
Tumors Established in a Defective Immune Environment Reprogram the Oncogenic Signaling Pathways to Escalate Tumor Antigenicity


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Cells
2.2. Tumor Harvesting, Digestion, and Re-Inoculation
2.3. Protein Preparation and LC-MS/MS Analysis
2.4. Bioinformatic Analysis
2.5. Immunofluorescence Staining and Analysis
2.6. Statistical Analysis
3. Results
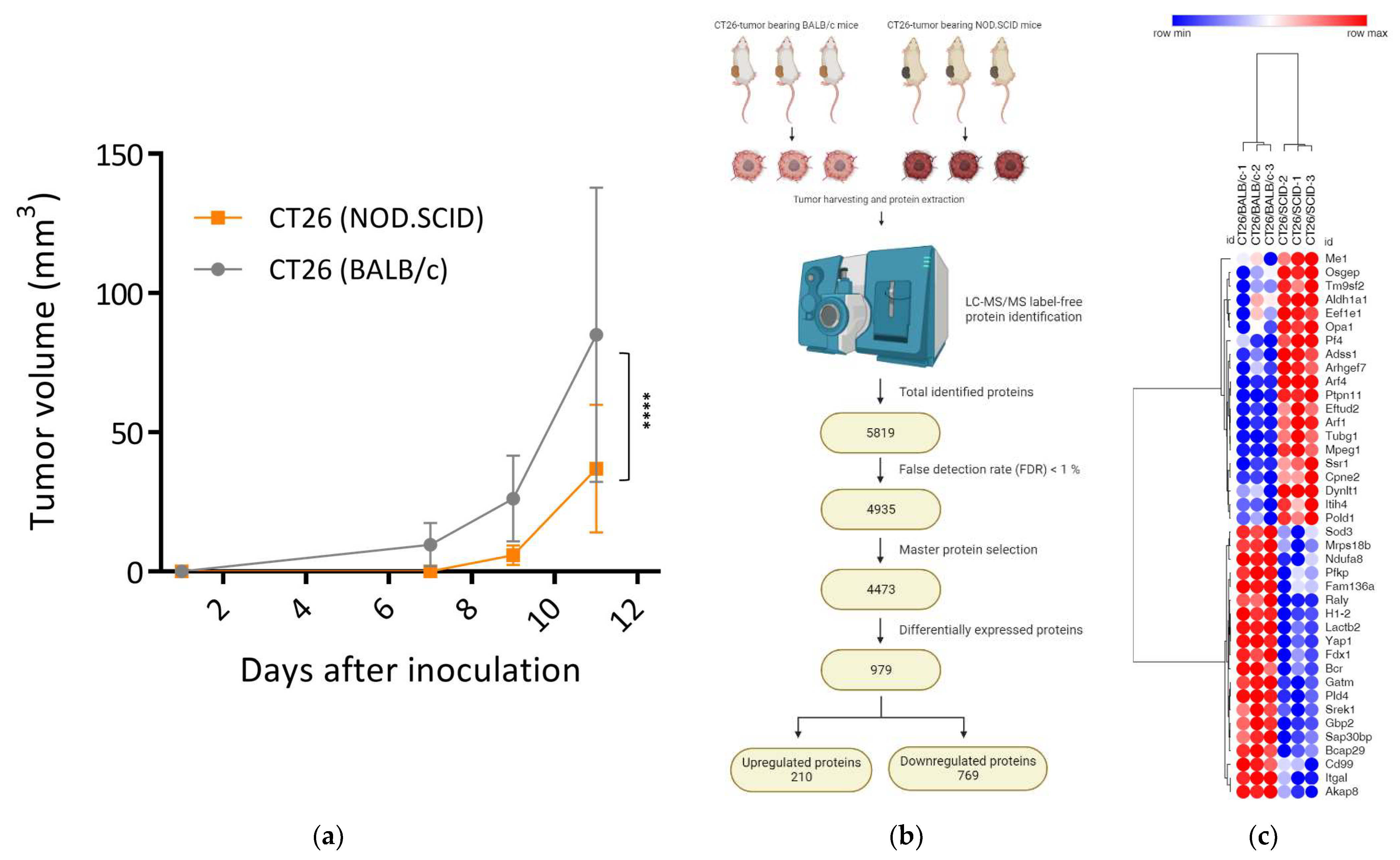
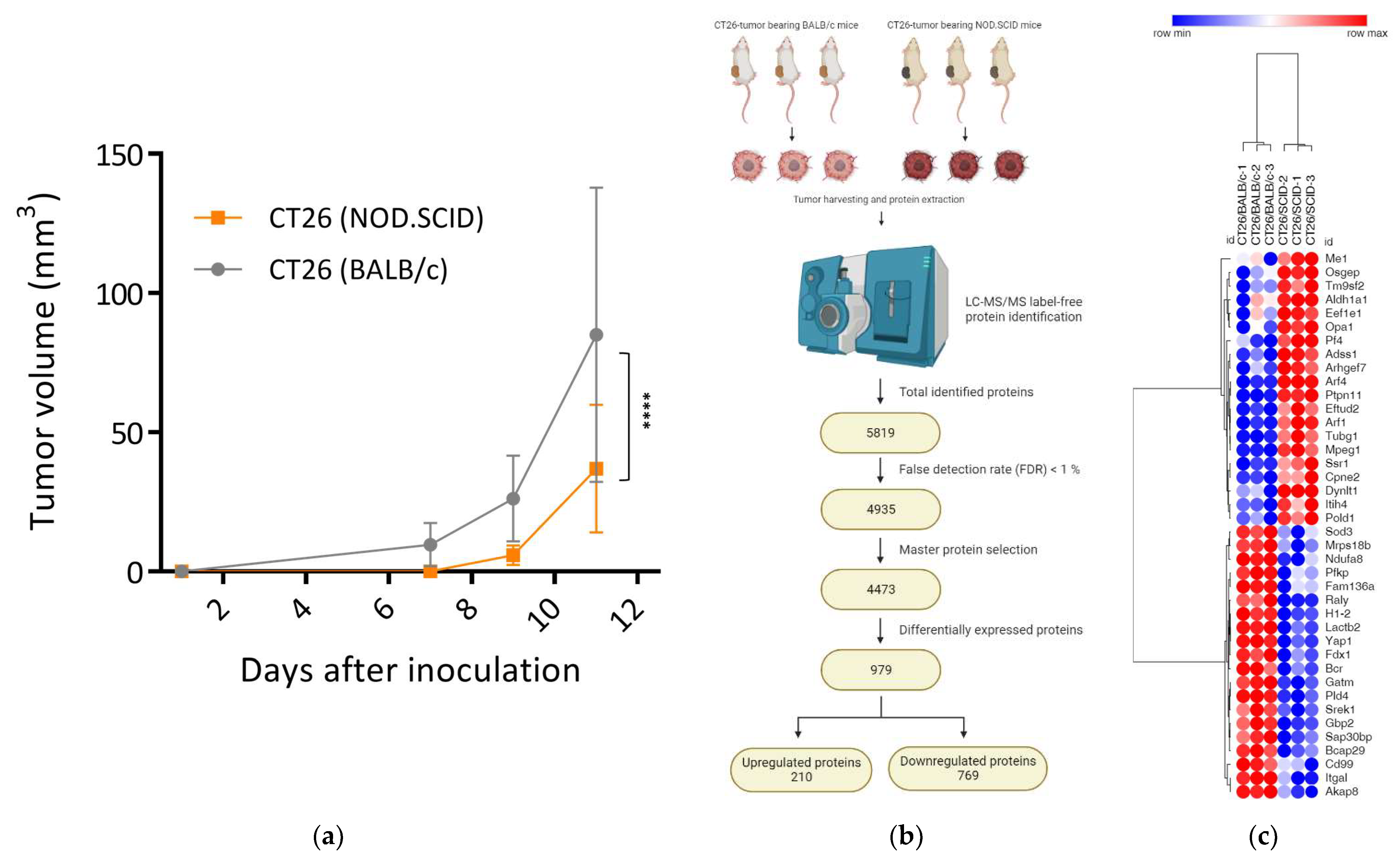
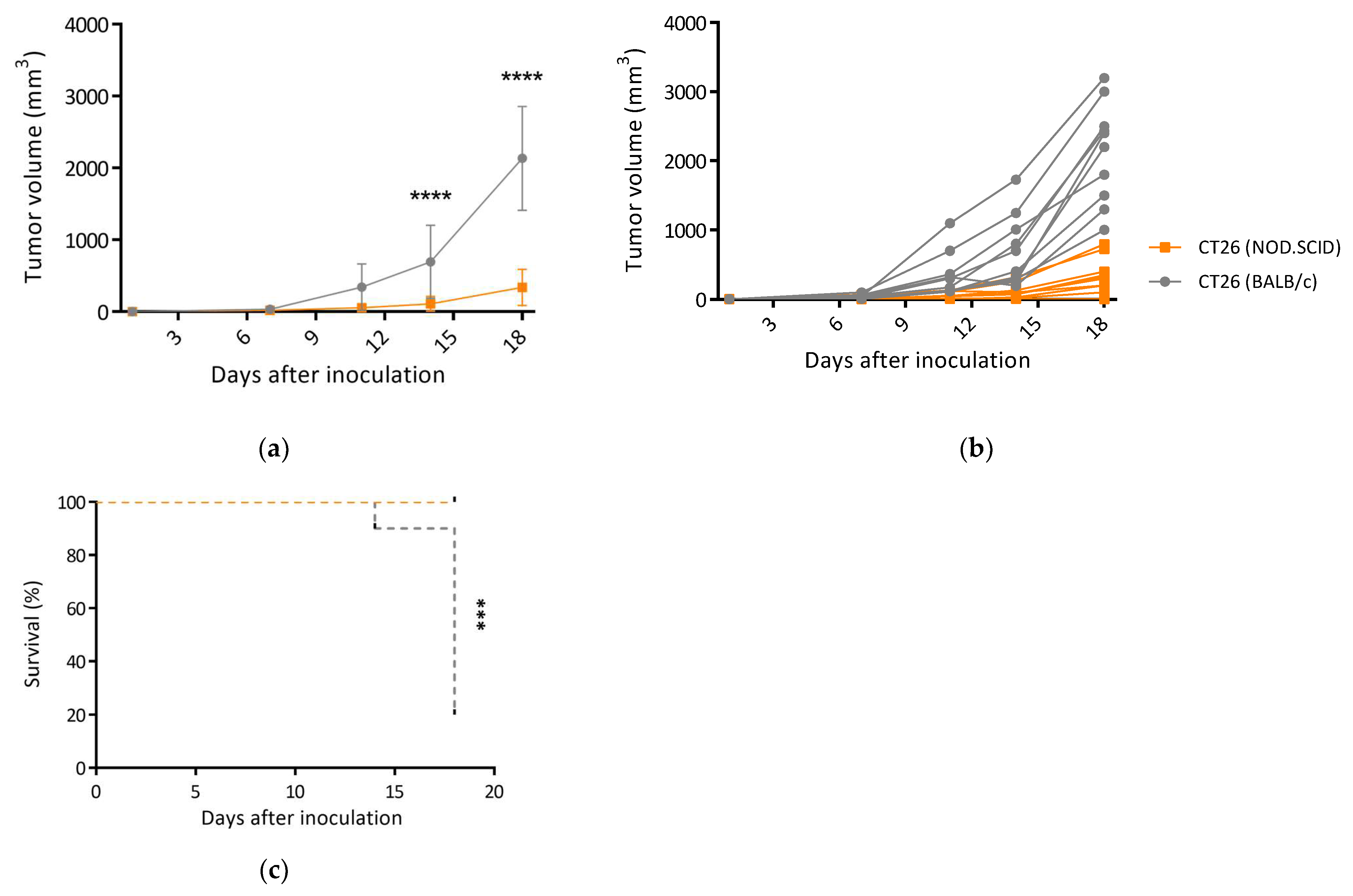
3.1. Tumors Developed in Immunocompromised Hosts Had a Decreased Growth Rate
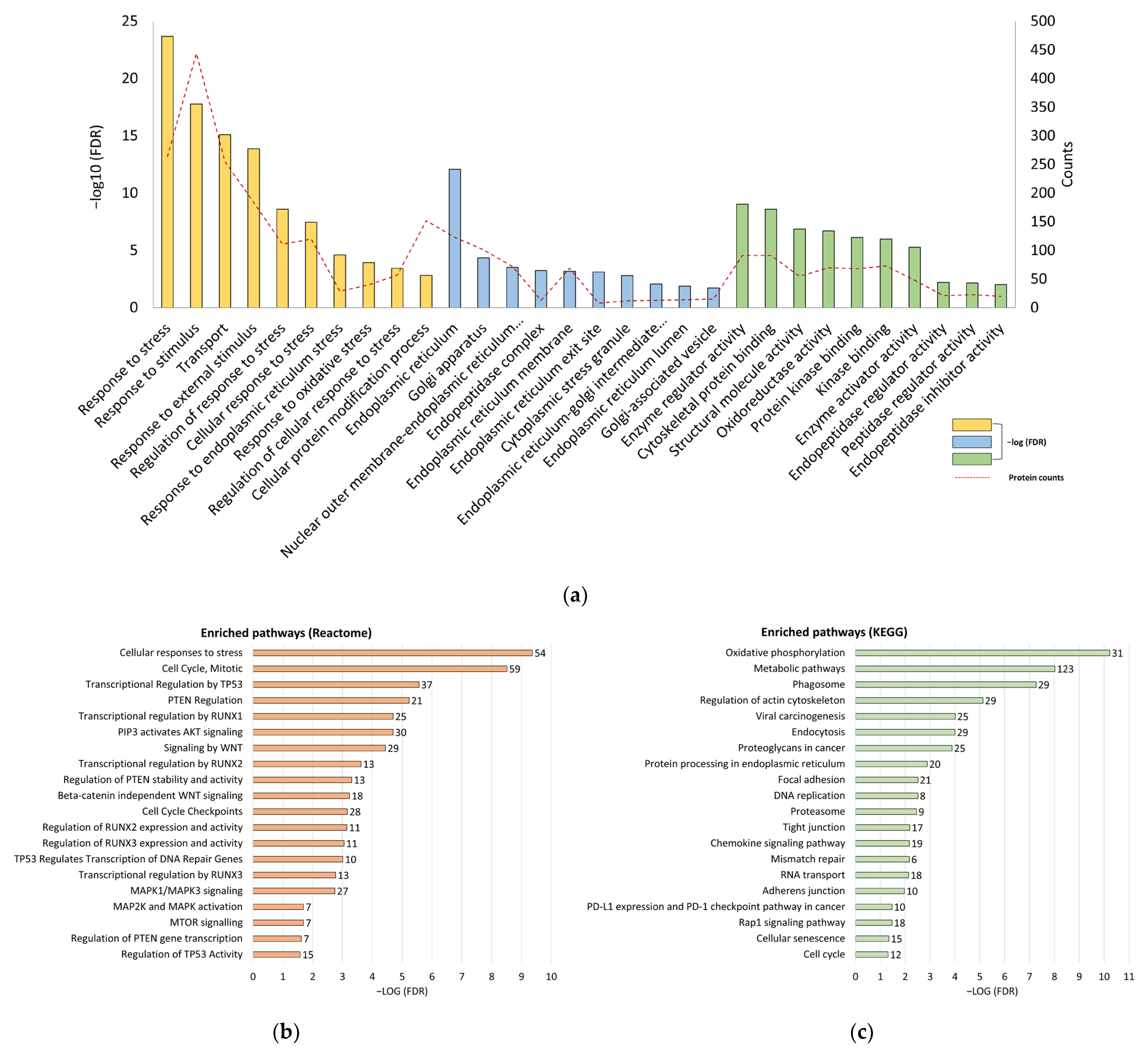
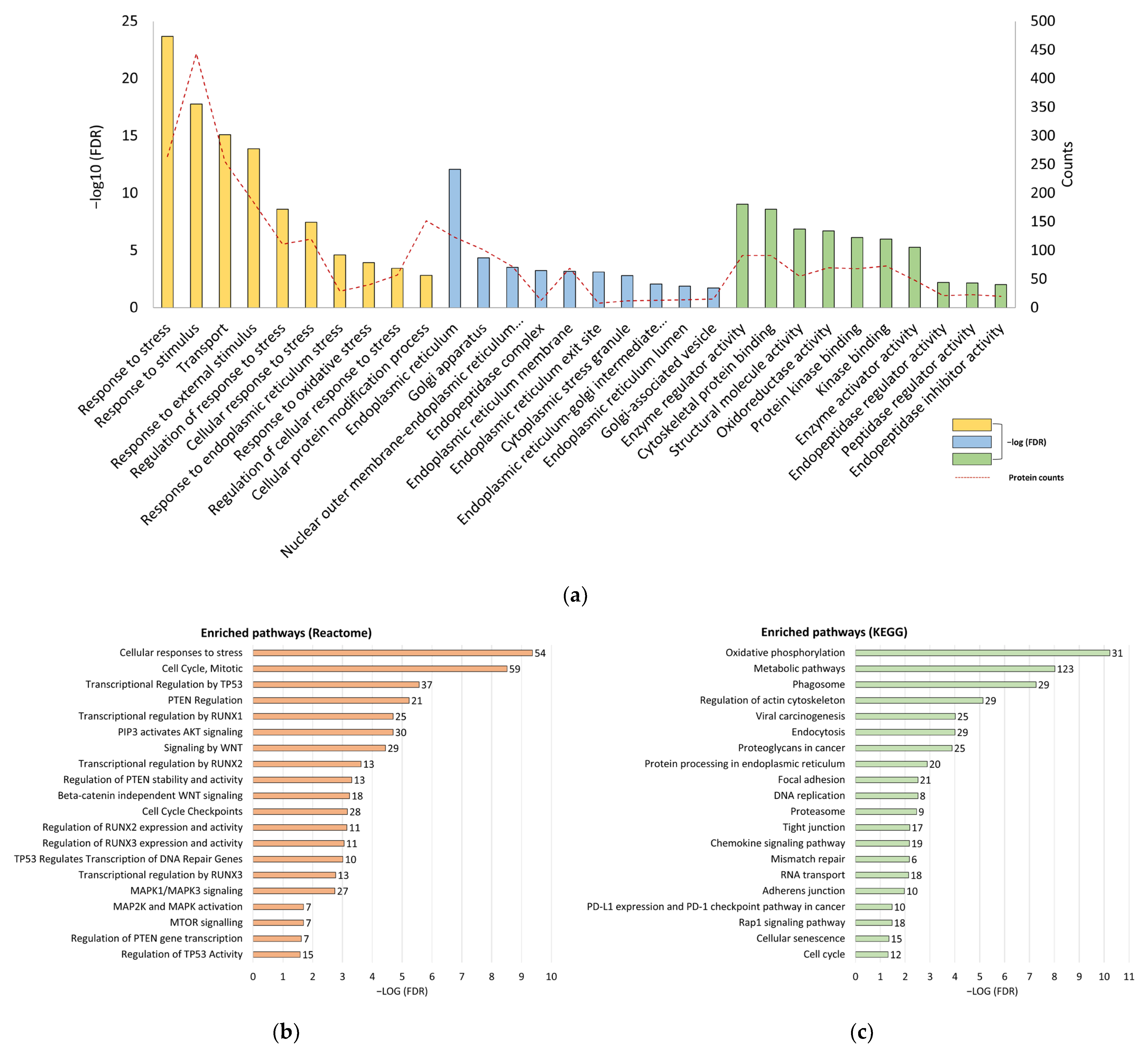
3.2. Silence of Tumor Suppressors and Reprogramming of Proliferative Signaling Pathways in CT26/SCID Tumors
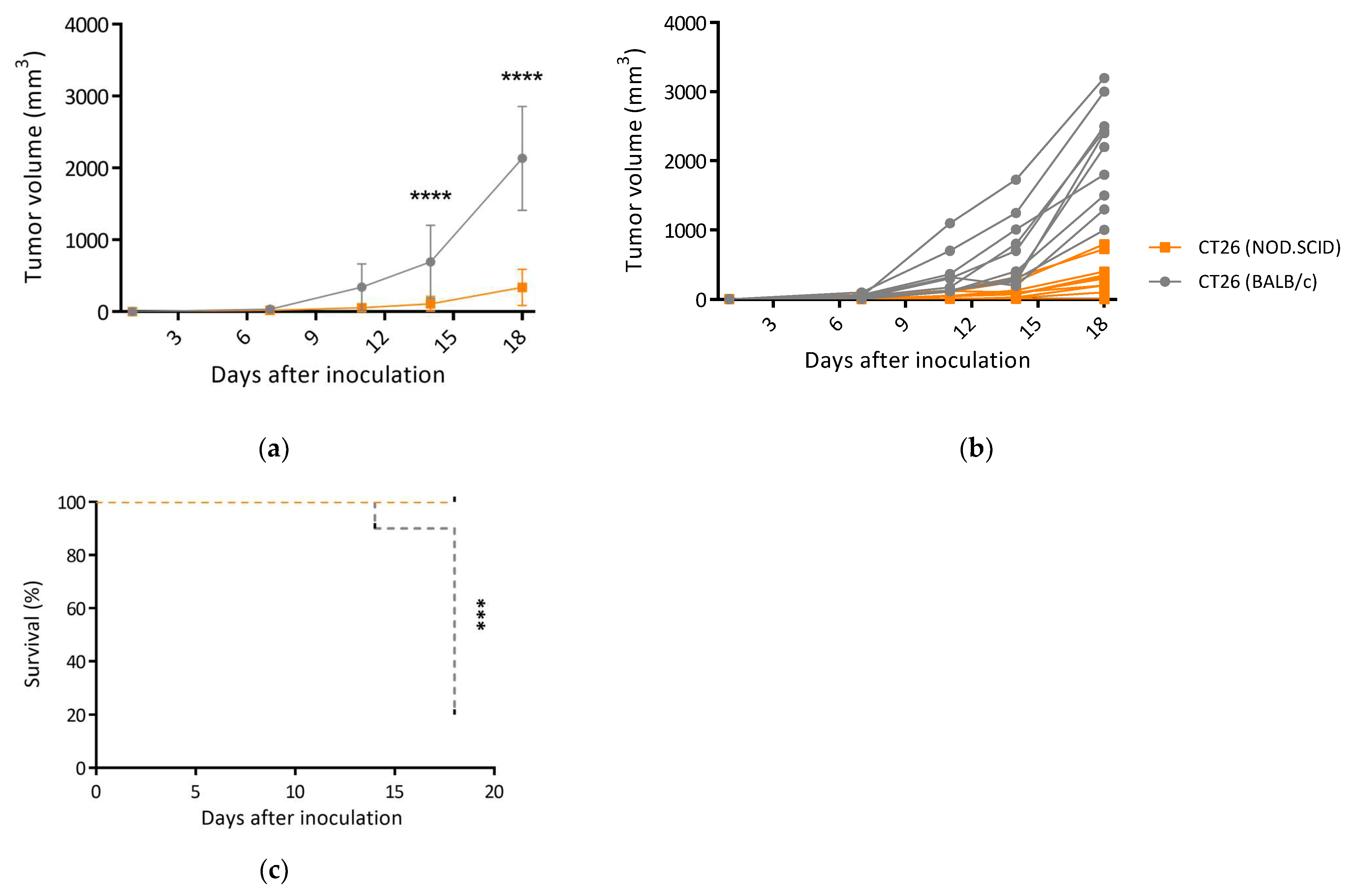
3.3. CT26/SCID Tumors Progressively Developed at the Endpoint
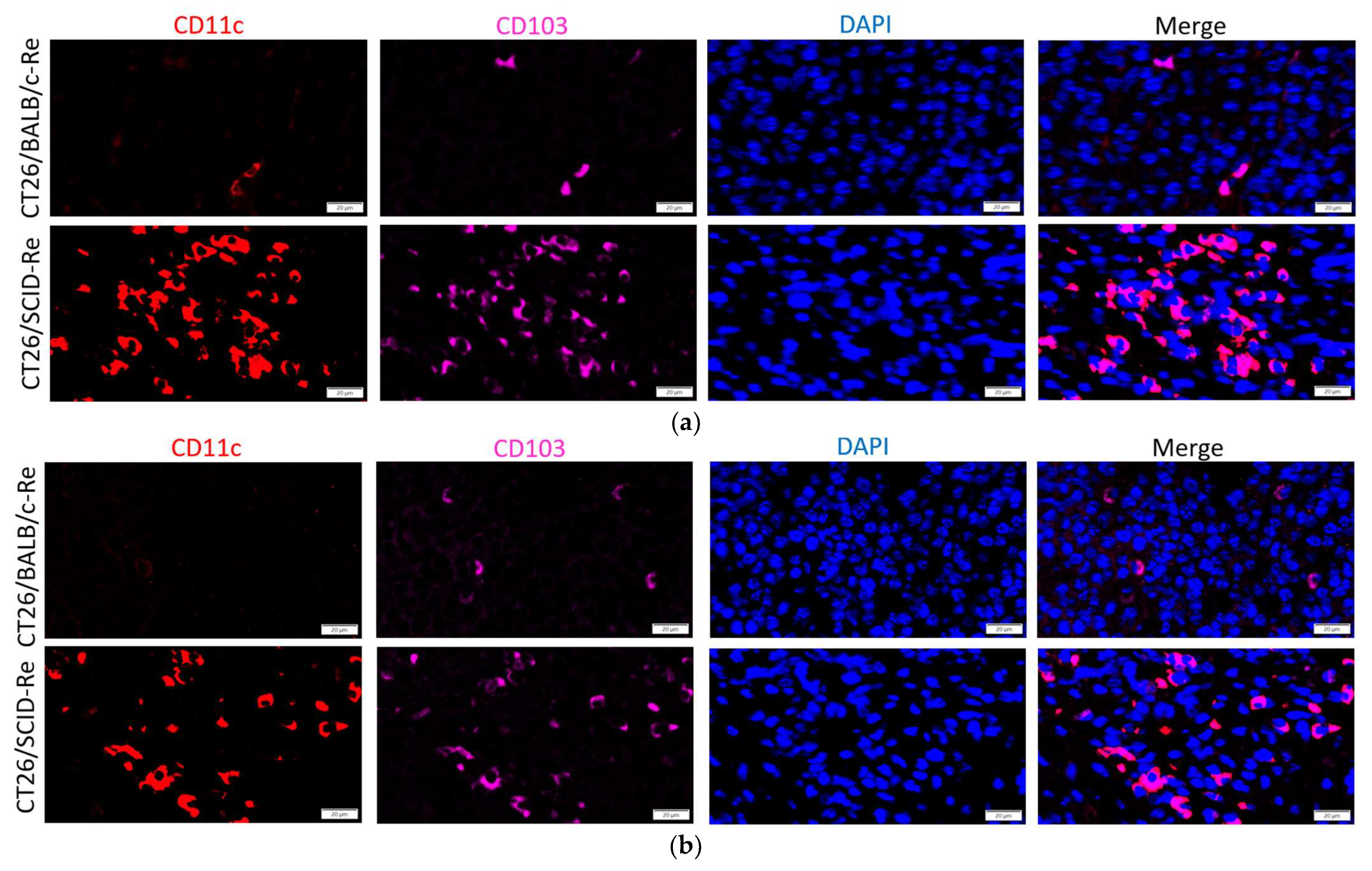
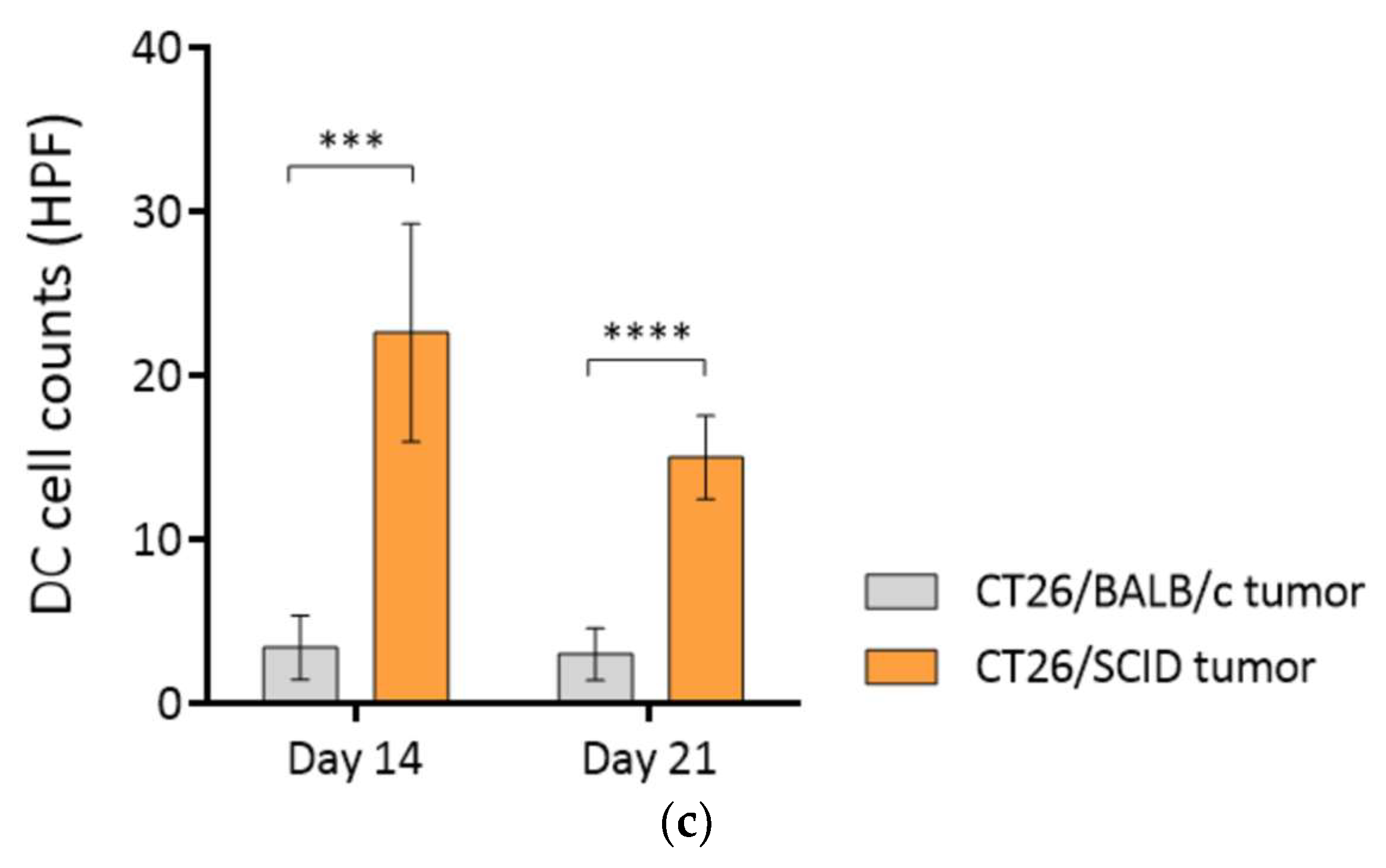
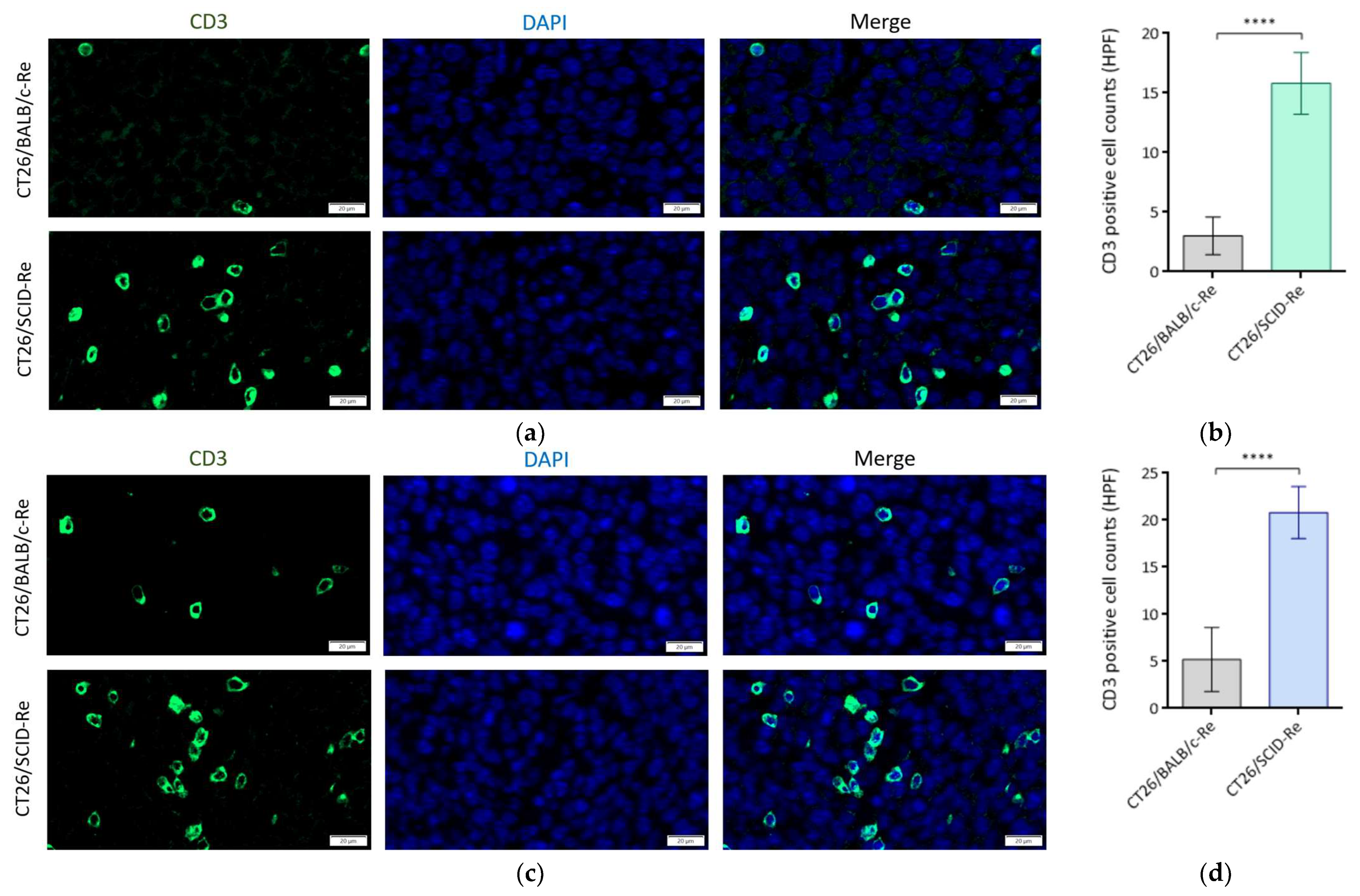
3.4. Highly Immunogenic CT26/SCID Tumors Trigger Robust Adaptive Immune Responses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sun, W.; Yang, J. Functional mechanisms for human tumor suppressors. J. Cancer 2010, 1, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Roberts, E.W.; Broz, M.L.; Binnewies, M.; Headley, M.B.; Nelson, A.E.; Wolf, D.M.; Kaisho, T.; Bogunovic, D.; Bhardwaj, N.; Krummel, M.F. Critical Role for CD103(+)/CD141(+) Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer Cell 2016, 30, 324–336. [Google Scholar] [CrossRef] [PubMed]
- Kaech, S.M.; Cui, W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat. Rev. Immunol. 2012, 12, 749–761. [Google Scholar] [CrossRef] [PubMed]
- Nolz, J.C. Molecular mechanisms of CD8(+) T cell trafficking and localization. Cell. Mol. Life Sci. 2015, 72, 2461–2473. [Google Scholar] [CrossRef] [PubMed]
- Jhunjhunwala, S.; Hammer, C.; Delamarre, L. Antigen presentation in cancer: Insights into tumour immunogenicity and immune evasion. Nat. Rev. Cancer 2021, 21, 298–312. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Jiang, P.; Wei, S.; Xu, X.; Wang, J. Regulatory T cells in tumor microenvironment: New mechanisms, potential therapeutic strategies and future prospects. Mol. Cancer 2020, 19, 116. [Google Scholar] [CrossRef] [PubMed]
- Berraondo, P.; Sanmamed, M.F.; Ochoa, M.C.; Etxeberria, I.; Aznar, M.A.; Pérez-Gracia, J.L.; Rodríguez-Ruiz, M.E.; Ponz-Sarvise, M.; Castañón, E.; Melero, I. Cytokines in clinical cancer immunotherapy. Br. J. Cancer 2019, 120, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Lora, A.; Algarra, I.; Garrido, F. MHC class I antigens, immune surveillance, and tumor immune escape. J. Cell Physiol. 2003, 195, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Ke, C.-H.; Chiu, Y.-H.; Huang, K.-C.; Lin, C.-S. Exposure of Immunogenic Tumor Antigens in Surrendered Immunity and the Significance of Autologous Tumor Cell-Based Vaccination in Precision Medicine. Int. J. Mol. Sci. 2023, 24, 147. [Google Scholar] [CrossRef]
- Paluskievicz, C.M.; Cao, X.; Abdi, R.; Zheng, P.; Liu, Y.; Bromberg, J.S. T Regulatory Cells and Priming the Suppressive Tumor Microenvironment. Front. Immunol. 2019, 10, 2453. [Google Scholar] [CrossRef]
- Spolski, R.; Li, P.; Leonard, W.J. Biology and regulation of IL-2: From molecular mechanisms to human therapy. Nat. Rev. Immunol. 2018, 18, 648–659. [Google Scholar] [CrossRef] [PubMed]
- Carmenate, T.; Ortíz, Y.; Enamorado, M.; García-Martínez, K.; Avellanet, J.; Moreno, E.; Graça, L.; León, K. Blocking IL-2 Signal In Vivo with an IL-2 Antagonist Reduces Tumor Growth through the Control of Regulatory T Cells. J. Immunol. 2018, 200, 3475. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, J.A.; Tomita, Y.; Jankowska-Gan, E.; Lema, D.A.; Arvedson, M.P.; Nair, A.; Bracamonte-Baran, W.; Zhou, Y.; Meyer, K.K.; Zhong, W.; et al. Treg-Cell-Derived IL-35-Coated Extracellular Vesicles Promote Infectious Tolerance. Cell Rep. 2020, 30, 1039–1051.e1035. [Google Scholar] [CrossRef] [PubMed]
- Ke, C.-H.; Wang, Y.-S.; Chiang, H.-C.; Wu, H.-Y.; Liu, W.-J.; Huang, C.-C.; Huang, Y.-C.; Lin, C.-S. Xenograft cancer vaccines prepared from immunodeficient mice increase tumor antigen diversity and host T cell efficiency against colorectal cancers. Cancer Lett. 2022, 526, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Ke, C.-H.; Tomiyasu, H.; Lin, Y.-L.; Huang, W.-H.; Huang, H.-H.; Chiang, H.-C.; Lin, C.-S. Canine transmissible venereal tumour established in immunodeficient mice reprograms the gene expression profiles associated with a favourable tumour microenvironment to enable cancer malignancy. BMC Vet. Res. 2022, 18, 4. [Google Scholar] [CrossRef] [PubMed]
- Escors, D. Tumour Immunogenicity, Antigen Presentation, and Immunological Barriers in Cancer Immunotherapy. New J. Sci. 2014, 2014, 734515. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, H.; Vesely, M.D.; Koboldt, D.C.; Rickert, C.G.; Uppaluri, R.; Magrini, V.J.; Arthur, C.D.; White, J.M.; Chen, Y.S.; Shea, L.K.; et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 2012, 482, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez de la Fuente, L.; Law, A.M.K.; Gallego-Ortega, D.; Valdes-Mora, F. Tumor dissociation of highly viable cell suspensions for single-cell omic analyses in mouse models of breast cancer. STAR Protoc. 2021, 2, 100841. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, T.; Saddawi-Konefka, R.; Vermi, W.; Koebel, C.M.; Arthur, C.; White, J.M.; Uppaluri, R.; Andrews, D.M.; Ngiow, S.F.; Teng, M.W.; et al. Cancer immunoediting by the innate immune system in the absence of adaptive immunity. J. Exp. Med. 2012, 209, 1869–1882. [Google Scholar] [CrossRef]
- Madda, R.; Chen, C.-M.; Wang, J.-Y.; Chen, C.-F.; Chao, K.-Y.; Yang, Y.-M.; Wu, H.-Y.; Chen, W.-M.; Wu, P.-K. Proteomic profiling and identification of significant markers from high-grade osteosarcoma after cryotherapy and irradiation. Sci. Rep. 2020, 10, 2105. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Liu, B.; Li, Z.; Li, Y.; Chen, X.; Ma, Y.; Yan, S.; Yang, X.; Zhong, L.; Wu, N. Proteomic Analyses Identify Differentially Expressed Proteins and Pathways Between Low-Risk and High-Risk Subtypes of Early-Stage Lung Adenocarcinoma and Their Prognostic Impacts. Mol. Cell. Proteom. 2021, 20, 100015. [Google Scholar] [CrossRef] [PubMed]
- Gene Ontology, C. The Gene Ontology (GO) project in 2006. Nucleic Acids Res. 2006, 34, D322–D326. [Google Scholar] [CrossRef] [PubMed]
- Dennis, G.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003, 4, R60. [Google Scholar] [CrossRef]
- Fabregat, A.; Sidiropoulos, K.; Viteri, G.; Forner, O.; Marin-Garcia, P.; Arnau, V.; D’Eustachio, P.; Stein, L.; Hermjakob, H. Reactome pathway analysis: A high-performance in-memory approach. BMC Bioinform. 2017, 18, 142. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.C.; Stucky, M.; Wakefield, C.; Melott, J.M.; Akbani, R.; Weinstein, J.N.; Broom, B.M. Interactive Clustered Heat Map Builder: An easy web-based tool for creating sophisticated clustered heat maps. F1000Research 2019, 8, ISCB Comm J-1750. [Google Scholar] [CrossRef] [PubMed]
- Franceschini, A.; Szklarczyk, D.; Frankild, S.; Kuhn, M.; Simonovic, M.; Roth, A.; Lin, J.; Minguez, P.; Bork, P.; von Mering, C.; et al. STRING v9.1: Protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013, 41, D808–D815. [Google Scholar] [CrossRef] [PubMed]
- Ke, C.-H.; Sio, K.-M.; Wang, S.-L.; Kuo, Y.; Huang, W.-H.; Lin, C.-S. The High Expression of Legumain in Canine Neoplasms: A Retrospective Analysis of 100 Cases. Animals 2022, 12, 504. [Google Scholar] [CrossRef]
- Wang, P.; Zhang, H.; Yang, J.; Li, Z.; Wang, Y.; Leng, X.; Ganapathy, S.; Isakson, P.; Chen, C.; Zhu, T. Mu-KRAS attenuates Hippo signaling pathway through PKCι to sustain the growth of pancreatic cancer. J. Cell. Physiol. 2020, 235, 408–420. [Google Scholar] [CrossRef] [PubMed]
- Vidotto, T.; Melo, C.M.; Castelli, E.; Koti, M.; Dos Reis, R.B.; Squire, J.A. Emerging role of PTEN loss in evasion of the immune response to tumours. Br. J. Cancer 2020, 122, 1732–1743. [Google Scholar] [CrossRef]
- Wellenstein, M.D.; de Visser, K.E. Cancer-Cell-Intrinsic Mechanisms Shaping the Tumor Immune Landscape. Immunity 2018, 48, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Rooney, M.S.; Shukla, S.A.; Wu, C.J.; Getz, G.; Hacohen, N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015, 160, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.R.; Chen, M.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor: New modes and prospects. Nat. Rev. Mol. Cell Biol. 2018, 19, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Naert, T.; Dimitrakopoulou, D.; Tulkens, D.; Demuynck, S.; Carron, M.; Noelanders, R.; Eeckhout, L.; Van Isterdael, G.; Deforce, D.; Vanhove, C.; et al. RBL1 (p107) functions as tumor suppressor in glioblastoma and small-cell pancreatic neuroendocrine carcinoma in Xenopus tropicalis. Oncogene 2020, 39, 2692–2706. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Chen, Z.; Qin, Z.; Qian, K.; Liu, T.; Zhang, Y. CDKN2AIP-induced cell senescence and apoptosis of testicular seminoma are associated with CARM1 and eIF4β. Acta Biochim. Biophys. Sin. 2022, 54, 604–614. [Google Scholar] [CrossRef]
- Bruno, A.; Noonan, D.M.; Valli, R.; Porta, G.; Taramelli, R.; Mortara, L.; Acquati, F. Human RNASET2: A Highly Pleiotropic and Evolutionary Conserved Tumor Suppressor Gene Involved in the Control of Ovarian Cancer Pathogenesis. Int. J. Mol. Sci. 2022, 23, 9074. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Balajee, A.S.; Su, T.; Cen, B.; Hei, T.K.; Weinstein, I.B. The HINT1 tumor suppressor regulates both gamma-H2AX and ATM in response to DNA damage. J. Cell Biol. 2008, 183, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Shao, C.; Qiu, Y.; Liu, J.; Feng, H.; Shen, S.; Saiyin, H.; Yu, W.; Wei, Y.; Yu, L.; Su, W.; et al. PARP12 (ARTD12) suppresses hepatocellular carcinoma metastasis through interacting with FHL2 and regulating its stability. Cell Death Dis. 2018, 9, 856. [Google Scholar] [CrossRef]
- Maehama, T.; Dixon, J.E. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 1998, 273, 13375–13378. [Google Scholar] [CrossRef]
- Lee, J.O.; Yang, H.; Georgescu, M.M.; Di Cristofano, A.; Maehama, T.; Shi, Y.; Dixon, J.E.; Pandolfi, P.; Pavletich, N.P. Crystal structure of the PTEN tumor suppressor: Implications for its phosphoinositide phosphatase activity and membrane association. Cell 1999, 99, 323–334. [Google Scholar] [CrossRef]
- Ozcan, U.; Ozcan, L.; Yilmaz, E.; Düvel, K.; Sahin, M.; Manning, B.D.; Hotamisligil, G.S. Loss of the tuberous sclerosis complex tumor suppressors triggers the unfolded protein response to regulate insulin signaling and apoptosis. Mol. Cell 2008, 29, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Oakes, S.A. Endoplasmic Reticulum Stress Signaling in Cancer Cells. Am. J. Pathol. 2020, 190, 934–946. [Google Scholar] [CrossRef] [PubMed]
- Dejeans, N.; Manié, S.; Hetz, C.; Bard, F.; Hupp, T.; Agostinis, P.; Samali, A.; Chevet, E. Addicted to secrete—Novel concepts and targets in cancer therapy. Trends Mol. Med. 2014, 20, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, N.R.; Rodvold, J.; Sepulveda, H.; Rossi, S.; Drew, A.F.; Zanetti, M. Transmission of endoplasmic reticulum stress and pro-inflammation from tumor cells to myeloid cells. Proc. Natl. Acad. Sci. USA 2011, 108, 6561–6566. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Oh, J.Y.; Kang, T.H.; Shin, H.S.; Cheng, M.A.; Farmer, E.; Wu, T.C.; Hung, C.F. Endoplasmic reticulum stress enhances the antigen-specific T cell immune responses and therapeutic antitumor effects generated by therapeutic HPV vaccines. J. Biomed. Sci. 2019, 26, 41. [Google Scholar] [CrossRef] [PubMed]
- Preynat-Seauve, O.; Coudurier, S.; Favier, A.; Marche, P.N.; Villiers, C. Oxidative stress impairs intracellular events involved in antigen processing and presentation to T cells. Cell Stress Chaperones 2003, 8, 162–171. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ke, C.-H.; Wu, H.-Y.; Wang, Y.-S.; Huang, W.-H.; Lin, C.-S. Tumors Established in a Defective Immune Environment Reprogram the Oncogenic Signaling Pathways to Escalate Tumor Antigenicity. Biomedicines 2024, 12, 846. https://doi.org/10.3390/biomedicines12040846
Ke C-H, Wu H-Y, Wang Y-S, Huang W-H, Lin C-S. Tumors Established in a Defective Immune Environment Reprogram the Oncogenic Signaling Pathways to Escalate Tumor Antigenicity. Biomedicines. 2024; 12(4):846. https://doi.org/10.3390/biomedicines12040846
Chicago/Turabian StyleKe, Chiao-Hsu, Hsin-Yi Wu, Yu-Shan Wang, Wei-Hsiang Huang, and Chen-Si Lin. 2024. "Tumors Established in a Defective Immune Environment Reprogram the Oncogenic Signaling Pathways to Escalate Tumor Antigenicity" Biomedicines 12, no. 4: 846. https://doi.org/10.3390/biomedicines12040846
APA StyleKe, C.-H., Wu, H.-Y., Wang, Y.-S., Huang, W.-H., & Lin, C.-S. (2024). Tumors Established in a Defective Immune Environment Reprogram the Oncogenic Signaling Pathways to Escalate Tumor Antigenicity. Biomedicines, 12(4), 846. https://doi.org/10.3390/biomedicines12040846