
Namodenoson at the Crossroad of Metabolic Dysfunction-Associated Steatohepatitis and Hepatocellular Carcinoma


Abstract
1. Introduction
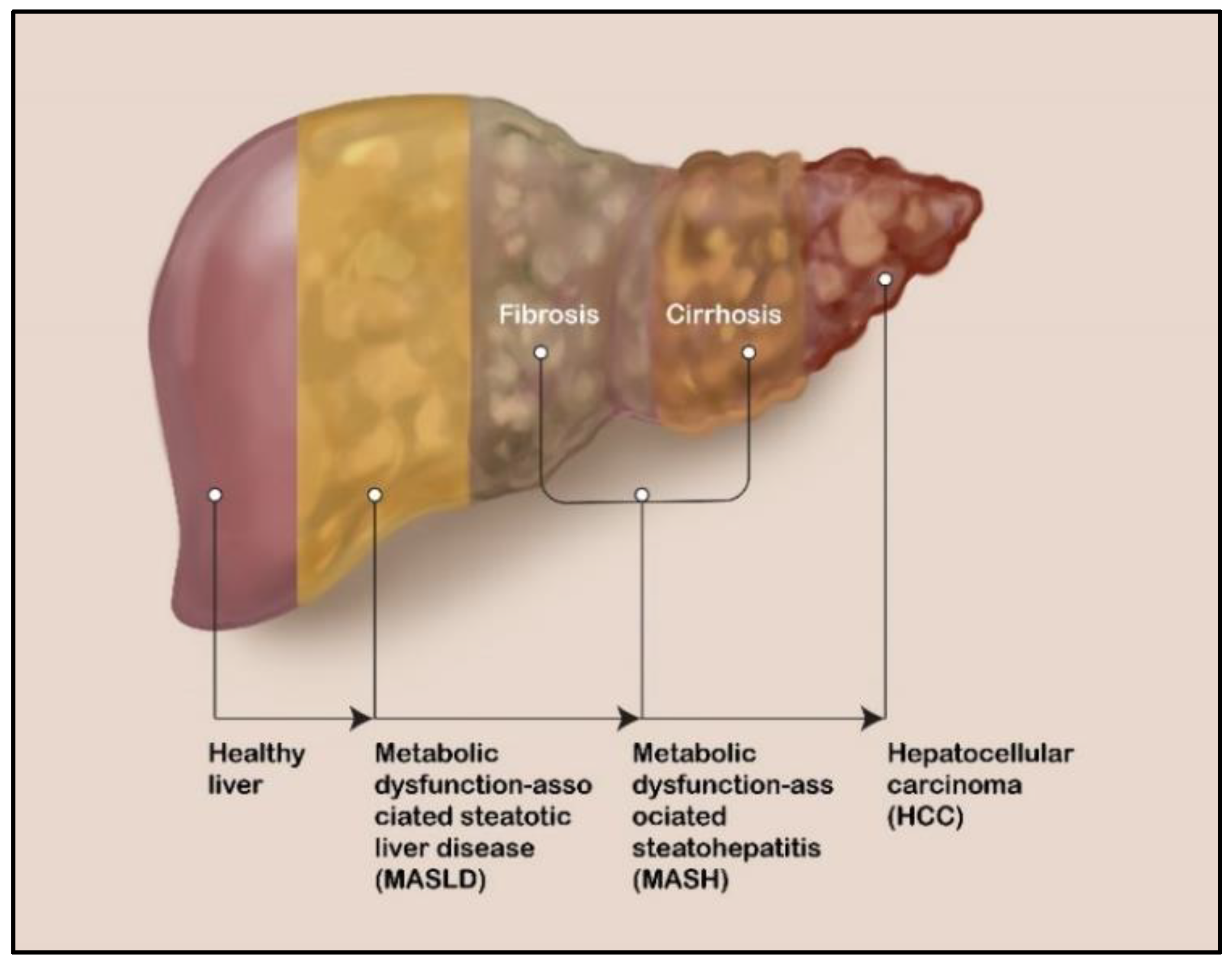
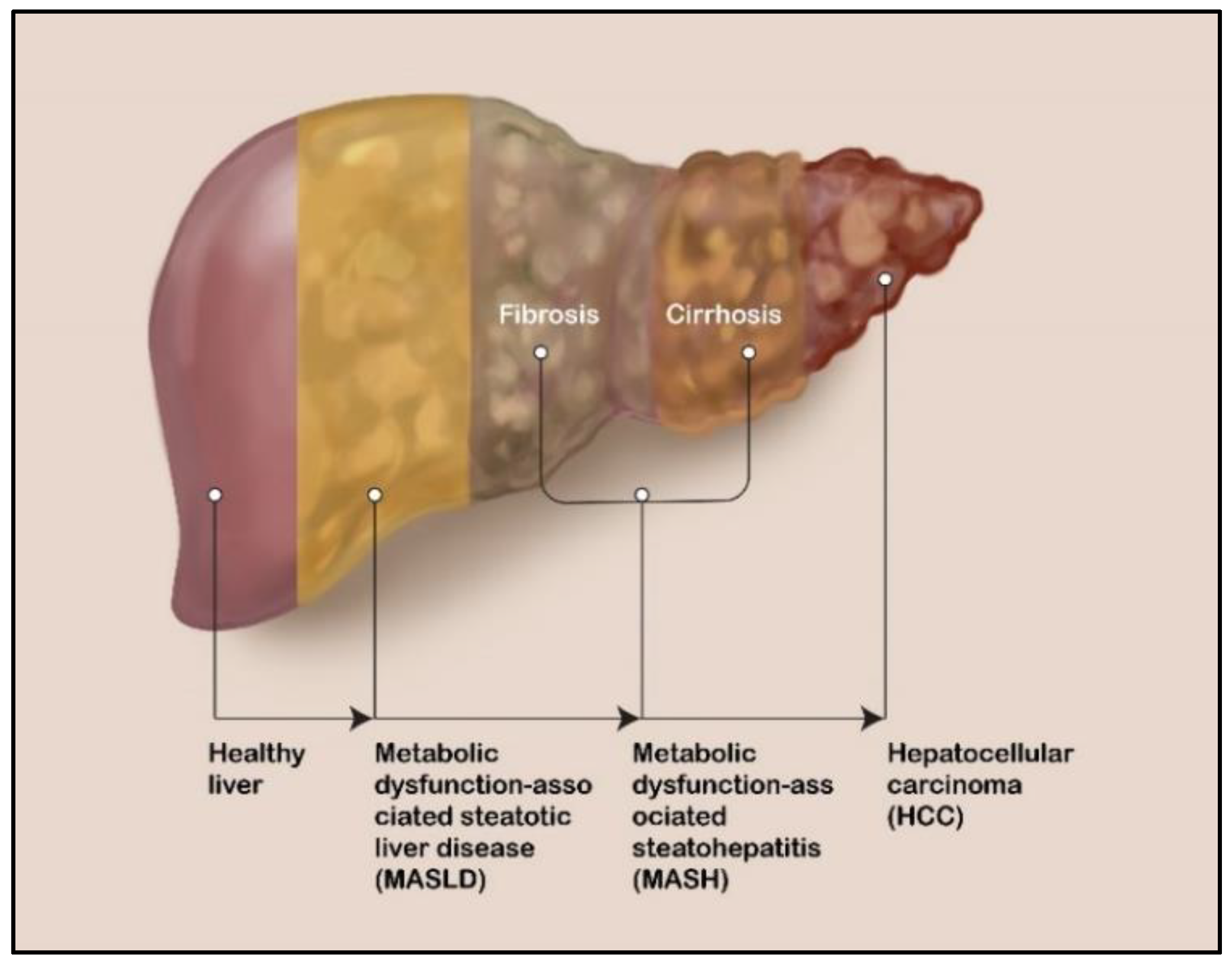
2. Liver Diseases: MASLD, MASH, and HCC
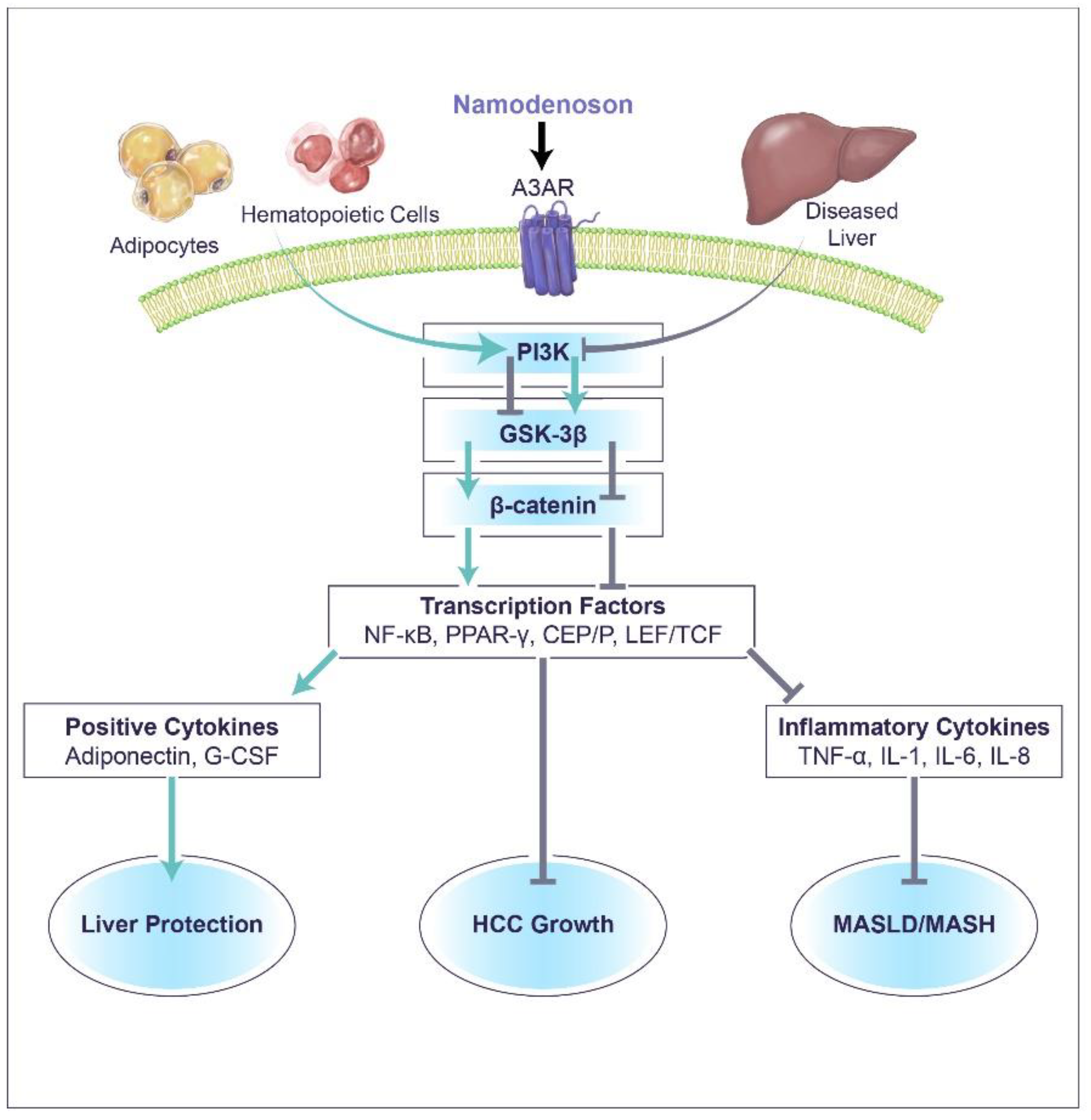
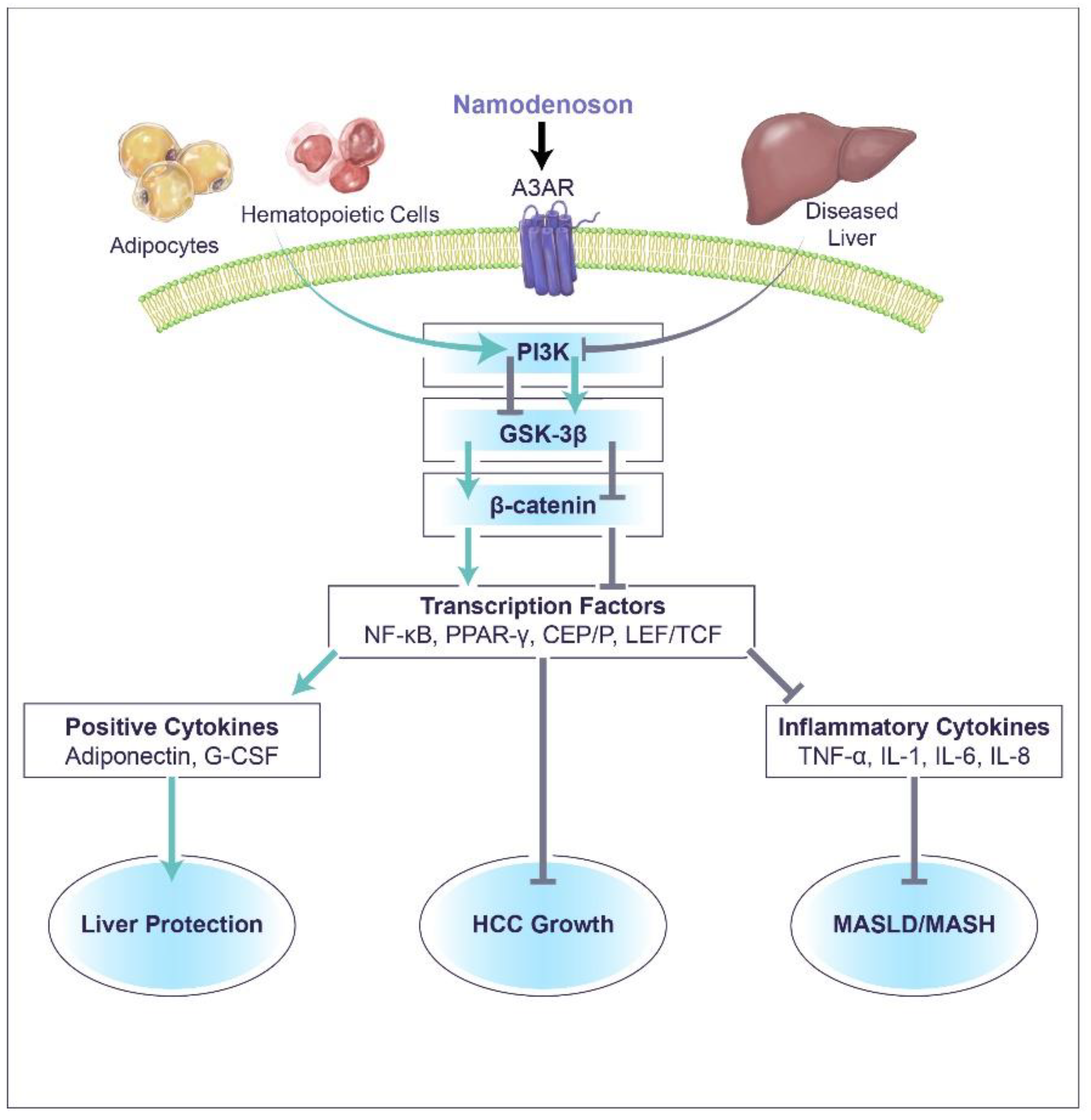
3. Namodenoson Molecular Mechanism of Action
3.1. The Effect of Namodenoson on Liver Inflammatory and Cancer Cells
3.2. The Effect of Namodenoson on Normal Cells
4. Namodenoson for the Treatment of MASLD/MASH
4.1. Preclinical Evidence
4.2. Clinical Evidence
5. Namodenoson for the Treatment of HCC
5.1. Preclinical Evidence
5.2. Clinical Evidence
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Poulsen, S.A.; Quinn, R.J. Adenosine receptors: New opportunities for future drugs. Bioorg. Med. Chem. 1998, 6, 619–641. [Google Scholar] [CrossRef] [PubMed]
- Layland, J.; Carrick, D.; Lee, M.; Oldroyd, K.; Berry, C. Adenosine: Physiology, pharmacology, and clinical applications. JACC Cardiovasc. Interv. 2014, 7, 581–591. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.V.; Downey, J.M. Adenosine: Trigger and mediator of cardioprotection. Basic Res. Cardiol. 2008, 103, 203–215. [Google Scholar] [CrossRef] [PubMed]
- Wardas, J. Neuroprotective role of adenosine in the CNS. Pol. J. Pharmacol. 2002, 54, 313–326. [Google Scholar] [PubMed]
- Effendi, W.I.; Nagano, T.; Kobayashi, K.; Nishimura, Y. Focusing on adenosine receptors as a potential targeted therapy in human diseases. Cells 2020, 9, 785. [Google Scholar] [CrossRef] [PubMed]
- Sheth, S.; Brito, R.; Mukherjea, D.; Rybak, L.P.; Ramkumar, V. Adenosine receptors: Expression, function and regulation. Int. J. Mol. Sci. 2014, 15, 2024–2052. [Google Scholar] [CrossRef] [PubMed]
- Sajjadi, F.G.; Firestein, G.S. cDNA cloning and sequence analysis of the human A3 adenosine receptor. Biochim. Biophys. Acta 1993, 1179, 105–107. [Google Scholar] [CrossRef] [PubMed]
- Gessi, S.; Cattabriga, E.; Avitabile, A.; Gafa, R.; Lanza, G.; Cavazzini, L.; Bianchi, N.; Gambari, R.; Feo, C.; Liboni, A.; et al. Elevated expression of A3 adenosine receptors in human colorectal cancer is reflected in peripheral blood cells. Clin. Cancer Res. 2004, 10, 5895–5901. [Google Scholar] [CrossRef] [PubMed]
- Bar-Yehuda, S.; Stemmer, S.M.; Madi, L.; Castel, D.; Ochaion, A.; Cohen, S.; Barer, F.; Zabutti, A.; Perez-Liz, G.; Del Valle, L.; et al. The A3 adenosine receptor agonist CF102 induces apoptosis of hepatocellular carcinoma via de-regulation of the Wnt and NF-kappaB signal transduction pathways. Int. J. Oncol. 2008, 33, 287–295. [Google Scholar]
- Madi, L.; Ochaion, A.; Rath-Wolfson, L.; Bar-Yehuda, S.; Erlanger, A.; Ohana, G.; Harish, A.; Merimski, O.; Barer, F.; Fishman, P. The A3 adenosine receptor is highly expressed in tumor versus normal cells: Potential target for tumor growth inhibition. Clin. Cancer Res. 2004, 10, 4472–4479. [Google Scholar] [CrossRef]
- Fishman, P.; Stemmer, S.M.; Bareket-Samish, A.; Silverman, M.H.; Kerns, W.D. Targeting the A3 adenosine receptor to treat hepatocellular carcinoma: Anti-cancer and hepatoprotective effects. Purinergic Signal. 2023, 19, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Rinella, M.E.; Lazarus, J.V.; Ratziu, V.; Francque, S.M.; Sanyal, A.J.; Kanwal, F.; Romero, D.; Abdelmalek, M.F.; Anstee, Q.M.; Arab, J.P.; et al. A multisociety Delphi consensus statement on new fatty liver disease nomenclature. Ann. Hepatol. 2024, 29, 101133. [Google Scholar] [CrossRef] [PubMed]
- Stemmer, S.M.; Benjaminov, O.; Medalia, G.; Ciuraru, N.B.; Silverman, M.H.; Bar-Yehuda, S.; Fishman, S.; Harpaz, Z.; Farbstein, M.; Cohen, S.; et al. CF102 for the treatment of hepatocellular carcinoma: A phase I/II, open-label, dose-escalation study. Oncologist 2013, 18, 25–26. [Google Scholar] [CrossRef] [PubMed]
- Stemmer, S.M.; Manojlovic, N.S.; Marinca, M.V.; Petrov, P.; Cherciu, N.; Ganea, D.; Ciuleanu, T.E.; Pusca, I.A.; Beg, M.S.; Purcell, W.T.; et al. Namodenoson in advanced hepatocellular carcinoma and Child-Pugh B cirrhosis: Randomized placebo-controlled clinical trial. Cancers 2021, 13, 187. [Google Scholar] [CrossRef] [PubMed]
- Safadi, R.; Braun, M.; Francis, A.; Milgrom, Y.; Massarwa, M.; Hakimian, D.; Hazou, W.; Issachar, A.; Harpaz, Z.; Farbstein, M.; et al. Randomised clinical trial: A phase 2 double-blind study of namodenoson in non-alcoholic fatty liver disease and steatohepatitis. Aliment. Pharmacol. Ther. 2021, 54, 1405–1415. [Google Scholar] [CrossRef] [PubMed]
- Paklar, N.; Mijic, M.; Filipec-Kanizaj, T. The outcomes of liver transplantation in severe metabolic dysfunction-associated steatotic liver disease patients. Biomedicines 2023, 11, 3096. [Google Scholar] [CrossRef]
- Miao, L.; Targher, G.; Byrne, C.D.; Cao, Y.Y.; Zheng, M.H. Current status and future trends of the global burden of MASLD. Trends Endocrinol. Metab. 2024. Epub ahead of print. [Google Scholar]
- Liu, J.; Tian, Y.; Fu, X.; Mu, C.; Yao, M.; Ni, Y.; Liu, Y.; Li, Z. Estimating global prevalence, incidence, and outcomes of non-alcoholic fatty liver disease from 2000 to 2021: Systematic review and meta-analysis. Chin. Med. J. 2022, 135, 1682–1691. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Golabi, P.; Paik, J.M.; Henry, A.; Van Dongen, C.; Henry, L. The global epidemiology of nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH): A systematic review. Hepatology 2023, 77, 1335–1347. [Google Scholar] [CrossRef]
- Fernando, D.H.; Forbes, J.M.; Angus, P.W.; Herath, C.B. Development and progression of non-alcoholic fatty liver disease: The role of advanced glycation end products. Int. J. Mol. Sci. 2019, 20, 5037. [Google Scholar] [CrossRef]
- Bellentani, S. The epidemiology of non-alcoholic fatty liver disease. Liver Int. 2017, 37 (Suppl. S1), 81–84. [Google Scholar] [CrossRef]
- Chew, N.W.S.; Ng, C.H.; Tan, D.J.H.; Kong, G.; Lin, C.; Chin, Y.H.; Lim, W.H.; Huang, D.Q.; Quek, J.; Fu, C.E.; et al. The global burden of metabolic disease: Data from 2000 to 2019. Cell Metab. 2023, 35, 414–428.e413. [Google Scholar] [CrossRef] [PubMed]
- Ampofo, A.G.; Boateng, E.B. Beyond 2020: Modelling obesity and diabetes prevalence. Diabetes Res. Clin. Pract. 2020, 167, 108362. [Google Scholar] [CrossRef] [PubMed]
- Teng, M.L.; Ng, C.H.; Huang, D.Q.; Chan, K.E.; Tan, D.J.; Lim, W.H.; Yang, J.D.; Tan, E.; Muthiah, M.D. Global incidence and prevalence of nonalcoholic fatty liver disease. Clin. Mol. Hepatol. 2023, 29, S32–S42. [Google Scholar] [CrossRef] [PubMed]
- Estes, C.; Anstee, Q.M.; Arias-Loste, M.T.; Bantel, H.; Bellentani, S.; Caballeria, J.; Colombo, M.; Craxi, A.; Crespo, J.; Day, C.P.; et al. Modeling NAFLD disease burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the period 2016–2030. J. Hepatol. 2018, 69, 896–904. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.Q.; El-Serag, H.B.; Loomba, R. Global epidemiology of NAFLD-related HCC: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Hester, D.; Golabi, P.; Paik, J.; Younossi, I.; Mishra, A.; Younossi, Z.M. Among Medicare patients with hepatocellular carcinoma, non-alcoholic fatty liver disease is the most common etiology and cause of mortality. J. Clin. Gastroenterol. 2020, 54, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Motta, B.M.; Masarone, M.; Torre, P.; Persico, M. From non-alcoholic steatohepatitis (NASH) to hepatocellular carcinoma (HCC): Epidemiology, incidence, predictions, risk factors, and prevention. Cancers 2023, 15, 5458. [Google Scholar] [CrossRef] [PubMed]
- Singal, A.G.; Kudo, M.; Bruix, J. Breakthroughs in hepatocellular carcinoma therapies. Clin. Gastroenterol. Hepatol. 2023, 21, 2135–2149. [Google Scholar] [CrossRef]
- Chidambaranathan-Reghupaty, S.; Fisher, P.B.; Sarkar, D. Hepatocellular carcinoma (hcc): Epidemiology, etiology and molecular classification. Adv. Cancer Res. 2021, 149, 1–61. [Google Scholar]
- Fishman, P.; Jacobson, K.A.; Ochaion, A.; Cohen, S.; Bar-Yehuda, S. The anti-cancer effect of A3 adenosine receptor agonists: A novel, targeted therapy. Immun. Endoc Metab. Agents Med. Chem. 2007, 7, 298–303. [Google Scholar] [CrossRef]
- Cohen, S.; Stemmer, S.M.; Zozulya, G.; Ochaion, A.; Patoka, R.; Barer, F.; Bar-Yehuda, S.; Rath-Wolfson, L.; Jacobson, K.A.; Fishman, P. CF102 an A3 adenosine receptor agonist mediates anti-tumor and anti-inflammatory effects in the liver. J. Cell Physiol. 2011, 226, 2438–2447. [Google Scholar] [CrossRef] [PubMed]
- Fishman, P.; Cohen, S.; Itzhak, I.; Amer, J.; Salhab, A.; Barer, F.; Safadi, R. The A3 adenosine receptor agonist, namodenoson, ameliorates nonalcoholic steatohepatitis in mice. Int. J. Mol. Med. 2019, 44, 2256–2264. [Google Scholar]
- Fishman, P.; Cohen, S.; Salhab, A.; Amer, J.; Itzhak, I.; Barer, F.; Safadi, R. Namodenoson anti-NAFLD/NASH activity is mediated via de-regulation of the Wnt/β-catenin pathway. Inflamm. Intest. Dis. 2019, 44, 2256–2264. [Google Scholar]
- Dituri, F.; Mazzocca, A.; Giannelli, G.; Antonaci, S. PI3K functions in cancer progression, anticancer immunity and immune evasion by tumors. Clin. Dev. Immunol. 2011, 2011, 947858. [Google Scholar] [CrossRef]
- Stark, A.K.; Sriskantharajah, S.; Hessel, E.M.; Okkenhaug, K. PI3K inhibitors in inflammation, autoimmunity and cancer. Curr. Opin. Pharmacol. 2015, 23, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Ren, T.; Tian, T.; Feng, X.; Ye, S.; Wang, H.; Wu, W.; Qiu, Y.; Yu, C.; He, Y.; Zeng, J.; et al. An adenosine A3 receptor agonist inhibits DSS-induced colitis in mice through modulation of the NF-kappaB signaling pathway. Sci. Rep. 2015, 5, 9047. [Google Scholar] [CrossRef]
- Ravani, A.; Vincenzi, F.; Bortoluzzi, A.; Padovan, M.; Pasquini, S.; Gessi, S.; Merighi, S.; Borea, P.A.; Govoni, M.; Varani, K. Role and function of A(2A) and A(3) adenosine receptors in patients with ankylosing spondylitis, psoriatic arthritis and rheumatoid arthritis. Int. J. Mol. Sci. 2017, 18, 697. [Google Scholar] [CrossRef] [PubMed]
- Ren, T.H.; Lv, M.M.; An, X.M.; Leung, W.K.; Seto, W.K. Activation of adenosine A3 receptor inhibits inflammatory cytokine production in colonic mucosa of patients with ulcerative colitis by down-regulating the nuclear factor-kappa b signaling. J. Dig. Dis. 2020, 21, 38–45. [Google Scholar] [CrossRef]
- Bar-Yehuda, S.; Madi, L.; Barak, D.; Mittelman, M.; Ardon, E.; Ochaion, A.; Cohn, S.; Fishman, P. Agonists to the a3 adenosine receptor induce g-csf production via nf-kappab activation: A new class of myeloprotective agents. Exp. Hematol. 2002, 30, 1390–1398. [Google Scholar] [CrossRef]
- Hofer, M.; Pospisil, M.; Sefc, L.; Dusek, L.; Vacek, A.; Hola, J.; Hoferova, Z.; Streitova, D. Activation of adenosine A(3) receptors supports hematopoiesis-stimulating effects of granulocyte colony-stimulating factor in sublethally irradiated mice. Int. J. Radiat. Biol. 2010, 86, 649–656. [Google Scholar] [CrossRef] [PubMed]
- Link, H. Current state and future opportunities in granulocyte colony-stimulating factor (G-CSF). Support. Care Cancer 2022, 30, 7067–7077. [Google Scholar] [CrossRef] [PubMed]
- Cetean, S.; Cainap, C.; Constantin, A.M.; Cainap, S.; Gherman, A.; Oprean, L.; Hangan, A.; Oprean, R. The importance of the granulocyte-colony stimulating factor in oncology. Clujul Med. 2015, 88, 468–472. [Google Scholar] [CrossRef]
- Gascon, P.; Awada, A.; Karihtala, P.; Lorenzen, S.; Minichsdorfer, C. Optimal use of granulocyte colony-stimulating factor prophylaxis to improve survival in cancer patients receiving treatment: An expert view. Wien. Klin. Wochenschr. 2023. Epub ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Sharma, A.K.; Narasimhan, R.L.; Bhalla, A.; Sharma, N.; Sharma, R. Granulocyte colony-stimulating factor in severe alcoholic hepatitis: A randomized pilot study. Am. J. Gastroenterol. 2014, 109, 1417–1423. [Google Scholar] [CrossRef] [PubMed]
- Engelmann, C.; Berg, T. G-CSF treatment in decompensated liver disease: A double-edged sword? Hepatol. Int. 2022, 16, 979–982. [Google Scholar] [CrossRef] [PubMed]
- Engelmann, C.; Herber, A.; Franke, A.; Bruns, T.; Reuken, P.; Schiefke, I.; Zipprich, A.; Zeuzem, S.; Goeser, T.; Canbay, A.; et al. Granulocyte-colony stimulating factor (G-CSF) to treat acute-on-chronic liver failure: A multicenter randomized trial (graft study). J. Hepatol. 2021, 75, 1346–1354. [Google Scholar] [CrossRef]
- Gamberi, T.; Magherini, F.; Modesti, A.; Fiaschi, T. Adiponectin signaling pathways in liver diseases. Biomedicines 2018, 6, 52. [Google Scholar] [CrossRef]
- Wang, Z.V.; Scherer, P.E. Adiponectin, the past two decades. J. Mol. Cell Biol. 2016, 8, 93–100. [Google Scholar] [CrossRef]
- Adolph, T.E.; Grander, C.; Grabherr, F.; Tilg, H. Adipokines and non-alcoholic fatty liver disease: Multiple interactions. Int. J. Mol. Sci. 2017, 18, 1649. [Google Scholar] [CrossRef]
- Zhang, L.; Yuan, Q.; Li, M.; Chai, D.; Deng, W.; Wang, W. The association of leptin and adiponectin with hepatocellular carcinoma risk and prognosis: A combination of traditional, survival, and dose-response meta-analysis. BMC Cancer 2020, 20, 1167. [Google Scholar] [CrossRef] [PubMed]
- Vachliotis, I.D.; Valsamidis, I.; Polyzos, S.A. Tumor necrosis factor-alpha and adiponectin in nonalcoholic fatty liver disease-associated hepatocellular carcinoma. Cancers 2023, 15, 5306. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, H.; Zhang, Z.; Huang, B.; Cheng, X.; Wang, D.; la Gahu, Z.; Xue, Z.; Da, Y.; Li, D.; et al. Adiponectin-derived active peptide ADP355 exerts anti-inflammatory and anti-fibrotic activities in thioacetamide-induced liver injury. Sci. Rep. 2016, 6, 19445. [Google Scholar] [CrossRef] [PubMed]
- Larsson, S.C.; Spyrou, N.; Mantzoros, C.S. Body fatness associations with cancer: Evidence from recent epidemiological studies and future directions. Metabolism 2022, 137, 155326. [Google Scholar] [CrossRef] [PubMed]
- Saxena, N.K.; Fu, P.P.; Nagalingam, A.; Wang, J.; Handy, J.; Cohen, C.; Tighiouart, M.; Sharma, D.; Anania, F.A. Adiponectin modulates C-jun N-terminal kinase and mammalian target of rapamycin and inhibits hepatocellular carcinoma. Gastroenterology 2010, 139, 1762–1773.e5. [Google Scholar] [CrossRef] [PubMed]
- Correnti, J.M.; Cook, D.; Aksamitiene, E.; Swarup, A.; Ogunnaike, B.; Vadigepalli, R.; Hoek, J.B. Adiponectin fine-tuning of liver regeneration dynamics revealed through cellular network modelling. J. Physiol. 2015, 593, 365–383. [Google Scholar] [CrossRef] [PubMed]
- Ezaki, H.; Yoshida, Y.; Saji, Y.; Takemura, T.; Fukushima, J.; Matsumoto, H.; Kamada, Y.; Wada, A.; Igura, T.; Kihara, S.; et al. Delayed liver regeneration after partial hepatectomy in adiponectin knockout mice. Biochem. Biophys. Res. Commun. 2009, 378, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]
- Ohana, G.; Cohen, S.; Rath-Wolfson, L.; Fishman, P. A3 adenosine receptor agonist, CF102, protects against hepatic ischemia/reperfusion injury following partial hepatectomy. Mol. Med. Rep. 2016, 14, 4335–4341. [Google Scholar] [CrossRef]
- Clinicaltrials.Gov. Description of the “Namodenoson in the Treatment of Non-Alcoholic Steatohepatitis (NASH)” Study (NCT04697810). Available online: https://classic.clinicaltrials.gov/ct2/show/NCT04697810?term=namodenoson&draw=2&rank=1 (accessed on 24 January 2024).
- Ciurescu, I.A.; Lencioni, R.; Stemmer, S.M.; Farbstein, M.; Harpaz, Z.; Silverman, M.H.; Fishman, P. Complete response induced by namodenoson in advanced hepatocellular carcinoma: A case report. In Proceedings of the ILCA Annual Conference 2022, Madrid, Spain, 1–4 September 2022. [Google Scholar]
- Clinicaltrials.Gov. Description of the “Namodenoson in the Treatment of Advanced Hepatocellular CARCINOMA in patients with Child-Pugh Class B7 Cirrhosis (LIVERATION)” Study (NCT05201404). Available online: https://clinicaltrials.gov/study/NCT05201404?intr=namodenoson&rank=2&tab=table (accessed on 28 January 2024).
- EMEA European Medicines Agency Announcement. Eu/3/15/1565–Orphan Designation for Treatment of Hepatocellular Carcinoma. Available online: https://www.ema.europa.eu/en/medicines/human/orphan-designations/eu-3-15-1565 (accessed on 30 March 2024).

{kind=link}
{kind=link}
| Author Reference | Disease | Phase, Study Design, n, Key Endpoints | Key Findings |
|---|---|---|---|
| Safadi et al. [15] | MASLD with or without MASH | Phase 2, randomized (1:1:1) double blind study of namodenoson 12.5 mg BID (n = 21) or 25 mg BID (n = 19) vs. placebo (n = 20). Main endpoints: ALT after 12 weeks, safety. |
|
| Stemmer et al. [13] | Advanced unresectable HCC | Phase 1/2 open-label dose-escalation study (n = 18, 6 at each dose level: 1, 5, and 25 mg BID). Main endpoint: Safety |
|
| Stemmer et al. [14] | HCC CPB patients who either progressed on, or could not tolerate, prior sorafenib treatment. | Phase 2, randomized (2:1) double blind study of namodenoson 25 mg BID (n = 50) vs. placebo (n = 28). Main endpoint: OS in the ITT population. |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Etzion, O.; Bareket-Samish, A.; Yardeni, D.; Fishman, P. Namodenoson at the Crossroad of Metabolic Dysfunction-Associated Steatohepatitis and Hepatocellular Carcinoma. Biomedicines 2024, 12, 848. https://doi.org/10.3390/biomedicines12040848
Etzion O, Bareket-Samish A, Yardeni D, Fishman P. Namodenoson at the Crossroad of Metabolic Dysfunction-Associated Steatohepatitis and Hepatocellular Carcinoma. Biomedicines. 2024; 12(4):848. https://doi.org/10.3390/biomedicines12040848
Chicago/Turabian StyleEtzion, Ohad, Avital Bareket-Samish, David Yardeni, and Pnina Fishman. 2024. "Namodenoson at the Crossroad of Metabolic Dysfunction-Associated Steatohepatitis and Hepatocellular Carcinoma" Biomedicines 12, no. 4: 848. https://doi.org/10.3390/biomedicines12040848
APA StyleEtzion, O., Bareket-Samish, A., Yardeni, D., & Fishman, P. (2024). Namodenoson at the Crossroad of Metabolic Dysfunction-Associated Steatohepatitis and Hepatocellular Carcinoma. Biomedicines, 12(4), 848. https://doi.org/10.3390/biomedicines12040848