dysmorphic Syndromes with Short Stature and Risk of Malignancy. Endocr. Regul. 2013, 47, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Montero-Bullón, J.-F.; González-Velasco, Ó.; Isidoro-García, M.; Lacal, J. Integrated in Silico MS-Based Phosphoproteomics and Network Enrichment Analysis of RASopathy Proteins. Orphanet J. Rare Dis. 2021, 16, 303. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.J.; van der Smagt, J.J.; Rosenfeld, J.A.; Pagnamenta, A.T.; Alswaid, A.; Baker, E.H.; Blair, E.; Borck, G.; Brinkmann, J.; Craigen, W.; et al. Autosomal Recessive Noonan Syndrome Associated with Biallelic LZTR1 Variants. Genet. Med. 2018, 20, 1175–1185. [Google Scholar] [CrossRef] [PubMed]
- Motta, M.; Fasano, G.; Gredy, S.; Brinkmann, J.; Bonnard, A.A.; Simsek-Kiper, P.O.; Gulec, E.Y.; Essaddam, L.; Utine, G.E.; Guarnetti Prandi, I.; et al. SPRED2 Loss-of-Function Causes a Recessive Noonan Syndrome-like Phenotype. Am. J. Hum. Genet. 2021, 108, 2112–2129. [Google Scholar] [CrossRef] [PubMed]
- Rauen, K.A.; Alsaegh, A.; Ben-Shachar, S.; Berman, Y.; Blakeley, J.; Cordeiro, I.; Elgersma, Y.; Evans, D.G.; Fisher, M.J.; Frayling, I.M.; et al. First International Conference on RASopathies and Neurofibromatoses in Asia: Identification and Advances of New Therapeutics. Am. J. Med. Genet. Part A 2019, 179, 1091–1097. [Google Scholar] [CrossRef]
- Hebron, K.E.; Hernandez, E.R.; Yohe, M.E. The RASopathies: From Pathogenetics to Therapeutics. Dis. Model. Mech. 2022, 15, dmm049107. [Google Scholar] [CrossRef]
- Weaver, K.N.; Gripp, K.W. Central Nervous System Involvement in Individuals with RASopathies. Am. J. Med. Genet. Part C Semin. Med. Genet. 2022, 190, 494–500. [Google Scholar] [CrossRef]
- Kim, Y.E.; Baek, S.T. Neurodevelopmental Aspects of Rasopathies. Mol. Cells 2019, 42, 441–447. [Google Scholar] [CrossRef]
- Borrie, S.C.; Brems, H.; Legius, E.; Bagni, C. Cognitive Dysfunctions in Intellectual Disabilities: The Contributions of the Ras-MAPK and PI3K-AKT-MTOR Pathways. Annu. Rev. Genomics Hum. Genet. 2017, 18, 115–142. [Google Scholar] [CrossRef]
- Kontaridis, M.I.; Roberts, A.E.; Schill, L.; Schoyer, L.; Stronach, B.; Andelfinger, G.; Aoki, Y.; Axelrad, M.E.; Bakker, A.; Bennett, A.M.; et al. The Seventh International RASopathies Symposium: Pathways to a Cure—Expanding Knowledge, Enhancing Research, and Therapeutic Discovery. Am. J. Med. Genet. Part A 2022, 188, 1915–1927. [Google Scholar] [CrossRef] [PubMed]
- Jindal, G.A.; Goyal, Y.; Burdine, R.D.; Rauen, K.A.; Shvartsman, S.Y. RASopathies: Unraveling Mechanisms with Animal Models. Dis. Model. Mech. 2015, 8, 769–782. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.; Lee, Y.S. The Impact of RASopathy-Associated Mutations on CNS Development in Mice and Humans. Mol. Brain 2019, 12, 96. [Google Scholar] [CrossRef] [PubMed]
- Dinsmore, C.J.; Soriano, P. MAPK and PI3K Signaling: At the Crossroads of Neural Crest Development. Dev. Biol. 2018, 444, S79–S97. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.; Lalli, G. Rho and Ras GTPases in Axon Growth, Guidance, and Branching. Cold Spring Harb. Lab. Press 2010, 2, a001818. [Google Scholar] [CrossRef]
- Han, Y.; Chen, M.; Wang, H. Chiari I Malformation in Patients with RASopathies. Child’s Nerv. Syst. 2021, 37, 1831–1836. [Google Scholar] [CrossRef] [PubMed]
- Ayaz, E.; Yıldırım, R.; Çelebi, C.; Ozalkak, S. Noonan Syndrome: Neuroimaging Findings and Morphometric Analysis of the Cranium Base and Posterior Fossa in Children. J. Neuroimaging 2023, 33, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Bayat, M.; Bayat, A. Neurological Manifestations of Neurofibromatosis: A Review. Neurol. Sci. 2020, 41, 2685–2690. [Google Scholar] [CrossRef] [PubMed]
- Zenker, M. Clinical Overview on RASopathies. Am. J. Med. Genet. Part C Semin. Med. Genet. 2022, 190, 414–424. [Google Scholar] [CrossRef]
- Ryu, H.H.; Lee, Y.S. Cell Type-Specific Roles of RAS-MAPK Signaling in Learning and Memory: Implications in Neurodevelopmental Disorders. Neurobiol. Learn. Mem. 2016, 135, 13–21. [Google Scholar] [CrossRef]
- Lazzaro, G.; Caciolo, C.; Menghini, D.; Cumbo, F.; Digilio, M.C.; Capolino, R.; Zampino, G.; Tartaglia, M.; Vicari, S.; Alfieri, P. Defining Language Disorders in Children and Adolescents with Noonan Syndrome. Mol. Genet. Genomic Med. 2020, 8, e1069. [Google Scholar] [CrossRef] [PubMed]
- Adviento, B.; Corbin, I.L.; Widjaja, F.; Desachy, G.; Enrique, N.; Rosser, T.; Risi, S.; Marco, E.J.; Hendren, R.L.; Bearden, C.E.; et al. Autism Traits in the RASopathies. J. Med. Genet. 2014, 51, 10–20. [Google Scholar] [CrossRef]
- Davies, O.M.T.; Bruckner, A.L.; McCalmont, T.; Mascarenhas, L.; Oza, V.; Williams, M.L.; Wine-Lee, L.; Shern, J.F.; Siegel, D.H. Cutaneous Mosaic RASopathies Associated with Rhabdomyosarcoma. Pediatr. Blood Cancer 2022, 69, e29639. [Google Scholar] [CrossRef] [PubMed]
- Ly, K.I.; Blakeley, J.O. The Diagnosis and Management of Neurofibromatosis Type 1. Med. Clin. North Am. 2019, 103, 1035–1054. [Google Scholar] [CrossRef] [PubMed]
- Staedtke, V.; Bai, R.Y.; Blakeley, J.O. Cancer of the Peripheral Nerve in Neurofibromatosis Type 1. Neurotherapeutics 2017, 14, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Báez-Flores, J.; Rodríguez-Martín, M.; Lacal, J. The Therapeutic Potential of Neurofibromin Signaling Pathways and Binding Partners. Commun. Biol. 2023, 6, 436. [Google Scholar] [CrossRef]
- Dard, L.; Bellance, N.; Lacombe, D.; Rossignol, R. RAS Signalling in Energy Metabolism and Rare Human Diseases. Biochim. Biophys. Acta—Bioenerg. 2018, 1859, 845–867. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.S.J.; Chopra, M.; Kim, R.H.; Parkin, P.C.; Barnett-Tapia, C. Incidence and Prevalence of Neurofibromatosis Type 1 and 2: A Systematic Review and Meta-Analysis. Orphanet J. Rare Dis. 2023, 18, 292. [Google Scholar] [CrossRef] [PubMed]
- Debrabant, J.; Plasschaert, E.; Caeyenberghs, K.; Vingerhoets, G.; Legius, E.; Janssens, S.; Van Waelvelde, H. Deficient Motor Timing in Children with Neurofibromatosis Type 1. Res. Dev. Disabil. 2014, 35, 3131–3138. [Google Scholar] [CrossRef]
- Lehtonen, A.; Howie, E.; Trump, D.; Huson, S.M. Behaviour in Children with Neurofibromatosis Type 1: Cognition, Executive Function, Attention, Emotion, and Social Competence. Dev. Med. Child Neurol. 2013, 55, 111–125. [Google Scholar] [CrossRef]
- Plasschaert, E.; Van Eylen, L.; Descheemaeker, M.J.; Noens, I.; Legius, E.; Steyaert, J. Executive Functioning Deficits in Children with Neurofibromatosis Type 1: The Influence of Intellectual and Social Functioning. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2016, 171, 348–362. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.M.; Acosta, M.T.; Garg, S.; Green, J.; Huson, S.; Legius, E.; North, K.N.; Payne, J.M.; Plasschaert, E.; Frazier, T.W.; et al. Disease Burden and Symptom Structure of Autism in Neurofibromatosis Type 1 A Study of the International NF1-ASD Consortium Team (INFACT) Supplemental Content. JAMA Psychiatry 2016, 73, 1276–1284. [Google Scholar] [CrossRef] [PubMed]
- Plasschaert, E.; Descheemaeker, M.J.; Van Eylen, L.; Noens, I.; Steyaert, J.; Legius, E. Prevalence of Autism Spectrum Disorder Symptoms in Children with Neurofibromatosis Type 1. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2015, 168, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Mainberger, F.; Langer, S.; Mall, V.; Jung, N.H. Impaired Synaptic Plasticity in RASopathies: A Mini-Review. J. Neural Transm. 2016, 123, 1133–1138. [Google Scholar] [CrossRef] [PubMed]
- Zimerman, M.; Wessel, M.J.; Timmermann, J.E.; Granström, S.; Gerloff, C.; Mautner, V.F.; Hummel, F.C. Impairment of Procedural Learning and Motor Intracortical Inhibition in Neurofibromatosis Type 1 Patients. EBioMedicine 2015, 2, 1430–1437. [Google Scholar] [CrossRef] [PubMed]
- Brems, H.; Beert, E.; de Ravel, T.; Legius, E. Mechanisms in the Pathogenesis of Malignant Tumours in Neurofibromatosis Type 1. Lancet Oncol. 2009, 10, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Daniel, R.; De Souza, M.R.; Pipek, L.Z.; Fagundes, C.F.; Solla, D.J.F.; Carlos, G.; Godoy, D.A.; Kolias, A.G.; Luis, R.; Amorim, O.; et al. External Validation of the Glasgow Coma Scale-Pupils in Low- to Middle-Income Country Patients with Traumatic Brain Injury: Could “ Motor Score-Pupil ” Have Higher Prognostic Value ? Surg. Neurol. Int. 2022, 13, 4–7. [Google Scholar] [CrossRef]
- Williams, K.B.; Largaespada, D.A. New Model Systems and the Development of Targeted Therapies for the Treatment of Neurofibromatosis Type 1-Associated Malignant Peripheral Nerve Sheath Tumors. Genes 2020, 11, 477. [Google Scholar] [CrossRef] [PubMed]
- Brems, H.; Chmara, M.; Sahbatou, M.; Denayer, E.; Taniguchi, K.; Kato, R.; Somers, R.; Messiaen, L.; De Schepper, S.; Fryns, J.P.; et al. Germline Loss-of-Function Mutations in SPRED1 Cause a Neurofibromatosis 1-like Phenotype. Nat. Genet. 2007, 39, 1120–1126. [Google Scholar] [CrossRef] [PubMed]
- Bergoug, M.; Doudeau, M.; Godin, F.; Mosrin, C.; Vallée, B.; Bénédetti, H. Neurofibromin Structure, Functions and Regulation. Cells 2020, 9, 2365. [Google Scholar] [CrossRef]
- Mo, J.; Moye, S.L.; McKay, R.M.; Le, L.Q. Neurofibromin and Suppression of Tumorigenesis: Beyond the GAP. Oncogene 2022, 41, 1235–1251. [Google Scholar] [CrossRef] [PubMed]
- Denayer, E.; Chmara, M.; Brems, H.; Kievit, A.M.; Van Bever, Y.; Van Den Ouweland, A.M.W.; Van Minkelen, R.; De Goede-Bolder, A.; Oostenbrink, R.; Lakeman, P.; et al. Legius Syndrome in Fourteen Families. Hum. Mutat. 2011, 32, 1985–1998. [Google Scholar] [CrossRef] [PubMed]
- Brems, H.; Pasmant, E.; Van Minkelen, R.; Wimmer, K.; Upadhyaya, M.; Legius, E.; Messiaen, L. Review and Update of SPRED1 Mutations Causing Legius Syndrome. Hum. Mutat. 2012, 33, 1538–1546. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, C.; McCormick, F. SPRED Proteins and Their Roles in Signal Transduction, Development, and Malignancy. Genes Dev. 2020, 34, 1410–1421. [Google Scholar] [CrossRef]
- Chłopek, M.; Lasota, J.; Thompson, L.D.R.; Szczepaniak, M.; Kuźniacka, A.; Hińcza, K.; Kubicka, K.; Kaczorowski, M.; Newford, M.; Liu, Y.; et al. Alterations in Key Signaling Pathways in Sinonasal Tract Melanoma. A Molecular Genetics and Immunohistochemical Study of 90 Cases and Comprehensive Review of the Literature. Mod. Pathol. 2022, 35, 1609–1617. [Google Scholar] [CrossRef] [PubMed]
- Der Van Burgt, I.; Brunner, H. Genetic Heterogeneity in Noonan Syndrome: Evidence for an Autosomal Recessive Form. Am. J. Med. Genet. 2000, 94, 46–51. [Google Scholar] [CrossRef]
- Pierpont, E.I.; Tworog-Dube, E.; Roberts, A.E. Attention Skills and Executive Functioning in Children with Noonan Syndrome and Their Unaffected Siblings. Dev. Med. Child Neurol. 2015, 57, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Romano, A.A.; Allanson, J.E.; Dahlgren, J.; Gelb, B.D.; Hall, B.; Pierpont, M.E.; Roberts, A.E.; Robinson, W.; Takemoto, C.M.; Noonan, J.A. Noonan Syndrome: Clinical Features, Diagnosis, and Management Guidelines. Pediatrics 2010, 126, 746–759. [Google Scholar] [CrossRef]
- Garg, S.; Brooks, A.; Burns, A.; Burkitt-Wright, E.; Kerr, B.; Huson, S.; Emsley, R.; Green, J. Autism Spectrum Disorder and Other Neurobehavioural Comorbidities in Rare Disorders of the Ras/MAPK Pathway. Dev. Med. Child Neurol. 2017, 59, 544–549. [Google Scholar] [CrossRef]
- De Ridder, W.; van Engelen, B.; van Alfen, N. Neurological Features of Noonan Syndrome and Related RASopathies: Pain and Nerve Enlargement Characterized by Nerve Ultrasound. Am. J. Med. Genet. Part A 2022, 188, 1801–1807. [Google Scholar] [CrossRef]
- Siegfried, A.; Cances, C.; Denuelle, M.; Loukh, N.; Tauber, M.; Cavé, H.; Delisle, M.B. Noonan Syndrome, PTPN11 Mutations, and Brain Tumors. A Clinical Report and Review of the Literature. Am. J. Med. Genet. Part A 2017, 173, 1061–1065. [Google Scholar] [CrossRef] [PubMed]
- Aoki, Y.; Niihori, T.; Kawame, H.; Kurosawa, K.; Ohashi, H.; Tanaka, Y.; Filocamo, M.; Kato, K.; Suzuki, Y.; Kure, S.; et al. Germline Mutations in HRAS Proto-Oncogene Cause Costello Syndrome. Nat. Genet. 2005, 37, 1038–1040. [Google Scholar] [CrossRef]
- Tidyman, W.E.; Rauen, K.A. Noonan, Costello and Cardio-Facio-Cutaneous Syndromes: Dysregulation of the Ras-MAPK Pathway. Expert Rev. Mol. Med. 2008, 10, e37. [Google Scholar] [CrossRef] [PubMed]
- Kratz, C.P.; Franke, L.; Peters, H.; Kohlschmidt, N.; Kazmierczak, B.; Finckh, U.; Bier, A.; Eichhorn, B.; Blank, C.; Kraus, C.; et al. Cancer Spectrum and Frequency among Children with Noonan, Costello, and Cardio-Facio-Cutaneous Syndromes. Br. J. Cancer 2015, 112, 1392–1397. [Google Scholar] [CrossRef] [PubMed]
- Axelrad, M.E.; Schwartz, D.D.; Katzenstein, J.M.; Hopkins, E.; Gripp, K.W. Neurocognitive, Adaptive, and Behavioral Functioning of Individuals with Costello Syndrome: A Review. Am. J. Med. Genet. Part C Semin. Med. Genet. 2011, 157, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Dileone, M.; Profice, P.; Pilato, F.; Alfieri, P.; Cesarini, L.; Mercuri, E.; Leoni, C.; Tartaglia, M.; Di Iorio, R.; Zampino, G.; et al. Enhanced Human Brain Associative Plasticity in Costello Syndrome. J. Physiol. 2010, 588, 3445–3456. [Google Scholar] [CrossRef] [PubMed]
- Dileone, M.; Ranieri, F.; Florio, L.; Capone, F.; Musumeci, G.; Leoni, C.; Mordillo-Mateos, L.; Tartaglia, M.; Zampino, G.; Di Lazzaro, V. Differential Effects of HRAS Mutation on LTP-Like Activity Induced by Different Protocols of Repetitive Transcranial Magnetic Stimulation. Brain Stimul. 2016, 9, 33–38. [Google Scholar] [CrossRef]
- Pierpont, M.E.M.; Magoulas, P.L.; Adi, S.; Kavamura, M.I.; Neri, G.; Noonan, J.; Pierpont, E.I.; Reinker, K.; Roberts, A.E.; Shankar, S.; et al. Cardio-Facio-Cutaneous Syndrome: Clinical Features, Diagnosis, and Management Guidelines. Pediatrics 2014, 134, e1149–e1162. [Google Scholar] [CrossRef] [PubMed]
- Hamdan, F.F.; Gauthier, J.; Spiegelman, D.; Noreau, A.; Yang, Y.; Pellerin, S.; Dobrzeniecka, S.; Côté, M.; Perreau-Linck, E.; Carmant, L.; et al. Mutations in SYNGAP1 in Autosomal Nonsyndromic Mental Retardation. N. Engl. J. Med. 2009, 360, 599–605. [Google Scholar] [CrossRef]
- Tidyman, W.E.; Rauen, K.A. Expansion of the RASopathies. Curr. Genet. Med. Rep. 2016, 4, 57–64. [Google Scholar] [CrossRef]
- Meili, F.; Wei, W.J.; Sin, W.-C.; Meyers, W.M.; Dascalu, I.; Callaghan, D.B.; Rogic, S.; Pavlidis, P.; Haas, K. Multi-Parametric Analysis of 57 SYNGAP1 Variants Reveal Impacts on GTPase Signaling, Localization, and Protein Stability. Am. J. Hum. Genet. 2021, 108, 148–162. [Google Scholar] [CrossRef]
- Mignot, C.; von Stülpnage, C.; Nava, C.; Ville, D.; Sanlaville, D.; Lesca, G.; Rastetter, A.; Gachet, B.; Marie, Y.; Korenke, G.C.; et al. Genetic and Neurodevelopmental Spectrum of SYNGAP1-Associated Intellectual Disability and Epilepsy. J. Med. Genet. 2016, 53, 511–522. [Google Scholar] [CrossRef]
- De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Cicek, A.E.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S.; et al. Synaptic, Transcriptional and Chromatin Genes Disrupted in Autism. Nature 2014, 515, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.M.; Moran, J.L.; Fromer, M.; Ruderfer, D.; Solovieff, N.; Roussos, P.; O’Dushlaine, C.; Chambert, K.; Bergen, S.E.; Kähler, A.; et al. A Polygenic Burden of Rare Disruptive Mutations in Schizophrenia. Nature 2014, 506, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Llamosas, N.; Arora, V.; Vij, R.; Kilinc, M.; Bijoch, L.; Rojas, C.; Reich, A.; Sridharan, B.P.; Willems, E.; Piper, D.R.; et al. SYNGAP1 Controls the Maturation of Dendrites, Synaptic Function, and Network Activity in Developing Human Neurons. J. Neurosci. 2020, 40, 7980–7994. [Google Scholar] [CrossRef]
- Paridaen, J.T.; Huttner, W.B. Neurogenesis during Development of the Vertebrate Central Nervous System. EMBO Rep. 2014, 15, 351–364. [Google Scholar] [CrossRef]
- Hanashima, C.; Toma, K. Switching Modes in Corticogenesis: Mechanisms of Neuronal Subtype Transitions and Integration in the Cerebral Cortex. Front. Neurosci. 2015, 9, 274. [Google Scholar]
- Motta, M.; Pannone, L.; Pantaleoni, F.; Bocchinfuso, G.; Radio, F.C.; Cecchetti, S.; Ciolfi, A.; Di Rocco, M.; Elting, M.W.; Brilstra, E.H.; et al. Enhanced MAPK1 Function Causes a Neurodevelopmental Disorder within the RASopathy Clinical Spectrum. Am. J. Hum. Genet. 2020, 107, 499–513. [Google Scholar] [CrossRef]
- Patterson, V.L.; Burdine, R.D. Swimming toward Solutions: Using Fish and Frogs as Models for Understanding RASopathies. Birth Defects Res. 2020, 112, 749–765. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-H.; Yoon, D.S. A Phenotype-Based RNAi Screening for Ras-ERK/MAPK Signaling-Associated Stem Cell Regulators in C. Elegans. Physiol. Behav. 2017, 176, 139–148. [Google Scholar] [CrossRef]
- McMullan, R.; Anderson, A.; Nurrish, S. Behavioral and Immune Responses to Infection Require Gαq- RhoA Signaling in C. Elegans. PLoS Pathog. 2012, 8, e1002530. [Google Scholar] [CrossRef] [PubMed]
- Tandon, P.; Conlon, F.; Furlow, J.D.; Horb, M.E. Expanding the Genetic Toolkit in Xenopus: Approaches and Opportunities for Human Disease Modeling. Dev. Biol. 2017, 426, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Wilmerding, A.; Bouteille, L.; Caruso, N.; Bidaut, G.; Etchevers, H.C.; Graba, Y.; Delfini, M.C. Sustained Experimental Activation of FGF8/ERK in the Developing Chicken Spinal Cord Models Early Events in ERK-Mediated Tumorigenesis. Neoplasia 2022, 24, 120–132. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, E.J.; Turner, K.J.; Fernandez, J.M.; Cifuentes, D.; Ghosh, M.; Ijaz, S.; Jain, R.A.; Kubo, F.; Bill, B.R.; Baier, H.; et al. Estrogens Suppress a Behavioral Phenotype in Zebrafish Mutants of the Autism Risk Gene, CNTNAP2. Neuron 2016, 89, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Ketley, A.; Chen, C.Z.; Li, X.; Arya, S.; Robinson, T.E.; Granados-Riveron, J.; Udosen, I.; Morris, G.E.; Holt, I.; Furling, D.; et al. High-Content Screening Identifies Small Molecules That Remove Nuclear Foci, Affect MBNL Distribution and CELF1 Protein Levels via a PKC-Independent Pathway in Myotonic Dystrophy Cell Lines. Hum. Mol. Genet. 2014, 23, 1551–1562. [Google Scholar] [CrossRef] [PubMed]
- Aoki, Y.; Niihori, T.; Banjo, T.; Okamoto, N.; Mizuno, S.; Kurosawa, K.; Ogata, T.; Takada, F.; Yano, M.; Ando, T.; et al. Gain-of-Function Mutations in RIT1 Cause Noonan Syndrome, a RAS/MAPK Pathway Syndrome. Am. J. Hum. Genet. 2013, 93, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Vissers, L.E.L.M.; Bonetti, M.; Paardekooper Overman, J.; Nillesen, W.M.; Frints, S.G.M.; De Ligt, J.; Zampino, G.; Justino, A.; Machado, J.C.; Schepens, M.; et al. Heterozygous Germline Mutations in A2ML1 Are Associated with a Disorder Clinically Related to Noonan Syndrome. Eur. J. Hum. Genet. 2015, 23, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Pandit, B.; Sarkozy, A.; Pennacchio, L.A.; Carta, C.; Oishi, K.; Martinelli, S.; Pogna, E.A.; Schackwitz, W.; Ustaszewska, A.; Landstrom, A.; et al. Gain-of-Function RAF1 Mutations Cause Noonan and LEOPARD Syndromes with Hypertrophic Cardiomyopathy. Nat. Genet. 2007, 39, 1007–1012. [Google Scholar] [CrossRef] [PubMed]
- Bonetti, M.; Rodriguez-Martinez, V.; Overman, J.P.; Overvoorde, J.; Van Eekelen, M.; Jopling, C.; Den Hertog, J. Distinct and Overlapping Functions of Ptpn11 Genes in Zebrafish Development. PLoS ONE 2014, 9, e94884. [Google Scholar] [CrossRef]
- Pierpont, M.E.; Brueckner, M.; Chung, W.K.; Garg, V.; Lacro, R.V.; Mcguire, A.L.; Mital, S.; Priest, J.R.; Pu, W.T.; Roberts, A.; et al. Genetic Basis for Congenital Heart Disease: Revisited: A scientific statement from the American Heart Association. Circulation 2018, 138, e653–e711. [Google Scholar] [CrossRef]
- Dang, M.; Fogley, R.; Zon, L.I. Identifying Novel Cancer Therapies Using Chemical Genetics and Zebrafish. In Cancer and Zebrafish: Mechanisms, Techniques, and Models; Springer: Cham, Switzerland, 2016; pp. 103–124. [Google Scholar] [CrossRef]
- Al-Hamaly, M.A.; Turner, L.T.; Rivera-Martinez, A.; Rodriguez, A.; Blackburn, J.S. Zebrafish Cancer Avatars: A Translational Platform for Analyzing Tumor Heterogeneity and Predicting Patient Outcomes. Int. J. Mol. Sci. 2023, 24, 2288. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Padmanabhan, A.; De Groh, E.D.; Lee, J.S.; Haidar, S.; Dahlberg, S.; Guo, F.; He, S.; Wolman, M.A.; Granato, M.; et al. Zebrafish Neurofibromatosis Type 1 Genes Have Redundant Functions in Tumorigenesis and Embryonic Development. Dis. Model. Mech. 2012, 5, 881–894. [Google Scholar] [CrossRef] [PubMed]
- Kobar, K.; Collett, K.; Prykhozhij, S.V.; Berman, J.N. Zebrafish Cancer Predisposition Models. Front. Cell Dev. Biol. 2021, 9, 660069. [Google Scholar] [CrossRef] [PubMed]
- Wolman, M.A.; deGroh, E.D.; McBride, S.M.; Jongens, T.A.; Granato, M.; Epstein, J.A. Modulation of CAMP and Ras Signaling Pathways Improves Distinct Behavioral Deficits in a Zebrafish Model of Neurofibromatosis Type 1. Cell Rep. 2014, 8, 1265–1270. [Google Scholar] [CrossRef] [PubMed]
- Holtkamp, N.; Mautner, V.F.; Friedrich, R.E.; Harder, A.; Hartmann, C.; Theallier-Janko, A.; Hoffmann, K.T.; von Deimling, A. Differentially Expressed Genes in Neurofibromatosis 1-Associated Neurofibromas and Malignant Peripheral Nerve Sheath Tumors. Acta Neuropathol. 2004, 107, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Ki, D.H.; He, S.; Rodig, S.; Look, A.T. Overexpression of PDGFRA Cooperates with Loss of NF1 and P53 to Accelerate the Molecular Pathogenesis of Malignant Peripheral Nerve Sheath Tumors. Oncogene 2016, 36, 1058–1068. [Google Scholar] [CrossRef] [PubMed]
- Reijnders, M.R.F.; Ansor, N.M.; Kousi, M.; Yue, W.W.; Tan, P.L.; Clarkson, K.; Clayton-Smith, J.; Corning, K.; Jones, J.R.; Lam, W.W.K.; et al. RAC1 Missense Mutations in Developmental Disorders with Diverse Phenotypes. Am. J. Hum. Genet. 2017, 101, 466–477. [Google Scholar] [CrossRef]
- Jopling, C.; Van Geemen, D.; Den Hertog, J. Shp2 Knockdown and Noonan/LEOPARD Mutant Shp2-Induced Gastrulation Defects. PLoS Genet. 2007, 3, 2468–2476. [Google Scholar] [CrossRef] [PubMed]
- Oishi, K.; Gaengel, K.; Krishnamoorthy, S.; Kamiya, K.; Kim, I.K.; Ying, H.; Weber, U.; Perkins, L.A.; Tartaglia, M.; Mlodzik, M.; et al. Transgenic Drosophila Models of Noonan Syndrome Causing PTPN11 Gain-of-Function Mutations. Hum. Mol. Genet. 2006, 15, 543–553. [Google Scholar] [CrossRef]
- Pagani, M.R.; Oishi, K.; Gelb, B.D.; Zhong, Y. The Phosphatase SHP2 Regulates the Spacing Effect for Long-Term Memory Induction. Cell 2009, 139, 186–198. [Google Scholar] [CrossRef]
- Krens, S.F.G.; He, S.; Lamers, G.E.M.; Meijer, A.H.; Bakkers, J.; Schmidt, T.; Spaink, H.P.; Snaar-Jagalska, B.E. Distinct Functions for ERK1 and ERK2 in Cell Migration Processes during Zebrafish Gastrulation. Dev. Biol. 2008, 319, 370–383. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Eckerle, S.; Onichtchouk, D.; Marrs, J.A.; Nitschke, R.; Driever, W. Pou5f1-Dependent EGF Expression Controls E-Cadherin Endocytosis, Cell Adhesion, and Zebrafish Epiboly Movements. Dev. Cell 2013, 24, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Tada, M.; Heisenberg, C.P. Convergent Extension: Using Collective Cell Migration and Cell Intercalation to Shape Embryos. Dev. 2012, 139, 3897–3904. [Google Scholar] [CrossRef] [PubMed]
- Solman, M.; Blokzijl-franke, S.; Piques, F.; Yan, C.; Strullu, M.; Kamel, S.M.; Ak, P.; Bakkers, J.; Langenau, M.; den Hertog, J.; et al. Inflammatory Response in Hematopoietic Stem and Progenitor Cells Triggered by Activating SHP2 Mutations Potentiates Leukemogenesis. Elife 2022, 11, e73040. [Google Scholar] [CrossRef] [PubMed]
- Cuevas-Navarro, A.; Rodriguez-Muñoz, L.; Grego-Bessa, J.; Cheng, A.; Rauen, K.A.; Urisman, A.; McCormick, F.; Jimenez, G.; Castel, P. Cross-Species Analysis of LZTR1 Loss-of-Function Mutants Demonstrates Dependency to RIT1 Orthologs. Elife 2022, 11, e76495. [Google Scholar] [CrossRef] [PubMed]
- Niihori, T.; Nagai, K.; Fujita, A.; Ohashi, H.; Okamoto, N.; Okada, S.; Harada, A.; Kihara, H.; Arbogast, T.; Funayama, R.; et al. Germline-Activating RRAS2 Mutations Cause Noonan Syndrome. Am. J. Hum. Genet. 2019, 104, 1233–1240. [Google Scholar] [CrossRef] [PubMed]
- Leung, G.K.C.; Luk, H.M.; Tang, V.H.M.; Gao, W.W.; Mak, C.C.Y.; Yu, M.H.C.; Wong, W.L.; Chu, Y.W.Y.; Yang, W.L.; Wong, W.H.S.; et al. Integrating Functional Analysis in the Next-Generation Sequencing Diagnostic Pipeline of RASopathies. Sci. Rep. 2018, 8, 2421. [Google Scholar] [CrossRef]
- Radu, M.; Semenova, G.; Kosoff, R.; Chernoff, J. Pak Signaling in the Development and Progression of Cancer. Nat. Rev. Cancer 2014, 14, 13–25. [Google Scholar] [CrossRef]
- Araiza-Olivera, D.; Feng, Y.; Semenova, G.; Prudnikova, T.Y.; Rhodes, J.; Chernoff, J. Suppression of RAC1-Driven Malignant Melanoma by Group A PAK Inhibitors. Oncogene 2018, 37, 944–952. [Google Scholar] [CrossRef]
- Nissim, S.; Leshchiner, I.; Mancias, J.D.; Greenblatt, M.B.; Maertens, O.; Cassa, C.A.; Rosenfeld, J.A.; Cox, A.G.; Wucherpfennig, J.I.; Kim, A.J.; et al. Mutations in RABL3 alter KRAS prenylation and are associated with Hereditary Pancreatic Cancer. Nat. Genet. 2019, 51, 1308–1314. [Google Scholar] [CrossRef]
- Santoriello, C.; Deflorian, G.; Pezzimenti, F.; Kawakami, K.; Lanfrancone, L.; Di Fagagna, F.D.A.; Mione, M. Expression of H-RASV12 in a Zebrafish Model of Costello Syndrome Causes Cellular Senescence in Adult Proliferating Cells. Dis. Model. Mech. 2009, 2, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Dard, L.; Hubert, C.; Esteves, P.; Blanchard, W.; About, G.B.; Baldasseroni, L.; Dumon, E.; Angelini, C.; Delourme, M.; Guyonnet-Dupéra, V.; et al. HRAS Germline Mutations Impair LKB1/AMPK Signaling and Mitochondrial Homeostasis in Costello Syndrome Models. J. Clin. Investig. 2022, 132, e131053. [Google Scholar] [CrossRef] [PubMed]
- Anastasaki, C.; Estep, A.L.; Marais, R.; Rauen, K.A.; Patton, E.E. Kinase-Activating and Kinase-Impaired Cardio-Facio-Cutaneous Syndrome Alleles Have Activity during Zebrafish Development and Are Sensitive to Small Molecule Inhibitors. Hum. Mol. Genet. 2009, 18, 2543–2554. [Google Scholar] [CrossRef]
- Hong, J.; Lee, J.G.; Sohn, K.C.; Lee, K.; Lee, S.; Lee, J.; Hong, J.; Choi, D.; Hong, Y.; Jin, H.S.; et al. IQ-Switch Is a QF-Based Innocuous, Silencing-Free, and Inducible Gene Switch System in Zebrafish. Commun. Biol. 2021, 4, 1405. [Google Scholar] [CrossRef] [PubMed]
- Jindal, G.A.; Goyal, Y.; Yamay, K.; Futran, A.S.; Kountouridis, I.; Balgobin, C.A.; Schüpbach, T.; Burdine, R.D.C.; Shvartsman, S.Y. In Vivo Severity Ranking of Ras Pathway Mutations Associated with Developmental Disorders. Proc. Natl. Acad. Sci. USA 2017, 114, 510–515. [Google Scholar] [CrossRef]
- Goyal, Y.; Jindal, G.A.; Pelliccia, J.L.; Yamaya, K.; Yeung, E.; Futran, A.S.; Burdine, R.D.; Schüpbach, T.; Shvartsman, S.Y. Divergent Effects of Intrinsically Active MEK Variants on Developmental Ras Signaling. Nat. Genet. 2017, 49, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Brumby, A.M.; Goulding, K.R.; Schlosser, T.; Loi, S.; Galea, R.; Khoo, P.; Bolden, J.E.; Aigaki, T.; Humbert, P.O.; Richardson, H.E. Identification of Novel Ras-Cooperating Oncogenes in Drosophila Melanogaster: A RhoGEF/Rho-Family/JNK Pathway Is a Central Driver of Tumorigenesis. Genetics 2011, 188, 105–125. [Google Scholar] [CrossRef]
- Hales, K.G.; Korey, C.A.; Larracuente, A.M.; Roberts, D.M. Genetics on the Fly: A Primer on the Drosophila Model System. Genetics 2015, 201, 815–842. [Google Scholar] [CrossRef]
- Bellen, H.J.; Shinya, Y. Morgan’s Legacy: Fruit Flies and the Functional Annotation of Conserved Genes Public Access. Cell 2015, 163, 12–14. [Google Scholar] [CrossRef]
- McGurk, L.; Berson, A.; Bonini, N.M. Drosophila as an in Vivo Model for Human Neurodegenerative Disease. Genetics 2015, 201, 377–402. [Google Scholar] [CrossRef]
- Bellen, H.J.; Tong, C.; Tsuda, H. 100 Years of Drosophila Research and Its Impact on Vetebrate Neuroscience: A History Lesson for the Future. Nat. Rev. Neurosci. 2010, 11, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Biggs, W.H.; Zavitz, K.H.; Dickson, B.; Van Der Straten, A.; Brunner, D.; Hafen, E.; Zipursky, S.L. The Drosophila Rolled Locus Encodes a MAP Kinase Required in the Sevenless Signal Transduction Pathway. EMBO J. 1994, 13, 1628–1635. [Google Scholar] [CrossRef] [PubMed]
- Cordeddu, V.; Yin, J.C.; Gunnarsson, C.; Virtanen, C.; Drunat, S.; Lepri, F.; De Luca, A.; Rossi, C.; Ciolfi, A.; Pugh, T.J.; et al. Activating Mutations Affecting the Dbl Homology Domain of SOS2 Cause Noonan Syndrome. Hum. Mutat. 2015, 36, 1080–1087. [Google Scholar] [CrossRef] [PubMed]
- Therrien, M.; Chang, H.C.; Solomon, N.M.; Karim, F.D.; Wassarman, D.A.; Rubin, G.M. KSR, a Novel Protein Kinase Required for RAS Signal Transduction. Cell 1995, 83, 879–888. [Google Scholar] [CrossRef] [PubMed]
- Karim, F.D.; Chang, H.C.; Therrien, M.; Wassarman, D.A.; Laverty, T.; Rubin, G.M. A Screen for Genes That Function Downstream of Ras1 during Drosophila Eye Development. Genetics 1996, 143, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Oishi, K.; Zhang, H.; Gault, W.J.; Wang, C.J.; Tan, C.C.; Kim, I.K.; Ying, H.; Rahman, T.; Pica, N.; Tartaglia, M.; et al. Phosphatase-Defective LEOPARD Syndrome Mutations in PTPN11 Gene Have Gain-of-Function Effects during Drosophila Development. Hum. Mol. Genet. 2009, 18, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.F.; Tong, J.; Hannan, F.; Luo, L.; Zhong, Y. A Neurofibromatosis-1-Regulated Pathway Is Required for Learning in Drosophila. Nature 2000, 403, 895–898. [Google Scholar] [CrossRef] [PubMed]
- Ho, I.S.; Hannan, F.; Guo, H.F.; Hakker, I.; Zhong, Y. Distinct Functional Domains of Neurofibromatosis Type 1 Regulate Immediate versus Long-Term Memory Formation. J. Neurosci. 2007, 27, 6852–6857. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.; Hannan, F.; Zhu, Y.; Bernards, A.; Zhong, Y. Neurofibromin Regulates G Protein-Stimulated Adenylyl Cyclase Activity. Nat. Neurosci. 2002, 5, 95–96. [Google Scholar] [CrossRef]
- Cui, Y.; Costa, R.M.; Murphy, G.G.; Elgersma, Y.; Zhu, Y.; Gutmann, D.H.; Parada, L.F.; Mody, I.; Silva, A.J. Neurofibromin Regulation of ERK Signaling Modulates GABA Release and Learning. Cell 2008, 135, 549–560. [Google Scholar] [CrossRef]
- Oliveira, A.F.; Yasuda, R. Neurofibromin Is the Major Ras Inactivator in Dendritic Spines. J. Neurosci. 2014, 34, 776–783. [Google Scholar] [CrossRef]
- Bigenzahn, J.W.; Collu, G.M.; Kartnig, F.; Pieraks, M.; Vladimer, G.I.; Heinz, L.X.; Sedlyarov, V.; Schischlik, F.; Fauster, A.; Rebsamen, M.; et al. LZTR1 Is a Regulator of RAS Ubiquitination and Signaling. Science 2018, 362, 1171–1177. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.A.; Gouzi, J.Y.; Long, J.B.; Huang, S.; Maher, R.C.; Xia, H.; Khalil, K.; Ray, A.; Van Vactor, D.; Bernards, R.; et al. Genetic and Functional Studies Implicate Synaptic Overgrowth and Ring Gland CAMP/PKA Signaling Defects in the Drosophila Melanogaster Neurofibromatosis-1 Growth Deficiency. PLoS Genet. 2013, 9, e1003958. [Google Scholar] [CrossRef] [PubMed]
- Gouzi, J.Y.; Moressis, A.; Walker, J.A.; Apostolopoulou, A.A.; Palmer, R.H.; Bernards, A.; Skoulakis, E.M.C. The Receptor Tyrosine Kinase Alk Controls Neurofibromin Functions in Drosophila Growth and Learning. PLoS Genet. 2011, 7, e1002281. [Google Scholar] [CrossRef] [PubMed]
- Das, T.K.; Gatto, J.; Mirmira, R.; Hourizadeh, E.; Kaufman, D.; Gelb, B.D.; Cagan, R. Drosophila RASopathy Models Identify Disease Subtype Differences and Biomarkers of Drug Efficacy. iScience 2021, 24, 102306. [Google Scholar] [CrossRef]
- Fields, R.D. Map the Other Brain. Nature 2013, 501, 25–27. [Google Scholar] [CrossRef]
- Apfeld, J.; Alper, S. What Can We Learn About Human Disease from the Nematode C. Elegans? In Methods in Molecular Biology; Humana Press: New York, NY, USA, 2018; Volume 1706, pp. 53–75. [Google Scholar] [CrossRef]
- Alexander, A.G.; Marfil, V.; Li, C. Use of C. Elegans as a Model to Study Alzheimer’s Disease and Other Neurodegenerative Diseases. Front. Genet. 2014, 5, 279. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, L.P.; Luke, C.J.; Perlmutter, D.H.; Silverman, G.A.; Pak, S.C. C. elegans in High-Throughput Drug Discovery. Adv. Drug Deliv. Rev. 2014, 69, 247–253. [Google Scholar] [CrossRef]
- Kwon, J.J.; Hahna, W.C. A Leucine-Rich Repeat Protein Provides a SHOC2 the RAS Circuit: A Structure-Function Perspective. Mol. Cell. Biol. 2021, 41, e00627-20. [Google Scholar] [CrossRef]
- Cordeddu, V.; Di Schiavi, E.; Pennacchio, L.A.; Ma’ayan, A.; Sarkozy, A.; Fodale, V.; Cecchetti, S.; Cardinale, A.; Martin, J.; Schackwitz, W.; et al. Mutation of SHOC2 Promotes Aberrant Protein N-Myristoylation and Causes Noonan-like Syndrome with Loose Anagen Hair. Nat. Genet. 2009, 41, 1022–1026. [Google Scholar] [CrossRef]
- Flex, E.; Jaiswal, M.; Pantaleoni, F.; Martinelli, S.; Strullu, M.; Fansa, E.K.; Caye, A.; De Luca, A.; Lepri, F.; Dvorsky, R.; et al. Activating Mutations in RRAS Underlie a Phenotype within the RASopathy Spectrum and Contribute to Leukaemogenesis. Hum. Mol. Genet. 2014, 23, 4315–4327. [Google Scholar] [CrossRef] [PubMed]
- Griffin, J.N.; Liu, K.J.; Sempou, E. Editorial: Xenopus Models of Organogenesis and Disease. Front. Physiol. 2020, 11, 10–12. [Google Scholar] [CrossRef] [PubMed]
- Umbhauer, M.; Marshall, C.J.; Mason, C.S.; Old, R.W.; Smith, J.C. Mesoderm Induction in Xenopus Caused by Activation of MAP Kinase. Nature 1995, 376, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.L.; Freeman, R.M.; O’Reilly, A.M.; Neel, B.G.; Sokol, S.Y. The SH2-Containing Protein-Tyrosine Phosphatase SH-PTP2 Is Required Upstream of MAP Kinase for Early Xenopus Development. Cell 1995, 80, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Klaman, L.D.; Chen, B.; Araki, T.; Harada, H.; Thomas, S.M.; George, E.L.; Neel, B.G. An Shp2/SFK/Ras/Erk Signaling Pathway Controls Trophoblast Stem Cell Survival. Dev. Cell 2006, 10, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Geary, L.; Labonne, C. FGF Mediated MAPK and PI3K/Akt Signals Make Distinct Contributions to Pluripotency and the Establishment of Neural Crest. Elife 2018, 7, e33845. [Google Scholar] [CrossRef]
- Popov, I.K.; Hiatt, S.M.; Whalen, S.; Keren, B.; Ruivenkamp, C.; Van Haeringen, A.; Chen, M.J.; Cooper, G.M.; Korf, B.R.; Chang, C. A YWHAZ Variant Associated with Cardiofaciocutaneous Syndrome Activates the RAF-ERK Pathway. Front. Physiol. 2019, 10, 388. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Bahn, M.; Hwan Kim, Y.; Shin, J.Y.; Cheong, S.W.; Ju, B.G.; Kim, W.S.; Yeo, C.Y. Xenopus Laevis FGF Receptor Substrate 3 (XFrs3) Is Important for Eye Development and Mediates Pax6 Expression in Lens Placode through Its Shp2-Binding Sites. Dev. Biol. 2015, 397, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Soundararajan, P.; Fawcett, J.P.; Rafuse, V.F. Guidance of Postural Motoneurons Requires MAPK/ERK Signaling Downstream of Fibroblast Growth Factor Receptor 1. J. Neurosci. 2010, 30, 6595–6606. [Google Scholar] [CrossRef]
- Li, S.; Mattar, P.; Dixit, R.; Lawn, S.O.; Wilkinson, G.; Kinch, C.; Eisenstat, D.; Kurrasch, D.M.; Chan, J.A.; Schuurmans, C. RAS/ERK Signaling Controls Proneural Genetic Programs in Cortical Development and Gliomagenesis. J. Neurosci. 2014, 34, 2169–2190. [Google Scholar] [CrossRef]
- Huang, X.; Cheng, H.W. Perspective: Chicken Models for Studying the Ontogenetic Origin of Neuropsychiatric Disorders. Biomedicines 2022, 10, 1155. [Google Scholar] [CrossRef]
- Abramyan, J.; Richman, J.M. Craniofacial Development: Discoveries Made in the Chicken Embryo. Int. J. Dev. Biol. 2018, 62, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Gammill, L.S.; Krull, C.E. Embryological and Genetic Manipulation of Chick Development. Methods Mol. Biol. 2011, 770, 119–137. [Google Scholar] [CrossRef]
- Zhao, X.; Bhattacharyya, A. Human Models Are Needed for Studying Human Neurodevelopmental Disorders. Am. J. Hum. Genet. 2018, 103, 829–857. [Google Scholar] [CrossRef] [PubMed]
- Toutain, P.L.; Ferran, A.; Bousquet-Mélou, A. Species Differences in Pharmacokinetics and Pharmacodynamics. Handb. Exp. Pharmacol. 2010, 199, 19–48. [Google Scholar] [CrossRef]
- Kiani, A.K.; Pheby, D.; Henehan, G.; Brown, R.; Sieving, P.; Sykora, P.; Marks, R.; Falsini, B.; Capodicasa, N.; Miertus, S.; et al. Ethical Considerations Regarding Animal Experimentation. J. Prev. Med. Hyg. 2022, 63, E255–E266. [Google Scholar] [CrossRef] [PubMed]
- Vaz, R.; Hofmeister, W.; Lindstrand, A. Zebrafish Models of Neurodevelopmental Disorders: Limitations and Benefits of Current Tools and Techniques. Int. J. Mol. Sci. 2019, 20, 1296. [Google Scholar] [CrossRef]
- Pandey, U.B.; Nichols, C.D. Human Disease Models in Drosophila Melanogaster and the Role of the Fly in Therapeutic Drug Discovery. Pharmacol. Rev. 2011, 63, 411–436. [Google Scholar] [CrossRef]
- Ishibashi, S.; Saldanha, F.Y.L.; Amaya, E. Xenopus as a Model Organism for Biomedical Research; Elsevier Inc.: Amsterdam, The Netherlands, 2017; ISBN 9780128030783. [Google Scholar]
- Andrews, D.D.T.; Franz-Odendaal, T.A. Organotypic Culture Method to Study the Development of Embryonic Chicken Tissues. J. Vis. Exp. 2018, 2018, e57619. [Google Scholar] [CrossRef]
- Givisiez, P.E.N.; Moreira Filho, A.L.B.; Santos, M.R.B.; Oliveira, H.B.; Ferket, P.R.; Oliveira, C.J.B.; Malheiros, R.D. Chicken Embryo Development: Metabolic and Morphological Basis for in Ovo Feeding Technology. Poult. Sci. 2020, 99, 6774–6782. [Google Scholar] [CrossRef]
- Gross, A.M.; Frone, M.; Gripp, K.W.; Gelb, B.D.; Schoyer, L.; Schill, L.; Stronach, B.; Biesecker, L.G.; Esposito, D.; Hernandez, E.R.; et al. Advancing RAS/RASopathy Therapies: An NCI-Sponsored Intramural and Extramural Collaboration for the Study of RASopathies. Am. J. Med. Genet. Part A 2020, 182, 866–876. [Google Scholar] [CrossRef]
- Anastasaki, C.; Rauen, K.A.; Patton, E.E. Continual Low-Level MEK Inhibition Ameliorates Cardio-Facio-Cutaneous Phenotypes in Zebrafish. Dis. Model. Mech. 2012, 5, 546–552. [Google Scholar] [CrossRef] [PubMed]
- Ki, D.H.; Oppel, F.; Durbin, A.D.; Look, A.T. Mechanisms Underlying Synergy between DNA Topoisomerase I-Targeted Drugs and MTOR Kinase Inhibitors in NF1-Associated Malignant Peripheral Nerve Sheath Tumors. Oncogene 2019, 38, 6585–6598. [Google Scholar] [CrossRef] [PubMed]
- Karam, C.S.; Jones, S.K.; Javitch, J.A. Come Fly with Me: An Overview of Dopamine Receptors in Drosophila Melanogaster. Basic Clin. Pharmacol. Toxicol. 2020, 126, 56–65. [Google Scholar] [CrossRef]
- Rauen, K.A.; Schoyer, L.; McCormick, F.; Lin, A.E.; Allanson, J.E.; Stevenson, D.A.; Gripp, K.W.; Neri, G.; Carey, J.C.; Legius, E.; et al. Proceedings from the 2009 Genetic Syndromes of the Ras/MAPK Pathway: From Bedside to Bench and Back. Am. J. Med. Genet. Part A 2010, 152, 4–24. [Google Scholar] [CrossRef] [PubMed]
- Iliadi, K.G.; Avivi, A.; Iliadi, N.N.; Knight, D.; Korol, A.B.; Nevo, E.; Taylor, P.; Moran, M.F.; Kamyshev, N.G.; Boulianne, G.L. Nemy Encodes a Cytochrome B561 That Is Required for Drosophila Learning and Memory. Proc. Natl. Acad. Sci. USA 2008, 105, 19986–19991. [Google Scholar] [CrossRef] [PubMed]
- Coleman, B.; Topalidou, I.; Ailion, M. Modulation of Gq-Rho Signaling by the Erk Mapk Pathway Controls Locomotion in Caenorhabditis Elegans. Genetics 2018, 209, 523–535. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Fan, Z.; Qiao, P.; Zhao, Y.; Wang, Y.; Jiang, D.; Wang, X.; Zhu, X.; Zhang, Y.; Huang, B.; et al. MiR-51 Regulates GABAergic Synapses by Targeting Rab GEF GLO-4 and Lysosomal Trafficking-Related GLO/AP-3 Pathway in Caenorhabditis Elegans. Dev. Biol. 2018, 436, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Y.; Zheng, R.Q.; Wang, Y.; Liu, Y.H.; Jiang, S.; Wang, X.Z.; He, K.; Pan, X.; Zhou, T.; Li, T.; et al. The Endogenous Metabolite Glycerophosphocholine Promotes Longevity and Fitness in Caenorhabditis Elegans. Metabolites 2022, 12, 177. [Google Scholar] [CrossRef]
- Bitner, R.S.; Bunnelle, W.H.; Anderson, D.J.; Briggs, C.A.; Buccafusco, J.; Curzon, P.; Decker, M.W.; Frost, J.M.; Gronlien, J.H.; Gubbins, E.; et al. Broad-Spectrum Efficacy across Cognitive Domains by A7 Nicotinic Acetylcholine Receptor Agonism Correlates with Activation of ERK1/2 and CREB Phosphorylation Pathways. J. Neurosci. 2007, 27, 10578–10587. [Google Scholar] [CrossRef]
- Ahir, B.K.; Sanders, A.P.; Rager, J.E.; Fry, R.C. Systems Biology and Birth Defects Prevention: Blockade of the Glucocorticoid Receptor Prevents Arsenic-Induced Birth Defects. Environ. Health Perspect. 2013, 121, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Larrivée, B.; Zhuang, Z.W.; Atri, D.; Moraes, F.; Prahst, C.; Eichmann, A.; Simons, M. Endothelial RAF1/ERK Activation Regulates Arterial Morphogenesis. Blood 2013, 121, 3988–3996. [Google Scholar] [CrossRef] [PubMed]

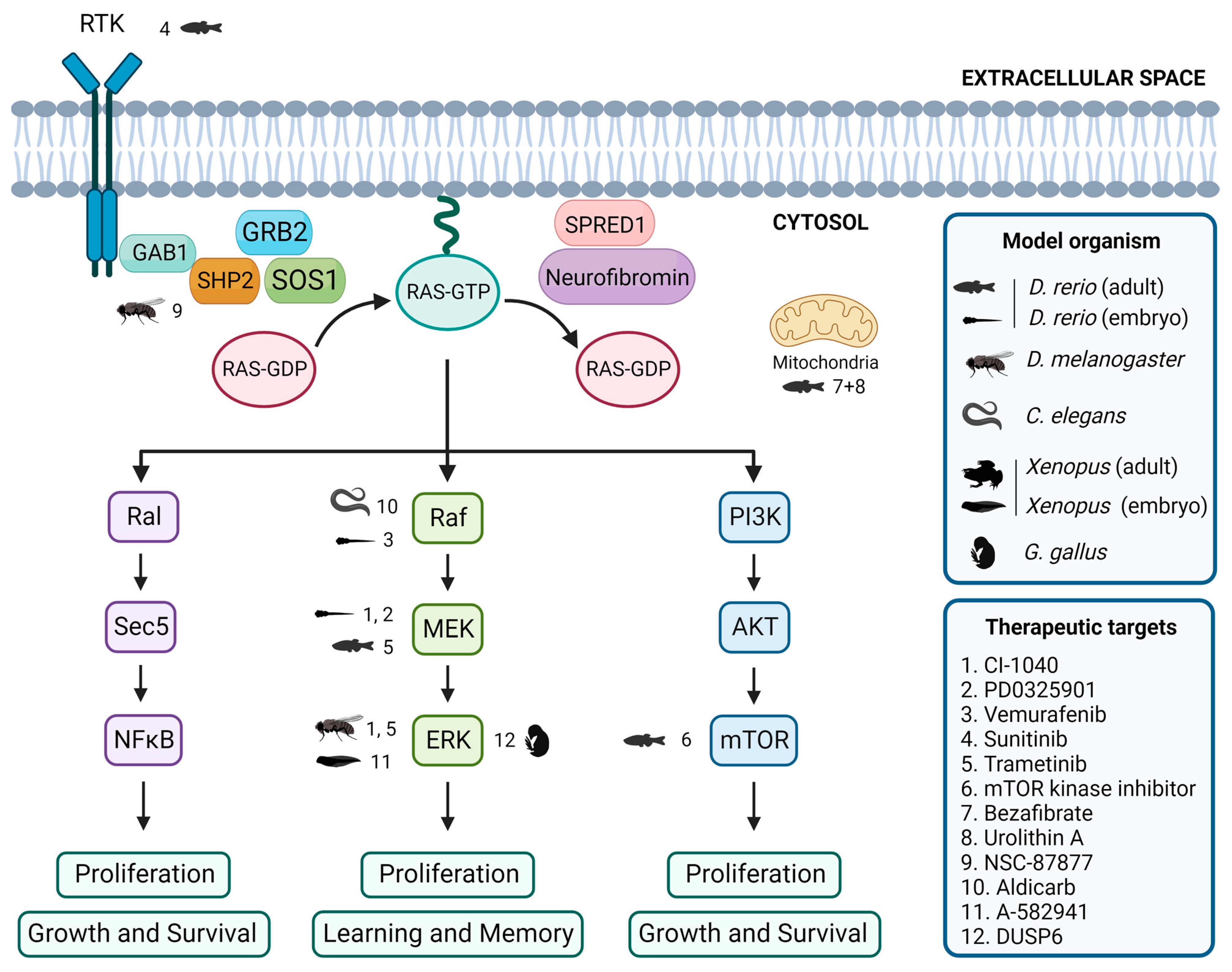
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).