
Two Signaling Modes Are Better than One: Flux-Independent Signaling by Ionotropic Glutamate Receptors Is Coming of Age

 ,
,  , ,
, ,  ,
,  and
and 
Abstract
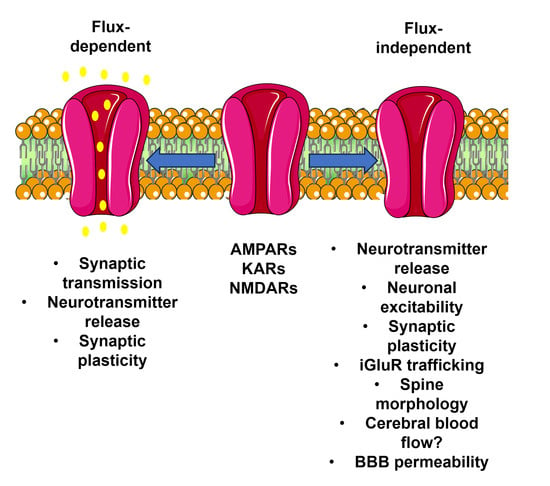
:
1. Introduction
2. Flux-Independent Signaling by iGluRs
3. Flux-Independent Signaling by AMPARs
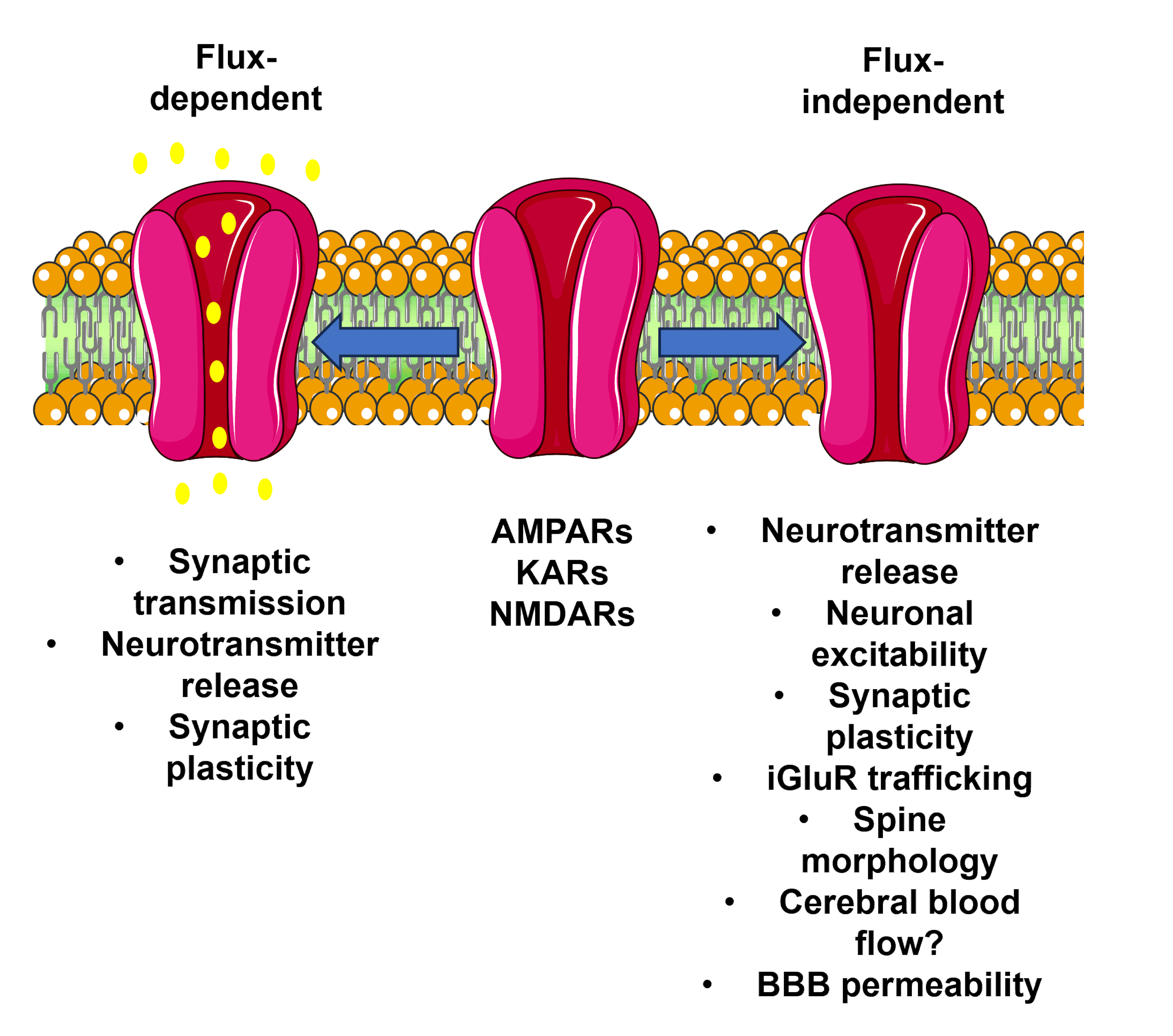
3.1. Flux-Independent Signaling by AMPARs Involves Multiple Metabotropic Signaling Pathways
3.2. The Molecular Determinants of Flux-Independent Signaling by AMPARs
3.3. Flux-Independent Signaling by AMPARs May Involve a Structural Component
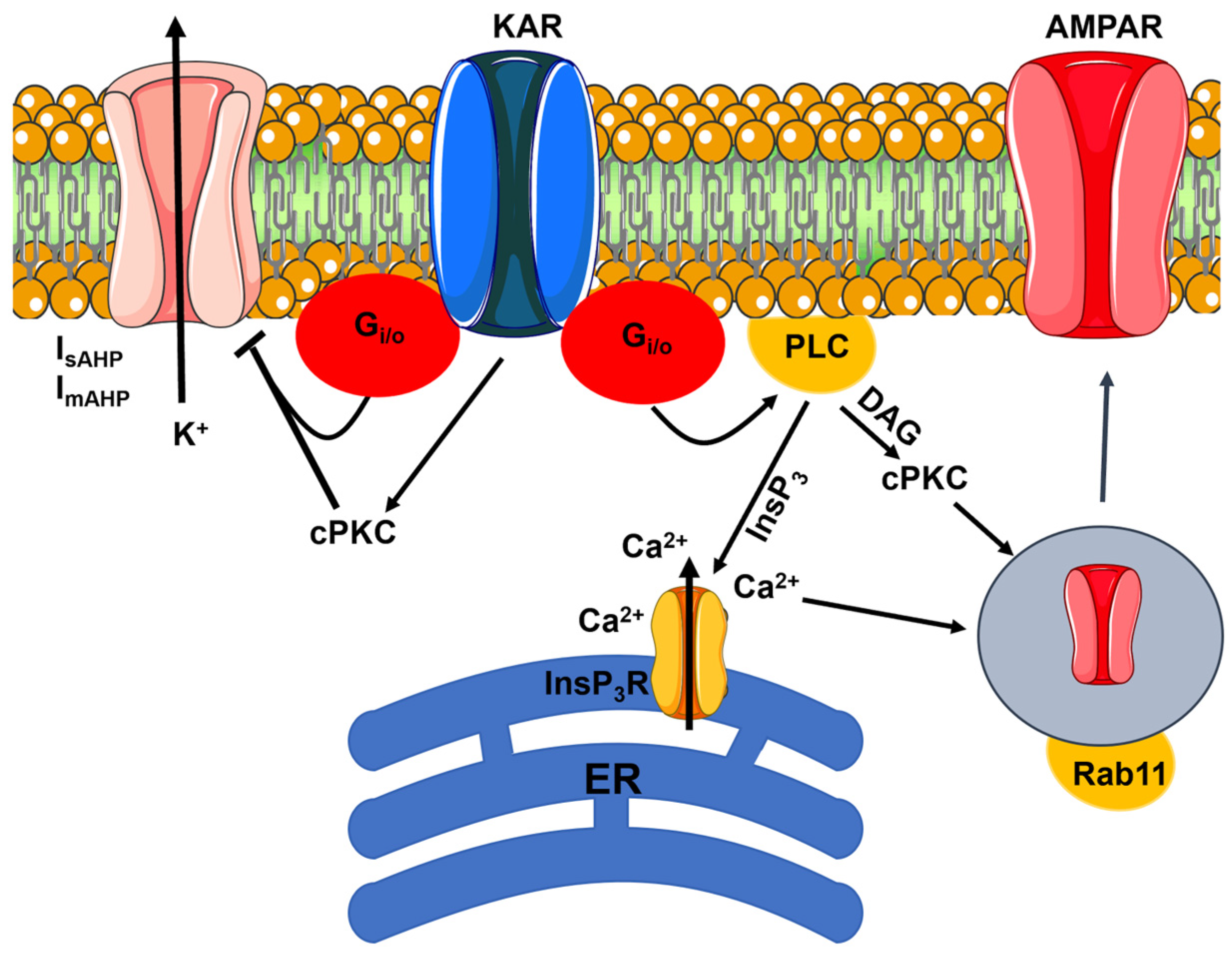
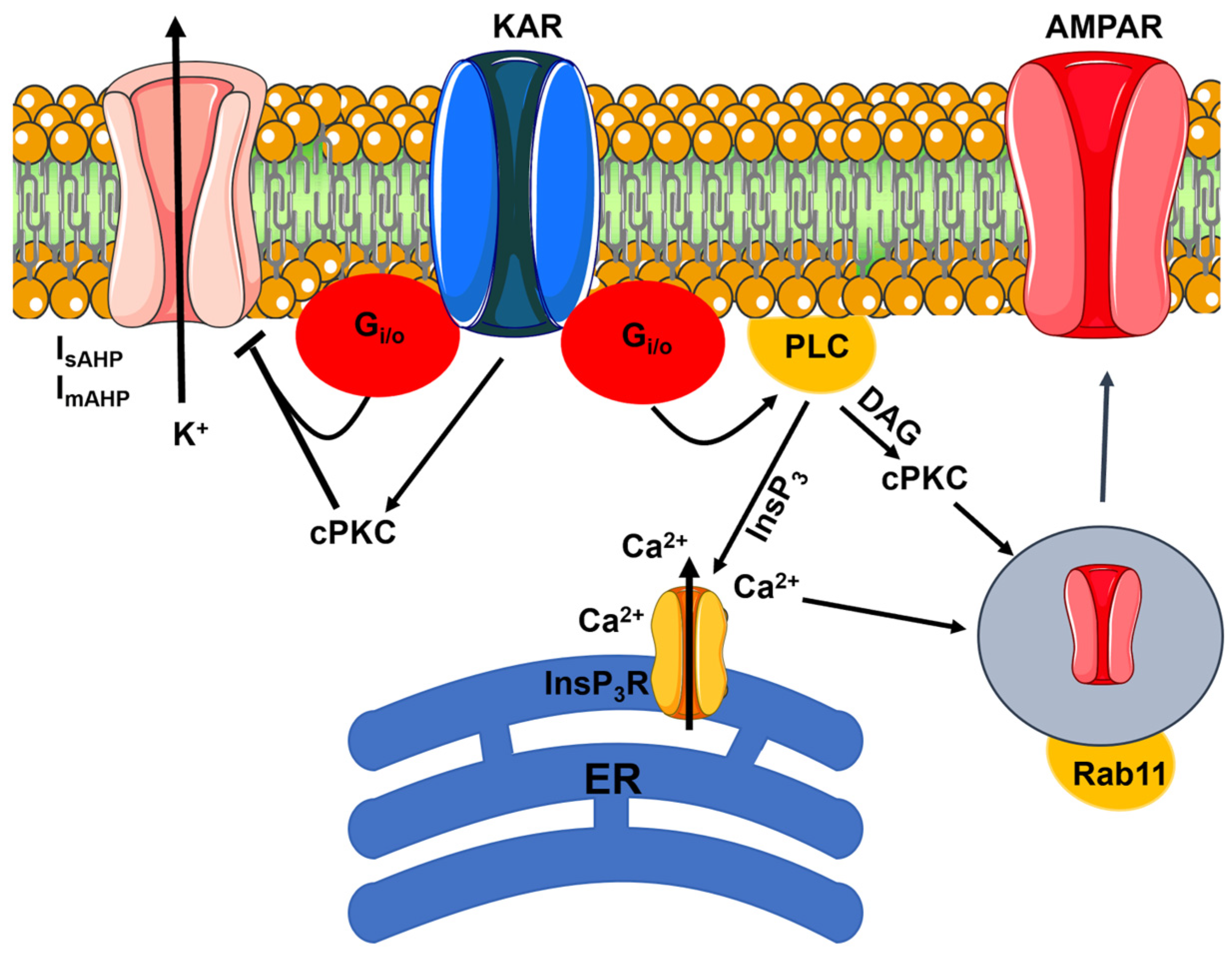
4. Flux-Independent Signaling by KARs
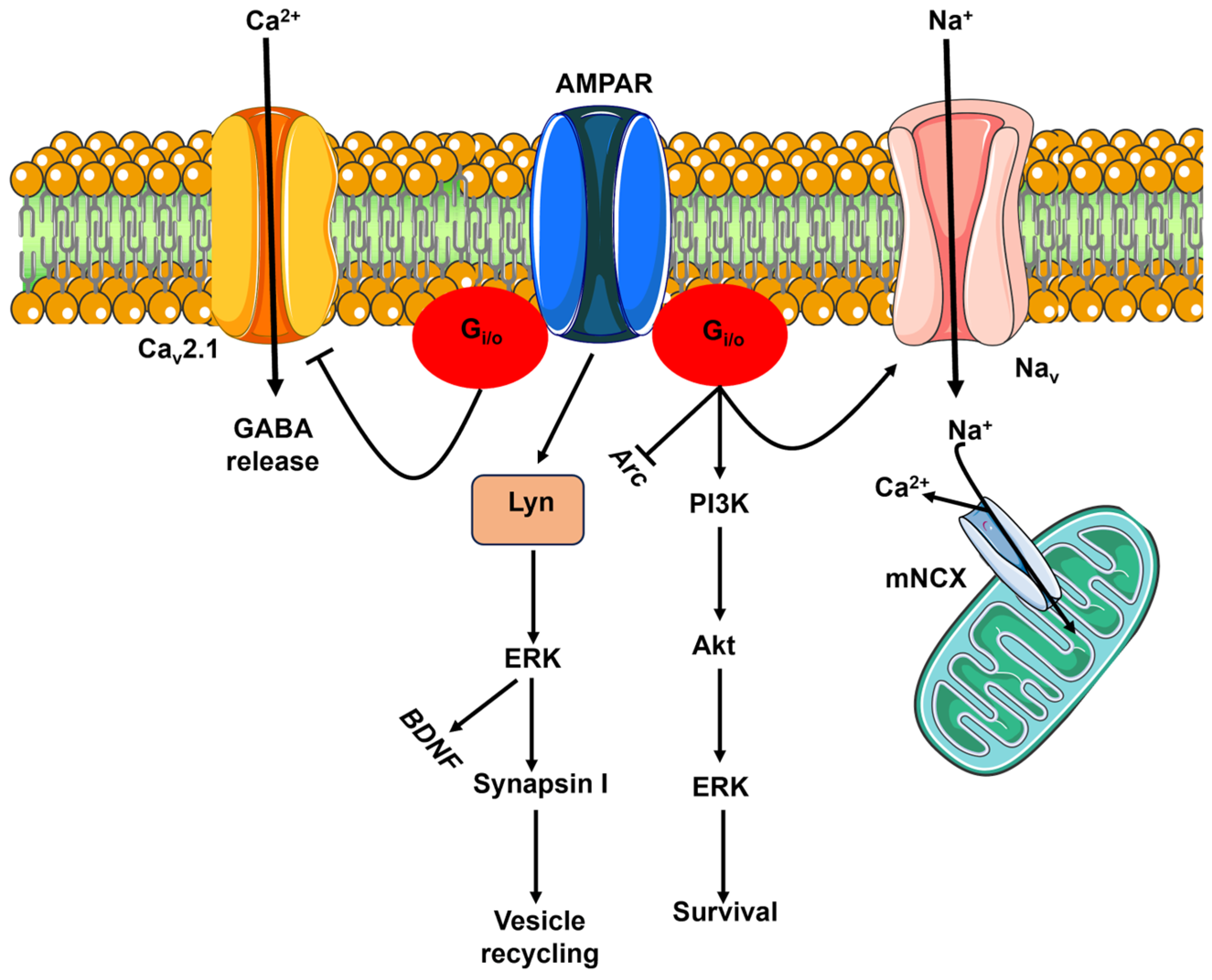
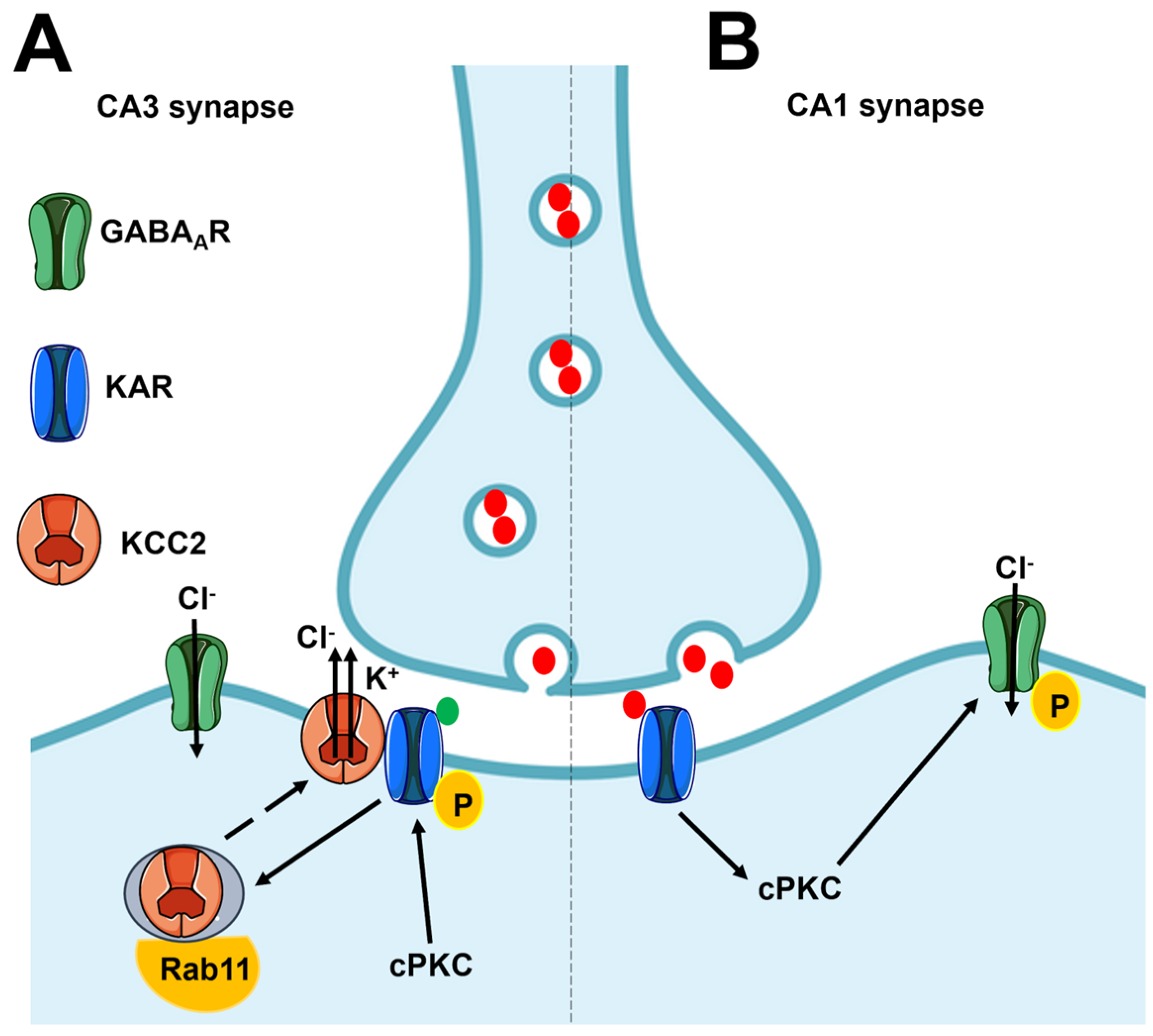
4.1. Flux-Independent Signaling by Presynaptic KARs Regulates GABA Release in the Hippocampus
4.2. Flux-Independent Signaling by Presynaptic KARs Regulates Glutamate Release in the Hippocampus
4.3. Flux-Independent Signaling by Presynaptic KARs Regulates Neurotransmitter Release in the Cerebellum

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Brain Region | Effect | Signaling Pathway(s) | Function | Ref. |
|---|---|---|---|---|
| Rat hippocampus CA1 interneurons | GABA ↓ | Gi/o proteins, PLC, cPKC | Regulation of PN excitability | [83,84,85] |
| Rat hippocampus SChC-CA1 Neonate | Glu ↓ | Gi/o proteins, cPKC | Synaptic maturation and plasticity | [102,104,105] |
| Rat hippocampus SChC-CA1 Adult | Glu ↓ | Gi/o proteins, cPKC | Unknown | [106,107] |
| Rat hippocampus MF-CA3 Neonate | Glu ↓ onto PNs | Gi/o proteins, cPKC | Hippocampus development | [101] |
| Rat hippocampus MF-CA3 Neonate | Glu ↑ onto GIs | Unknown | Hippocampus development | [101] |
| Rat hippocampus CA3 (A/C) | Glu ↓ | Gi/o proteins | Vesicle release | [120] |
| Mouse hippocampus MF-CA3 Adult | Glu ↓ | Gi/o proteins, AC inhibition | Unknown | [112,113] |
| Mouse cerebellum PF-PuC Adult | Glu ↓ | Gi/o proteins, AC inhibition | Synaptic maturation | [75,80,121] |
| Mouse amygdala MGN-LA Adult | Glu ↓ | PKA | Plasticity and oscillations at the theta and gamma bands? | [122] |
| Rat globus pallidus | Glu ↓ | Gi/o proteins, cPKC | Unknown | [123] |
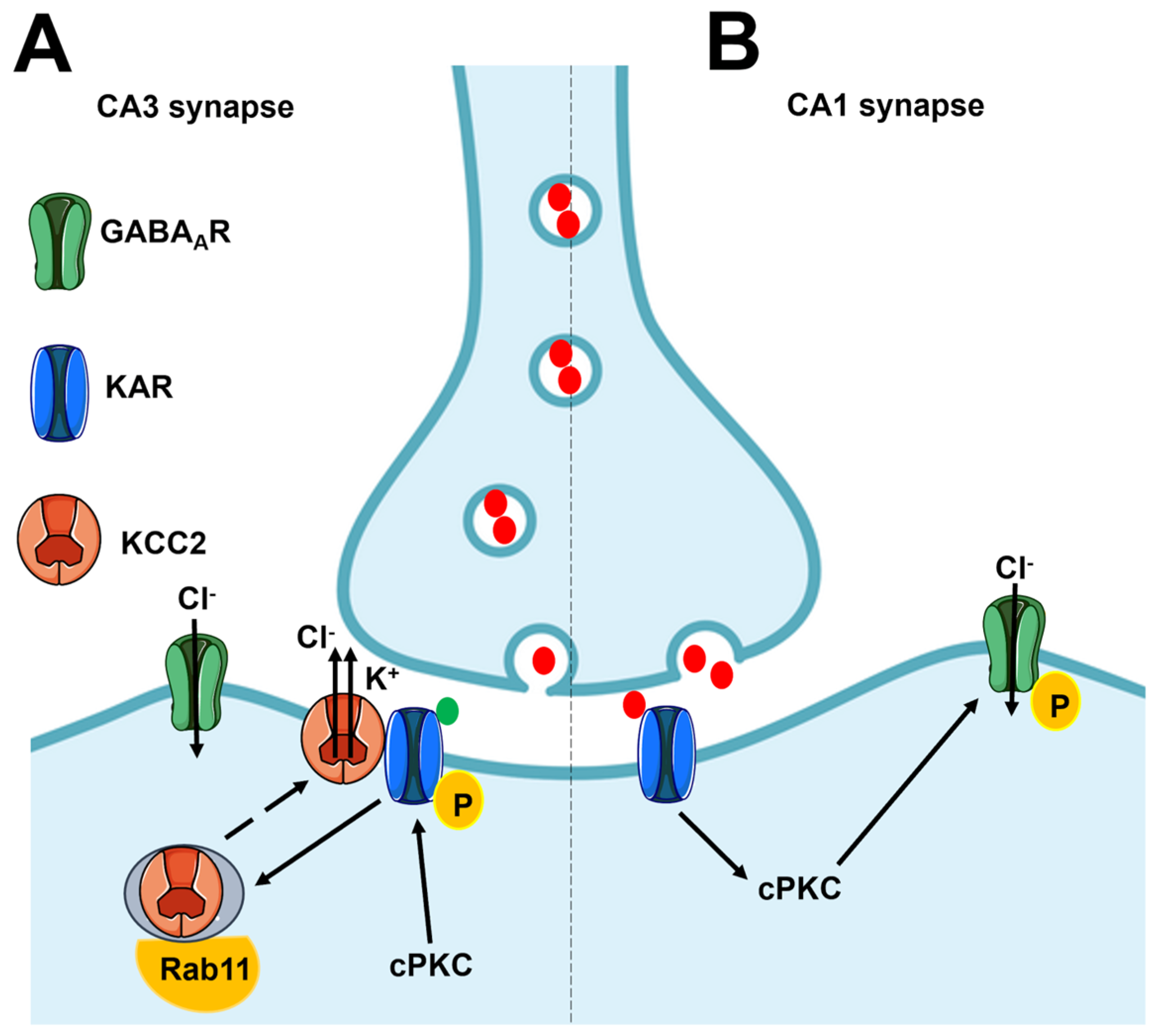
4.4. Flux-Independent Signaling by Postsynaptic KARs Regulates Neuronal Excitability and Synaptic Plasticity in the Hippocampus
4.5. Flux-Independent Signaling by KARs Regulates Axon Growth and Synaptic Differentiation
4.6. Flux-Independent Signaling by KARs: Future Perspectives
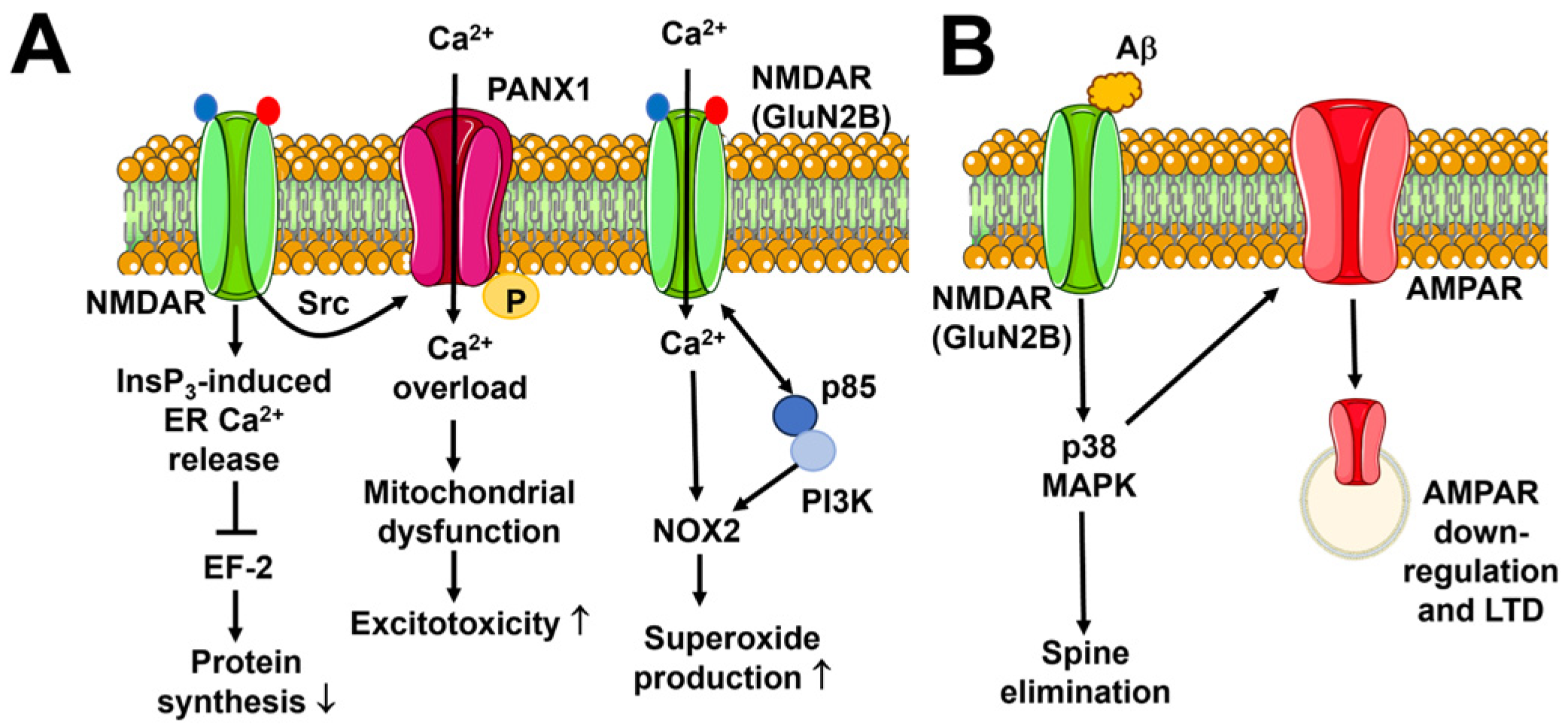
5. Flux-Independent Signaling by NMDARs
5.1. Flux-Independent Signaling by NMDARs in Neuronal Physiology
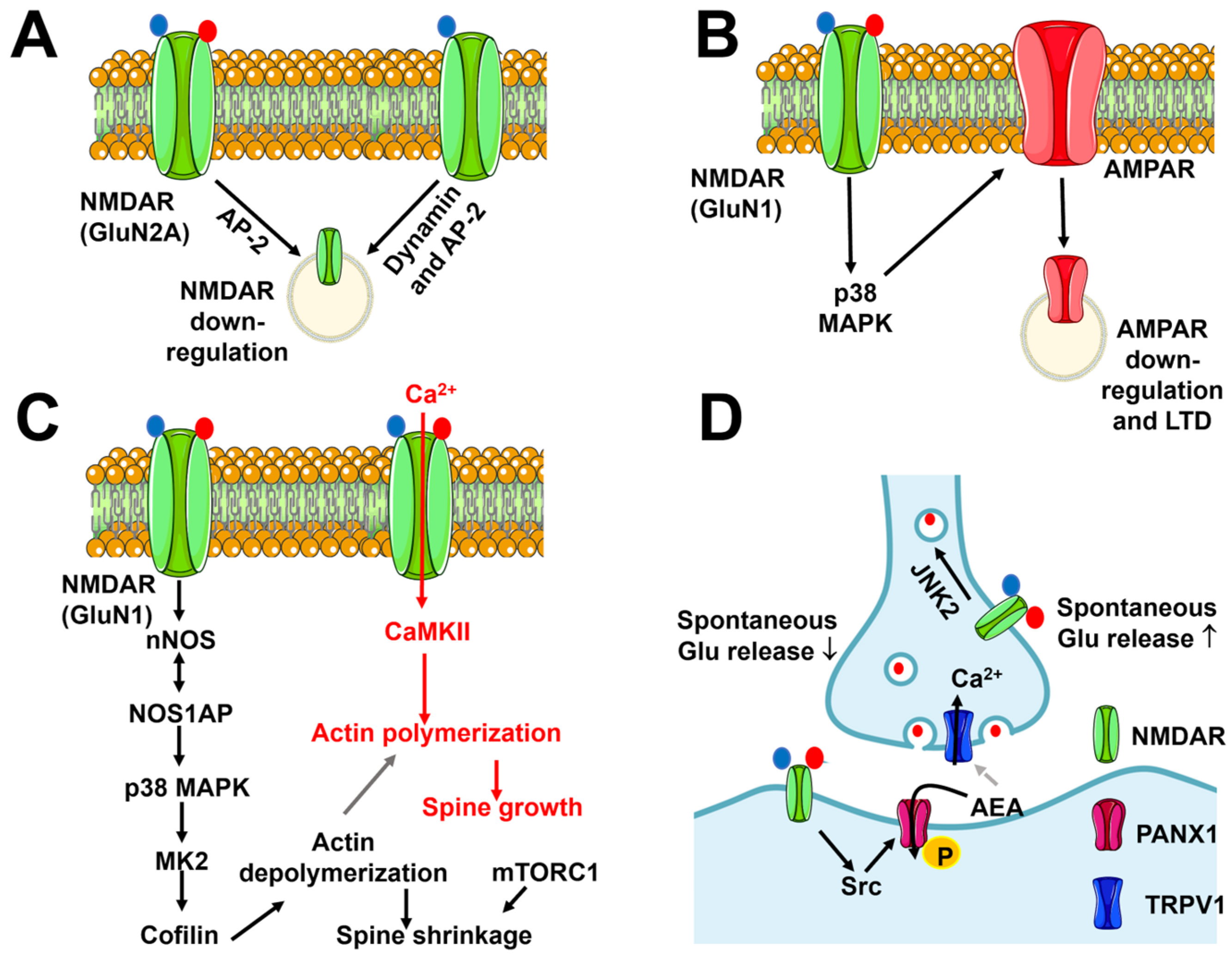
5.1.1. Flux-Independent Signaling by NMDARs Controls NMDAR Internalization and Trafficking
5.1.2. Flux-Independent Signaling by NMDARs Controls Synaptic Plasticity
5.1.3. Flux-Independent Signaling by NMDARs Bidirectionally Regulates Spine Morphology
5.1.4. Flux-Independent Signaling by NMDARs Regulates Spontaneous Glutamate Release
5.1.5. Flux-Independent Signaling by NMDARs in Neuronal Physiology: Future Perspectives
5.2. Flux-Independent Signaling by NMDARs in Brain Astrocytes and Microvascular Endothelial Cells
5.2.1. Flux-Independent Signaling by NMDARs in Brain Astrocytes
5.2.2. Flux-Independent Signaling by NMDARs in Human CECs
6. Flux-Independent Signaling by NMDARs in Brain Disorders
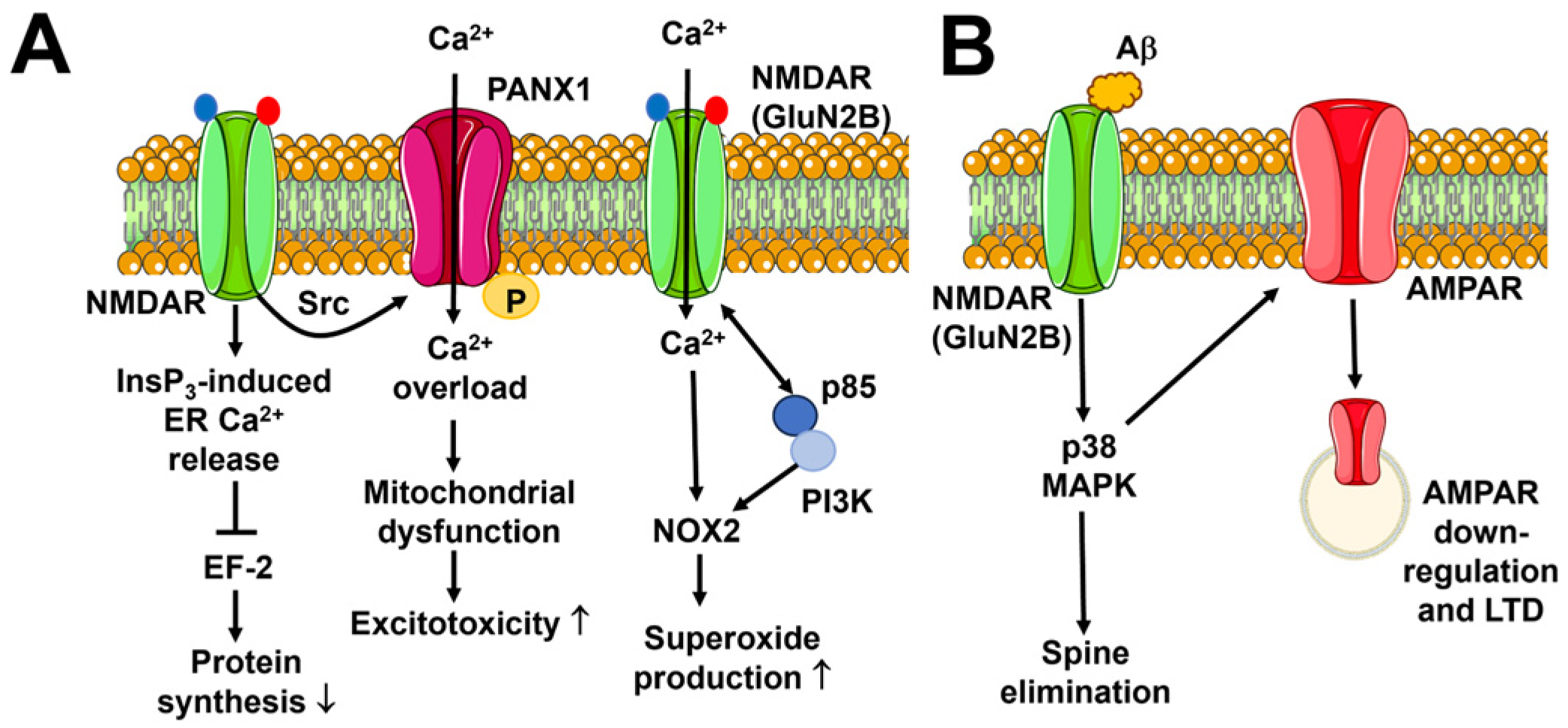
6.1. Neuronal Excitotoxicity
6.2. Alzheimer’s Disease
6.3. Schizophrenia
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Eccles, J.C.; McGeer, P.L. Ionotropic and metabotropic neurotransmission. Trends Neurosci. 1979, 2, P39–P40. [Google Scholar] [CrossRef]
- Purves, D.; Augustine, G.J.; Fitzpatrick, D.; Katz, L.C.; LaMantia, L.S.; McNamara, J.O.; Williams, S.M. Neuroscience, 2nd ed.; Two Families of Postsynaptic Receptors; Sinauer Associates: Sunderland, MA, USA, 2001. Available online: https://www.ncbi.nlm.nih.gov/books/NBK10855/ (accessed on 1 February 2024).
- Roth, B.L. Molecular pharmacology of metabotropic receptors targeted by neuropsychiatric drugs. Nat. Struct. Mol. Biol. 2019, 26, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Gray, J.A.; Zito, K.; Hell, J.W. Non-ionotropic signaling by the NMDA receptor: Controversy and opportunity. F1000Research 2016, 5, F1000 Faculty Rev-1010. [Google Scholar] [CrossRef] [PubMed]
- Dore, K.; Stein, I.S.; Brock, J.A.; Castillo, P.E.; Zito, K.; Sjostrom, P.J. Unconventional NMDA Receptor Signaling. J. Neurosci. 2017, 37, 10800–10807. [Google Scholar] [CrossRef] [PubMed]
- Park, D.K.; Stein, I.S.; Zito, K. Ion flux-independent NMDA receptor signaling. Neuropharmacology 2022, 210, 109019. [Google Scholar] [CrossRef] [PubMed]
- Montes de Oca Balderas, P. Meeting report: Flux-independent signaling by ionotropic receptors: Unforeseen roles, complexities, and challenges. J. Biol. Chem. 2022, 298, 102330. [Google Scholar] [CrossRef] [PubMed]
- Valbuena, S.; Lerma, J. Non-canonical Signaling, the Hidden Life of Ligand-Gated Ion Channels. Neuron 2016, 92, 316–329. [Google Scholar] [CrossRef] [PubMed]
- Soda, T.; Brunetti, V.; Berra-Romani, R.; Moccia, F. The Emerging Role of N-Methyl-D-Aspartate (NMDA) Receptors in the Cardiovascular System: Physiological Implications, Pathological Consequences, and Therapeutic Perspectives. Int. J. Mol. Sci. 2023, 24, 3914. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Fiorio Pla, A.; Lim, D.; Lodola, F.; Gerbino, A. Intracellular Ca2+ signalling: Unexpected new roles for the usual suspect. Front. Physiol. 2023, 14, 1210085. [Google Scholar] [CrossRef]
- Negri, S.; Scolari, F.; Vismara, M.; Brunetti, V.; Faris, P.; Terribile, G.; Sancini, G.; Berra-Romani, R.; Moccia, F. GABA(A) and GABA(B) Receptors Mediate GABA-Induced Intracellular Ca2+ Signals in Human Brain Microvascular Endothelial Cells. Cells 2022, 11, 3860. [Google Scholar] [CrossRef]
- Sinclair, P.; Kabbani, N. Ionotropic and metabotropic responses by alpha 7 nicotinic acetylcholine receptors. Pharmacol. Res. 2023, 197, 106975. [Google Scholar] [CrossRef] [PubMed]
- Reiner, A.; Levitz, J. Glutamatergic Signaling in the Central Nervous System: Ionotropic and Metabotropic Receptors in Concert. Neuron 2018, 98, 1080–1098. [Google Scholar] [CrossRef] [PubMed]
- Bazzari, A.H.; Parri, H.R. Neuromodulators and Long-Term Synaptic Plasticity in Learning and Memory: A Steered-Glutamatergic Perspective. Brain Sci. 2019, 9, 300. [Google Scholar] [CrossRef] [PubMed]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate receptor ion channels: Structure, regulation, and function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef] [PubMed]
- Wollmuth, L.P. Ion permeation in ionotropic glutamate receptors: Still dynamic after all these years. Curr. Opin. Physiol. 2018, 2, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.B.; Yi, F.; Perszyk, R.E.; Furukawa, H.; Wollmuth, L.P.; Gibb, A.J.; Traynelis, S.F. Structure, function, and allosteric modulation of NMDA receptors. J. Gen. Physiol. 2018, 150, 1081–1105. [Google Scholar] [CrossRef] [PubMed]
- Niswender, C.M.; Conn, P.J. Metabotropic glutamate receptors: Physiology, pharmacology, and disease. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 295–322. [Google Scholar] [CrossRef] [PubMed]
- Yuzaki, M.; Aricescu, A.R. A GluD Coming-Of-Age Story. Trends Neurosci. 2017, 40, 138–150. [Google Scholar] [CrossRef] [PubMed]
- Naur, P.; Hansen, K.B.; Kristensen, A.S.; Dravid, S.M.; Pickering, D.S.; Olsen, L.; Vestergaard, B.; Egebjerg, J.; Gajhede, M.; Traynelis, S.F.; et al. Ionotropic glutamate-like receptor delta2 binds D-serine and glycine. Proc. Natl. Acad. Sci. USA 2007, 104, 14116–14121. [Google Scholar] [CrossRef]
- Burada, A.P.; Vinnakota, R.; Bharti, P.; Dutta, P.; Dubey, N.; Kumar, J. Emerging insights into the structure and function of ionotropic glutamate delta receptors. Br. J. Pharmacol. 2022, 179, 3612–3627. [Google Scholar] [CrossRef]
- Ady, V.; Perroy, J.; Tricoire, L.; Piochon, C.; Dadak, S.; Chen, X.; Dusart, I.; Fagni, L.; Lambolez, B.; Levenes, C. Type 1 metabotropic glutamate receptors (mGlu1) trigger the gating of GluD2 delta glutamate receptors. EMBO Rep. 2014, 15, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Benamer, N.; Marti, F.; Lujan, R.; Hepp, R.; Aubier, T.G.; Dupin, A.A.M.; Frebourg, G.; Pons, S.; Maskos, U.; Faure, P.; et al. GluD1, linked to schizophrenia, controls the burst firing of dopamine neurons. Mol. Psychiatry 2018, 23, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Montes de Oca Balderas, P. Flux-Independent NMDAR Signaling: Molecular Mediators, Cellular Functions, and Complexities. Int. J. Mol. Sci. 2018, 19, 3800. [Google Scholar] [CrossRef] [PubMed]
- Geiger, J.R.; Melcher, T.; Koh, D.S.; Sakmann, B.; Seeburg, P.H.; Jonas, P.; Monyer, H. Relative abundance of subunit mRNAs determines gating and Ca2+ permeability of AMPA receptors in principal neurons and interneurons in rat CNS. Neuron 1995, 15, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Jonas, P.; Racca, C.; Sakmann, B.; Seeburg, P.H.; Monyer, H. Differences in Ca2+ permeability of AMPA-type glutamate receptor channels in neocortical neurons caused by differential GluR-B subunit expression. Neuron 1994, 12, 1281–1289. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Shi, Y.; Jackson, A.C.; Bjorgan, K.; During, M.J.; Sprengel, R.; Seeburg, P.H.; Nicoll, R.A. Subunit composition of synaptic AMPA receptors revealed by a single-cell genetic approach. Neuron 2009, 62, 254–268. [Google Scholar] [CrossRef]
- Wenthold, R.J.; Petralia, R.S.; Blahos, J., II; Niedzielski, A.S. Evidence for multiple AMPA receptor complexes in hippocampal CA1/CA2 neurons. J. Neurosci. 1996, 16, 1982–1989. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Chen, S.; Swensen, A.C.; Qian, W.J.; Gouaux, E. Architecture and subunit arrangement of native AMPA receptors elucidated by cryo-EM. Science 2019, 364, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.S.; Huang, T.H.; Lai, M.C.; Huang, C.W. The Role of Glutamate Receptors in Epilepsy. Biomedicines 2023, 11, 783. [Google Scholar] [CrossRef]
- Wang, Y.; Small, D.L.; Stanimirovic, D.B.; Morley, P.; Durkin, J.P. AMPA receptor-mediated regulation of a Gi-protein in cortical neurons. Nature 1997, 389, 502–504. [Google Scholar] [CrossRef]
- Limatola, C.; Ciotti, M.T.; Mercanti, D.; Santoni, A.; Eusebi, F. Signaling pathways activated by chemokine receptor CXCR2 and AMPA-type glutamate receptors and involvement in granule cells survival. J. Neuroimmunol. 2002, 123, 9–17. [Google Scholar] [CrossRef]
- Marin, P.; Fagni, L.; Torrens, Y.; Alcaraz, G.; Couraud, F.; Bockaert, J.; Glowinski, J.; Premont, J. AMPA receptor activation induces association of G-beta protein with the alpha subunit of the sodium channel in neurons. Eur. J. Neurosci. 2001, 14, 1953–1960. [Google Scholar] [CrossRef]
- Gudz, T.I.; Komuro, H.; Macklin, W.B. Glutamate stimulates oligodendrocyte progenitor migration mediated via an alphav integrin/myelin proteolipid protein complex. J. Neurosci. 2006, 26, 2458–2466. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Brunetti, V.; Soda, T.; Berra-Romani, R.; Scarpellino, G. Cracking the Endothelial Calcium (Ca2+) Code: A Matter of Timing and Spacing. Int. J. Mol. Sci. 2023, 24, 16765. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.R.; Pintchovski, S.A.; Chin, J.; Peebles, C.L.; Mitra, S.; Finkbeiner, S. AMPA receptors regulate transcription of the plasticity-related immediate-early gene Arc. Nat. Neurosci. 2006, 9, 887–895. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, X.; Xiao, B.; Luo, Z.; Long, H. Mechanisms and Functions of Activity-Regulated Cytoskeleton-Associated Protein in Synaptic Plasticity. Mol. Neurobiol. 2023, 60, 5738–5754. [Google Scholar] [CrossRef] [PubMed]
- Mapelli, L.; Soda, T.; D’Angelo, E.; Prestori, F. The Cerebellar Involvement in Autism Spectrum Disorders: From the Social Brain to Mouse Models. Int. J. Mol. Sci. 2022, 23, 3894. [Google Scholar] [CrossRef] [PubMed]
- Satake, S.; Saitow, F.; Rusakov, D.; Konishi, S. AMPA receptor-mediated presynaptic inhibition at cerebellar GABAergic synapses: A characterization of molecular mechanisms. Eur. J. Neurosci. 2004, 19, 2464–2474. [Google Scholar] [CrossRef] [PubMed]
- Takago, H.; Nakamura, Y.; Takahashi, T. G protein-dependent presynaptic inhibition mediated by AMPA receptors at the calyx of Held. Proc. Natl. Acad. Sci. USA 2005, 102, 7368–7373. [Google Scholar] [CrossRef]
- Kawai, F.; Sterling, P. AMPA receptor activates a G-protein that suppresses a cGMP-gated current. J. Neurosci. 1999, 19, 2954–2959. [Google Scholar] [CrossRef]
- Hayashi, T.; Umemori, H.; Mishina, M.; Yamamoto, T. The AMPA receptor interacts with and signals through the protein tyrosine kinase Lyn. Nature 1999, 397, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Schenk, U.; Menna, E.; Kim, T.; Passafaro, M.; Chang, S.; De Camilli, P.; Matteoli, M. A novel pathway for presynaptic mitogen-activated kinase activation via AMPA receptors. J. Neurosci. 2005, 25, 1654–1663. [Google Scholar] [CrossRef]
- Kamalova, A.; Nakagawa, T. AMPA receptor structure and auxiliary subunits. J. Physiol. 2021, 599, 453–469. [Google Scholar] [CrossRef] [PubMed]
- Matthews, P.M.; Pinggera, A.; Kampjut, D.; Greger, I.H. Biology of AMPA receptor interacting proteins—From biogenesis to synaptic plasticity. Neuropharmacology 2021, 197, 108709. [Google Scholar] [CrossRef]
- Shanks, N.F.; Savas, J.N.; Maruo, T.; Cais, O.; Hirao, A.; Oe, S.; Ghosh, A.; Noda, Y.; Greger, I.H.; Yates, J.R., 3rd; et al. Differences in AMPA and kainate receptor interactomes facilitate identification of AMPA receptor auxiliary subunit GSG1L. Cell Rep. 2012, 1, 590–598. [Google Scholar] [CrossRef] [PubMed]
- Perozzo, A.M.; Schwenk, J.; Kamalova, A.; Nakagawa, T.; Fakler, B.; Bowie, D. GSG1L-containing AMPA receptor complexes are defined by their spatiotemporal expression, native interactome and allosteric sites. Nat. Commun. 2023, 14, 6799. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Zuccolo, E.; Soda, T.; Tanzi, F.; Guerra, G.; Mapelli, L.; Lodola, F.; D’Angelo, E. Stim and Orai proteins in neuronal Ca2+ signaling and excitability. Front. Cell. Neurosci. 2015, 9, 153. [Google Scholar] [CrossRef]
- Serwach, K.; Gruszczynska-Biegala, J. Target Molecules of STIM Proteins in the Central Nervous System. Front. Mol. Neurosci. 2020, 13, 617422. [Google Scholar] [CrossRef]
- Astesana, V.; Faris, P.; Ferrari, B.; Siciliani, S.; Lim, D.; Biggiogera, M.; De Pascali, S.A.; Fanizzi, F.P.; Roda, E.; Moccia, F.; et al. [Pt(O,O′-acac)(gamma-acac)(DMS)]: Alternative Strategies to Overcome Cisplatin-Induced Side Effects and Resistance in T98G Glioma Cells. Cell. Mol. Neurobiol. 2021, 41, 563–587. [Google Scholar] [CrossRef] [PubMed]
- Guerra, G.; Lucariello, A.; Perna, A.; Botta, L.; De Luca, A.; Moccia, F. The Role of Endothelial Ca2+ Signaling in Neurovascular Coupling: A View from the Lumen. Int. J. Mol. Sci. 2018, 19, 938. [Google Scholar] [CrossRef]
- Toth, A.B.; Hori, K.; Novakovic, M.M.; Bernstein, N.G.; Lambot, L.; Prakriya, M. CRAC channels regulate astrocyte Ca2+ signaling and gliotransmitter release to modulate hippocampal GABAergic transmission. Sci. Signal. 2019, 12, eaaw5450. [Google Scholar] [CrossRef] [PubMed]
- Zuccolo, E.; Laforenza, U.; Negri, S.; Botta, L.; Berra-Romani, R.; Faris, P.; Scarpellino, G.; Forcaia, G.; Pellavio, G.; Sancini, G.; et al. Muscarinic M5 receptors trigger acetylcholine-induced Ca2+ signals and nitric oxide release in human brain microvascular endothelial cells. J. Cell. Physiol. 2019, 234, 4540–4562. [Google Scholar] [CrossRef] [PubMed]
- Gruszczynska-Biegala, J.; Sladowska, M.; Kuznicki, J. AMPA Receptors Are Involved in Store-Operated Calcium Entry and Interact with STIM Proteins in Rat Primary Cortical Neurons. Front. Cell. Neurosci. 2016, 10, 251. [Google Scholar] [CrossRef] [PubMed]
- Saglietti, L.; Dequidt, C.; Kamieniarz, K.; Rousset, M.C.; Valnegri, P.; Thoumine, O.; Beretta, F.; Fagni, L.; Choquet, D.; Sala, C.; et al. Extracellular interactions between GluR2 and N-cadherin in spine regulation. Neuron 2007, 54, 461–477. [Google Scholar] [CrossRef]
- Passafaro, M.; Nakagawa, T.; Sala, C.; Sheng, M. Induction of dendritic spines by an extracellular domain of AMPA receptor subunit GluR2. Nature 2003, 424, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Scheuss, V.; Bonhoeffer, T. Function of dendritic spines on hippocampal inhibitory neurons. Cereb. Cortex 2014, 24, 3142–3153. [Google Scholar] [CrossRef] [PubMed]
- Keck, T.; Scheuss, V.; Jacobsen, R.I.; Wierenga, C.J.; Eysel, U.T.; Bonhoeffer, T.; Hubener, M. Loss of sensory input causes rapid structural changes of inhibitory neurons in adult mouse visual cortex. Neuron 2011, 71, 869–882. [Google Scholar] [CrossRef] [PubMed]
- Morita, I.; Kakuda, S.; Takeuchi, Y.; Itoh, S.; Kawasaki, N.; Kizuka, Y.; Kawasaki, T.; Oka, S. HNK-1 glyco-epitope regulates the stability of the glutamate receptor subunit GluR2 on the neuronal cell surface. J. Biol. Chem. 2009, 284, 30209–30217. [Google Scholar] [CrossRef]
- Morita, I.; Kakuda, S.; Takeuchi, Y.; Kawasaki, T.; Oka, S. HNK-1 (human natural killer-1) glyco-epitope is essential for normal spine morphogenesis in developing hippocampal neurons. Neuroscience 2009, 164, 1685–1694. [Google Scholar] [CrossRef]
- Morita, I.; Kizuka, Y.; Kakuda, S.; Oka, S. Expression and function of the HNK-1 carbohydrate. J. Biochem. 2008, 143, 719–724. [Google Scholar] [CrossRef]
- Medvedev, N.I.; Rodriguez-Arellano, J.J.; Popov, V.I.; Davies, H.A.; Tigaret, C.M.; Schoepfer, R.; Stewart, M.G. The glutamate receptor 2 subunit controls post-synaptic density complexity and spine shape in the dentate gyrus. Eur. J. Neurosci. 2008, 27, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Hu, J.; Passafaro, M.; Xie, W.; Jia, Z. GluA2 (GluR2) regulates metabotropic glutamate receptor-dependent long-term depression through N-cadherin-dependent and cofilin-mediated actin reorganization. J. Neurosci. 2011, 31, 819–833. [Google Scholar] [CrossRef] [PubMed]
- Senn, C.; Kutsche, M.; Saghatelyan, A.; Bosl, M.R.; Lohler, J.; Bartsch, U.; Morellini, F.; Schachner, M. Mice deficient for the HNK-1 sulfotransferase show alterations in synaptic efficacy and spatial learning and memory. Mol. Cell. Neurosci. 2002, 20, 712–729. [Google Scholar] [CrossRef] [PubMed]
- Hamad, M.I.; Ma-Hogemeier, Z.L.; Riedel, C.; Conrads, C.; Veitinger, T.; Habijan, T.; Schulz, J.N.; Krause, M.; Wirth, M.J.; Hollmann, M.; et al. Cell class-specific regulation of neocortical dendrite and spine growth by AMPA receptor splice and editing variants. Development 2011, 138, 4301–4313. [Google Scholar] [CrossRef] [PubMed]
- Ripley, B.; Otto, S.; Tiglio, K.; Williams, M.E.; Ghosh, A. Regulation of synaptic stability by AMPA receptor reverse signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Tracy, T.E.; Yan, J.J.; Chen, L. Acute knockdown of AMPA receptors reveals a trans-synaptic signal for presynaptic maturation. EMBO J. 2011, 30, 1577–1592. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Hirata, T.; Yonekawa, C.; Takeichi, K.; Fukamizu, A.; Nakagawa, T.; Kizuka, Y. Region-specific upregulation of HNK-1 glycan in the PRMT1-deficient brain. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2020, 1864, 129509. [Google Scholar] [CrossRef]
- Ezra-Nevo, G.; Prestori, F.; Locatelli, F.; Soda, T.; Ten Brinke, M.M.; Engel, M.; Boele, H.J.; Botta, L.; Leshkowitz, D.; Ramot, A.; et al. Cerebellar Learning Properties Are Modulated by the CRF Receptor. J. Neurosci. 2018, 38, 6751–6765. [Google Scholar] [CrossRef] [PubMed]
- Soda, T.; Mapelli, L.; Locatelli, F.; Botta, L.; Goldfarb, M.; Prestori, F.; D’Angelo, E. Hyperexcitability and Hyperplasticity Disrupt Cerebellar Signal Transfer in the IB2 KO Mouse Model of Autism. J. Neurosci. 2019, 39, 2383–2397. [Google Scholar] [CrossRef]
- Tapella, L.; Soda, T.; Mapelli, L.; Bortolotto, V.; Bondi, H.; Ruffinatti, F.A.; Dematteis, G.; Stevano, A.; Dionisi, M.; Ummarino, S.; et al. Deletion of calcineurin from GFAP-expressing astrocytes impairs excitability of cerebellar and hippocampal neurons through astroglial Na+/K+ ATPase. Glia 2020, 68, 543–560. [Google Scholar] [CrossRef]
- Locatelli, F.; Soda, T.; Montagna, I.; Tritto, S.; Botta, L.; Prestori, F.; D’Angelo, E. Calcium Channel-Dependent Induction of Long-Term Synaptic Plasticity at Excitatory Golgi Cell Synapses of Cerebellum. J. Neurosci. 2021, 41, 3307–3319. [Google Scholar] [CrossRef] [PubMed]
- Masoli, S.; Tognolina, M.; Laforenza, U.; Moccia, F.; D’Angelo, E. Parameter tuning differentiates granule cell subtypes enriching transmission properties at the cerebellum input stage. Commun. Biol. 2020, 3, 222. [Google Scholar] [CrossRef]
- Pressey, J.C.; Woodin, M.A. Kainate receptor regulation of synaptic inhibition in the hippocampus. J. Physiol. 2021, 599, 485–492. [Google Scholar] [CrossRef]
- Falcon-Moya, R.; Rodriguez-Moreno, A. Metabotropic actions of kainate receptors modulating glutamate release. Neuropharmacology 2021, 197, 108696. [Google Scholar] [CrossRef] [PubMed]
- Selvakumar, P.; Lee, J.; Khanra, N.; He, C.; Munguba, H.; Kiese, L.; Broichhagen, J.; Reiner, A.; Levitz, J.; Meyerson, J.R. Structural and compositional diversity in the kainate receptor family. Cell Rep. 2021, 37, 109891. [Google Scholar] [CrossRef] [PubMed]
- Lerma, J.; Marques, J.M. Kainate receptors in health and disease. Neuron 2013, 80, 292–311. [Google Scholar] [CrossRef] [PubMed]
- Swanson, G.T.; Feldmeyer, D.; Kaneda, M.; Cull-Candy, S.G. Effect of RNA editing and subunit co-assembly single-channel properties of recombinant kainate receptors. J. Physiol. 1996, 492 Pt 1, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Gurung, S.; Evans, A.J.; Wilkinson, K.A.; Henley, J.M. ADAR2-mediated Q/R editing of GluK2 regulates kainate receptor upscaling in response to suppression of synaptic activity. J. Cell Sci. 2018, 131, jcs222273. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.W.; Tang, Z.Q. Focusing on the Emerging Role of Kainate Receptors in the Dorsal Cochlear Nucleus (DCN) and Cerebellum. Int. J. Mol. Sci. 2023, 24, 1718. [Google Scholar] [CrossRef]
- Hadzic, M.; Jack, A.; Wahle, P. Ionotropic glutamate receptors: Which ones, when, and where in the mammalian neocortex. J. Comp. Neurol. 2017, 525, 976–1033. [Google Scholar] [CrossRef]
- Castillo, P.E.; Malenka, R.C.; Nicoll, R.A. Kainate receptors mediate a slow postsynaptic current in hippocampal CA3 neurons. Nature 1997, 388, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Moreno, A.; Lerma, J. Kainate receptor modulation of GABA release involves a metabotropic function. Neuron 1998, 20, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Cunha, R.A.; Malva, J.O.; Ribeiro, J.A. Pertussis toxin prevents presynaptic inhibition by kainate receptors of rat hippocampal [(3)H]GABA release. FEBS Lett. 2000, 469, 159–162. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Moreno, A.; Lopez-Garcia, J.C.; Lerma, J. Two populations of kainate receptors with separate signaling mechanisms in hippocampal interneurons. Proc. Natl. Acad. Sci. USA 2000, 97, 1293–1298. [Google Scholar] [CrossRef] [PubMed]
- Cunha, R.A.; Malva, J.O.; Ribeiro, J.A. Kainate receptors coupled to G(i)/G(o) proteins in the rat hippocampus. Mol. Pharmacol. 1999, 56, 429–433. [Google Scholar] [CrossRef] [PubMed]
- Igumenova, T.I. Dynamics and Membrane Interactions of Protein Kinase C. Biochemistry 2015, 54, 4953–4968. [Google Scholar] [CrossRef] [PubMed]
- Negri, S.; Faris, P.; Soda, T.; Moccia, F. Endothelial signaling at the core of neurovascular coupling: The emerging role of endothelial inward-rectifier K+ (Kir2.1) channels and N-methyl-d-aspartate receptors in the regulation of cerebral blood flow. Int. J. Biochem. Cell Biol. 2021, 135, 105983. [Google Scholar] [CrossRef] [PubMed]
- Sandal, M.; Paltrinieri, D.; Carloni, P.; Musiani, F.; Giorgetti, A. Structure/function relationships of phospholipases C Beta. Curr. Protein Pept. Sci. 2013, 14, 650–657. [Google Scholar] [CrossRef]
- Zuccolo, E.; Di Buduo, C.; Lodola, F.; Orecchioni, S.; Scarpellino, G.; Kheder, D.A.; Poletto, V.; Guerra, G.; Bertolini, F.; Balduini, A.; et al. Stromal Cell-Derived Factor-1alpha Promotes Endothelial Colony-Forming Cell Migration Through the Ca2+-Dependent Activation of the Extracellular Signal-Regulated Kinase 1/2 and Phosphoinositide 3-Kinase/AKT Pathways. Stem Cells Dev. 2018, 27, 23–34. [Google Scholar] [CrossRef]
- Moccia, F.; Bonetti, E.; Dragoni, S.; Fontana, J.; Lodola, F.; Romani, R.B.; Laforenza, U.; Rosti, V.; Tanzi, F. Hematopoietic progenitor and stem cells circulate by surfing on intracellular Ca2+ waves: A novel target for cell-based therapy and anti-cancer treatment? Curr. Signal Transduct. Ther. 2012, 7, 161–176. [Google Scholar] [CrossRef]
- Zuccolo, E.; Kheder, D.A.; Lim, D.; Perna, A.; Nezza, F.D.; Botta, L.; Scarpellino, G.; Negri, S.; Martinotti, S.; Soda, T.; et al. Glutamate triggers intracellular Ca2+ oscillations and nitric oxide release by inducing NAADP- and InsP3 -dependent Ca2+ release in mouse brain endothelial cells. J. Cell. Physiol. 2019, 234, 3538–3554. [Google Scholar] [CrossRef]
- Rozas, J.L.; Paternain, A.V.; Lerma, J. Noncanonical signaling by ionotropic kainate receptors. Neuron 2003, 39, 543–553. [Google Scholar] [CrossRef]
- Melyan, Z.; Lancaster, B.; Wheal, H.V. Metabotropic regulation of intrinsic excitability by synaptic activation of kainate receptors. J. Neurosci. 2004, 24, 4530–4534. [Google Scholar] [CrossRef] [PubMed]
- Daw, M.I.; Pelkey, K.A.; Chittajallu, R.; McBain, C.J. Presynaptic kainate receptor activation preserves asynchronous GABA release despite the reduction in synchronous release from hippocampal cholecystokinin interneurons. J. Neurosci. 2010, 30, 11202–11209. [Google Scholar] [CrossRef] [PubMed]
- Cossart, R.; Tyzio, R.; Dinocourt, C.; Esclapez, M.; Hirsch, J.C.; Ben-Ari, Y.; Bernard, C. Presynaptic kainate receptors that enhance the release of GABA on CA1 hippocampal interneurons. Neuron 2001, 29, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Frerking, M.; Malenka, R.C.; Nicoll, R.A. Synaptic activation of kainate receptors on hippocampal interneurons. Nat. Neurosci. 1998, 1, 479–486. [Google Scholar] [CrossRef]
- Bonfardin, V.D.; Fossat, P.; Theodosis, D.T.; Oliet, S.H. Glia-dependent switch of kainate receptor presynaptic action. J. Neurosci. 2010, 30, 985–995. [Google Scholar] [CrossRef]
- Schmitz, D.; Mellor, J.; Frerking, M.; Nicoll, R.A. Presynaptic kainate receptors at hippocampal mossy fiber synapses. Proc. Natl. Acad. Sci. USA 2001, 98, 11003–11008. [Google Scholar] [CrossRef]
- Jiang, L.; Xu, J.; Nedergaard, M.; Kang, J. A kainate receptor increases the efficacy of GABAergic synapses. Neuron 2001, 30, 503–513. [Google Scholar] [CrossRef]
- Lauri, S.E.; Segerstrale, M.; Vesikansa, A.; Maingret, F.; Mulle, C.; Collingridge, G.L.; Isaac, J.T.; Taira, T. Endogenous activation of kainate receptors regulates glutamate release and network activity in the developing hippocampus. J. Neurosci. 2005, 25, 4473–4484. [Google Scholar] [CrossRef]
- Lauri, S.E.; Vesikansa, A.; Segerstrale, M.; Collingridge, G.L.; Isaac, J.T.; Taira, T. Functional maturation of CA1 synapses involves activity-dependent loss of tonic kainate receptor-mediated inhibition of glutamate release. Neuron 2006, 50, 415–429. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, H.; Ozawa, S. Kainate receptor-mediated inhibition of presynaptic Ca2+ influx and EPSP in area CA1 of the rat hippocampus. J. Physiol. 1998, 509 Pt 3, 833–845. [Google Scholar] [CrossRef]
- Sallert, M.; Malkki, H.; Segerstrale, M.; Taira, T.; Lauri, S.E. Effects of the kainate receptor agonist ATPA on glutamatergic synaptic transmission and plasticity during early postnatal development. Neuropharmacology 2007, 52, 1354–1365. [Google Scholar] [CrossRef] [PubMed]
- Vesikansa, A.; Sallert, M.; Taira, T.; Lauri, S.E. Activation of kainate receptors controls the number of functional glutamatergic synapses in the area CA1 of rat hippocampus. J. Physiol. 2007, 583, 145–157. [Google Scholar] [CrossRef]
- Frerking, M.; Schmitz, D.; Zhou, Q.; Johansen, J.; Nicoll, R.A. Kainate receptors depress excitatory synaptic transmission at CA3-->CA1 synapses in the hippocampus via a direct presynaptic action. J. Neurosci. 2001, 21, 2958–2966. [Google Scholar] [CrossRef]
- Partovi, D.; Frerking, M. Presynaptic inhibition by kainate receptors converges mechanistically with presynaptic inhibition by adenosine and GABAB receptors. Neuropharmacology 2006, 51, 1030–1037. [Google Scholar] [CrossRef]
- Andrade-Talavera, Y.; Duque-Feria, P.; Negrete-Diaz, J.V.; Sihra, T.S.; Flores, G.; Rodriguez-Moreno, A. Presynaptic kainate receptor-mediated facilitation of glutamate release involves Ca2+ -calmodulin at mossy fiber-CA3 synapses. J. Neurochem. 2012, 122, 891–899. [Google Scholar] [CrossRef]
- Breustedt, J.; Schmitz, D. Assessing the role of GLUK5 and GLUK6 at hippocampal mossy fiber synapses. J. Neurosci. 2004, 24, 10093–10098. [Google Scholar] [CrossRef] [PubMed]
- Contractor, A.; Sailer, A.W.; Darstein, M.; Maron, C.; Xu, J.; Swanson, G.T.; Heinemann, S.F. Loss of kainate receptor-mediated heterosynaptic facilitation of mossy-fiber synapses in KA2−/− mice. J. Neurosci. 2003, 23, 422–429. [Google Scholar] [CrossRef]
- Pinheiro, P.S.; Perrais, D.; Coussen, F.; Barhanin, J.; Bettler, B.; Mann, J.R.; Malva, J.O.; Heinemann, S.F.; Mulle, C. GluR7 is an essential subunit of presynaptic kainate autoreceptors at hippocampal mossy fiber synapses. Proc. Natl. Acad. Sci. USA 2007, 104, 12181–12186. [Google Scholar] [CrossRef]
- Negrete-Diaz, J.V.; Sihra, T.S.; Delgado-Garcia, J.M.; Rodriguez-Moreno, A. Kainate receptor-mediated inhibition of glutamate release involves protein kinase A in the mouse hippocampus. J. Neurophysiol. 2006, 96, 1829–1837. [Google Scholar] [CrossRef] [PubMed]
- Negrete-Diaz, J.V.; Sihra, T.S.; Delgado-Garcia, J.M.; Rodriguez-Moreno, A. Kainate receptor-mediated presynaptic inhibition converges with presynaptic inhibition mediated by Group II mGluRs and long-term depression at the hippocampal mossy fiber-CA3 synapse. J. Neural Transm. 2007, 114, 1425–1431. [Google Scholar] [CrossRef] [PubMed]
- Contractor, A.; Swanson, G.T.; Sailer, A.; O’Gorman, S.; Heinemann, S.F. Identification of the kainate receptor subunits underlying modulation of excitatory synaptic transmission in the CA3 region of the hippocampus. J. Neurosci. 2000, 20, 8269–8278. [Google Scholar] [CrossRef] [PubMed]
- Valbuena, S.; Lerma, J. Kainate Receptors, Homeostatic Gatekeepers of Synaptic Plasticity. Neuroscience 2021, 456, 17–26. [Google Scholar] [CrossRef]
- Nair, J.D.; Wilkinson, K.A.; Yucel, B.P.; Mulle, C.; Vissel, B.; Mellor, J.; Henley, J.M. GluK2 Q/R editing regulates kainate receptor signaling and long-term potentiation of AMPA receptors. iScience 2023, 26, 107708. [Google Scholar] [CrossRef]
- Leenders, A.G.; Sheng, Z.H. Modulation of neurotransmitter release by the second messenger-activated protein kinases: Implications for presynaptic plasticity. Pharmacol. Ther. 2005, 105, 69–84. [Google Scholar] [CrossRef]
- Rutkowska-Wlodarczyk, I.; Aller, M.I.; Valbuena, S.; Bologna, J.C.; Prezeau, L.; Lerma, J. A proteomic analysis reveals the interaction of GluK1 ionotropic kainate receptor subunits with Go proteins. J. Neurosci. 2015, 35, 5171–5179. [Google Scholar] [CrossRef]
- Sherwood, J.L.; Amici, M.; Dargan, S.L.; Culley, G.R.; Fitzjohn, S.M.; Jane, D.E.; Collingridge, G.L.; Lodge, D.; Bortolotto, Z.A. Differences in kainate receptor involvement in hippocampal mossy fibre long-term potentiation depending on slice orientation. Neurochem. Int. 2012, 61, 482–489. [Google Scholar] [CrossRef]
- Delaney, A.J.; Jahr, C.E. Kainate receptors differentially regulate release at two parallel fiber synapses. Neuron 2002, 36, 475–482. [Google Scholar] [CrossRef]
- Falcon-Moya, R.; Losada-Ruiz, P.; Sihra, T.S.; Rodriguez-Moreno, A. Cerebellar Kainate Receptor-Mediated Facilitation of Glutamate Release Requires Ca2+-Calmodulin and PKA. Front. Mol. Neurosci. 2018, 11, 195. [Google Scholar] [CrossRef]
- Falcon-Moya, R.; Losada-Ruiz, P.; Rodriguez-Moreno, A. Kainate Receptor-Mediated Depression of Glutamate Release Involves Protein Kinase A in the Cerebellum. Int. J. Mol. Sci. 2019, 20, 4124. [Google Scholar] [CrossRef] [PubMed]
- Crepel, F. Role of presynaptic kainate receptors at parallel fiber-purkinje cell synapses in induction of cerebellar LTD: Interplay with climbing fiber input. J. Neurophysiol. 2009, 102, 965–973. [Google Scholar] [CrossRef]
- Salmen, B.; Beed, P.S.; Ozdogan, T.; Maier, N.; Johenning, F.W.; Winterer, J.; Breustedt, J.; Schmitz, D. GluK1 inhibits calcium dependent and independent transmitter release at associational/commissural synapses in area CA3 of the hippocampus. Hippocampus 2012, 22, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Negrete-Diaz, J.V.; Duque-Feria, P.; Andrade-Talavera, Y.; Carrion, M.; Flores, G.; Rodriguez-Moreno, A. Kainate receptor-mediated depression of glutamatergic transmission involving protein kinase A in the lateral amygdala. J. Neurochem. 2012, 121, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.T.; Pare, J.F.; Raju, D.V.; Smith, Y. Localization and function of pre- and postsynaptic kainate receptors in the rat globus pallidus. Eur. J. Neurosci. 2006, 23, 374–386. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, V.; Pressey, J.C.; Acton, B.A.; Uvarov, P.; Huang, M.Y.; Chevrier, J.; Puchalski, A.; Li, C.M.; Ivakine, E.A.; Airaksinen, M.S.; et al. Kainate receptors coexist in a functional complex with KCC2 and regulate chloride homeostasis in hippocampal neurons. Cell Rep. 2014, 7, 1762–1770. [Google Scholar] [CrossRef] [PubMed]
- Garand, D.; Mahadevan, V.; Woodin, M.A. Ionotropic and metabotropic kainate receptor signalling regulates Cl− homeostasis and GABAergic inhibition. J. Physiol. 2019, 597, 1677–1690. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, V.; Dargaei, Z.; Ivakine, E.A.; Hartmann, A.M.; Ng, D.; Chevrier, J.; Ormond, J.; Nothwang, H.G.; McInnes, R.R.; Woodin, M.A. Neto2-null mice have impaired GABAergic inhibition and are susceptible to seizures. Front. Cell. Neurosci. 2015, 9, 368. [Google Scholar] [CrossRef] [PubMed]
- Pressey, J.C.; Mahadevan, V.; Khademullah, C.S.; Dargaei, Z.; Chevrier, J.; Ye, W.; Huang, M.; Chauhan, A.K.; Meas, S.J.; Uvarov, P.; et al. A kainate receptor subunit promotes the recycling of the neuron-specific K(+)-Cl− co-transporter KCC2 in hippocampal neurons. J. Biol. Chem. 2017, 292, 6190–6201. [Google Scholar] [CrossRef]
- Jiang, L.; Kang, D.; Kang, J. Potentiation of tonic GABAergic inhibition by activation of postsynaptic kainate receptors. Neuroscience 2015, 298, 448–454. [Google Scholar] [CrossRef]
- Sahu, G.; Turner, R.W. The Molecular Basis for the Calcium-Dependent Slow Afterhyperpolarization in CA1 Hippocampal Pyramidal Neurons. Front. Physiol. 2021, 12, 759707. [Google Scholar] [CrossRef] [PubMed]
- Sah, P. Ca2+-activated K+ currents in neurones: Types, physiological roles and modulation. Trends Neurosci. 1996, 19, 150–154. [Google Scholar] [CrossRef] [PubMed]
- Melyan, Z.; Wheal, H.V.; Lancaster, B. Metabotropic-mediated kainate receptor regulation of IsAHP and excitability in pyramidal cells. Neuron 2002, 34, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Fisahn, A.; Contractor, A.; Traub, R.D.; Buhl, E.H.; Heinemann, S.F.; McBain, C.J. Distinct roles for the kainate receptor subunits GluR5 and GluR6 in kainate-induced hippocampal gamma oscillations. J. Neurosci. 2004, 24, 9658–9668. [Google Scholar] [CrossRef]
- Fisahn, A.; Heinemann, S.F.; McBain, C.J. The kainate receptor subunit GluR6 mediates metabotropic regulation of the slow and medium AHP currents in mouse hippocampal neurones. J. Physiol. 2005, 562, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, A.; Sachidhanandam, S.; Utvik, J.K.; Coussen, F.; Mulle, C. Distinct subunits in heteromeric kainate receptors mediate ionotropic and metabotropic function at hippocampal mossy fiber synapses. J. Neurosci. 2005, 25, 11710–11718. [Google Scholar] [CrossRef] [PubMed]
- Segerstrale, M.; Juuri, J.; Lanore, F.; Piepponen, P.; Lauri, S.E.; Mulle, C.; Taira, T. High firing rate of neonatal hippocampal interneurons is caused by attenuation of afterhyperpolarizing potassium currents by tonically active kainate receptors. J. Neurosci. 2010, 30, 6507–6514. [Google Scholar] [CrossRef] [PubMed]
- Orav, E.; Dowavic, I.; Huupponen, J.; Taira, T.; Lauri, S.E. NETO1 Regulates Postsynaptic Kainate Receptors in CA3 Interneurons During Circuit Maturation. Mol. Neurobiol. 2019, 56, 7473–7489. [Google Scholar] [CrossRef] [PubMed]
- Carta, M.; Opazo, P.; Veran, J.; Athane, A.; Choquet, D.; Coussen, F.; Mulle, C. CaMKII-dependent phosphorylation of GluK5 mediates plasticity of kainate receptors. EMBO J. 2013, 32, 496–510. [Google Scholar] [CrossRef]
- Chamberlain, S.E.; Sadowski, J.H.; Teles-Grilo Ruivo, L.M.; Atherton, L.A.; Mellor, J.R. Long-term depression of synaptic kainate receptors reduces excitability by relieving inhibition of the slow afterhyperpolarization. J. Neurosci. 2013, 33, 9536–9545. [Google Scholar] [CrossRef]
- Bureau, I.; Bischoff, S.; Heinemann, S.F.; Mulle, C. Kainate receptor-mediated responses in the CA1 field of wild-type and GluR6-deficient mice. J. Neurosci. 1999, 19, 653–663. [Google Scholar] [CrossRef] [PubMed]
- Petrovic, M.M.; Viana da Silva, S.; Clement, J.P.; Vyklicky, L.; Mulle, C.; Gonzalez-Gonzalez, I.M.; Henley, J.M. Metabotropic action of postsynaptic kainate receptors triggers hippocampal long-term potentiation. Nat. Neurosci. 2017, 20, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Chandra, N.; Awasthi, R.; Ozdogan, T.; Johenning, F.W.; Imbrosci, B.; Morris, G.; Schmitz, D.; Barkai, E. A Cellular Mechanism Underlying Enhanced Capability for Complex Olfactory Discrimination Learning. eNeuro 2019, 6, ENEURO.0198-18.2019. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, R.; Yuan, Q.; Barkai, E. Reversing Aging: Decline in Complex Olfactory Learning Can be Rectified by Restoring Intrinsic Plasticity of Hippocampal CA1 Pyramidal Neurons. Adv. Biol. 2023, 8, e2300323. [Google Scholar] [CrossRef] [PubMed]
- Tashiro, A.; Dunaevsky, A.; Blazeski, R.; Mason, C.A.; Yuste, R. Bidirectional regulation of hippocampal mossy fiber filopodial motility by kainate receptors: A two-step model of synaptogenesis. Neuron 2003, 38, 773–784. [Google Scholar] [CrossRef] [PubMed]
- Marques, J.M.; Rodrigues, R.J.; Valbuena, S.; Rozas, J.L.; Selak, S.; Marin, P.; Aller, M.I.; Lerma, J. CRMP2 tethers kainate receptor activity to cytoskeleton dynamics during neuronal maturation. J. Neurosci. 2013, 33, 18298–18310. [Google Scholar] [CrossRef]
- Dhingra, S.; Yadav, J.; Kumar, J. Structure, Function, and Regulation of the Kainate Receptor. Subcell. Biochem. 2022, 99, 317–350. [Google Scholar] [CrossRef] [PubMed]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA receptor subunit diversity: Impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 2013, 14, 383–400. [Google Scholar] [CrossRef] [PubMed]
- Papouin, T.; Ladepeche, L.; Ruel, J.; Sacchi, S.; Labasque, M.; Hanini, M.; Groc, L.; Pollegioni, L.; Mothet, J.P.; Oliet, S.H. Synaptic and extrasynaptic NMDA receptors are gated by different endogenous coagonists. Cell 2012, 150, 633–646. [Google Scholar] [CrossRef]
- Rosenberg, D.; Artoul, S.; Segal, A.C.; Kolodney, G.; Radzishevsky, I.; Dikopoltsev, E.; Foltyn, V.N.; Inoue, R.; Mori, H.; Billard, J.M.; et al. Neuronal D-serine and glycine release via the Asc-1 transporter regulates NMDA receptor-dependent synaptic activity. J. Neurosci. 2013, 33, 3533–3544. [Google Scholar] [CrossRef]
- Ferreira, J.S.; Papouin, T.; Ladepeche, L.; Yao, A.; Langlais, V.C.; Bouchet, D.; Dulong, J.; Mothet, J.P.; Sacchi, S.; Pollegioni, L.; et al. Co-agonists differentially tune GluN2B-NMDA receptor trafficking at hippocampal synapses. Elife 2017, 6, e25492. [Google Scholar] [CrossRef] [PubMed]
- Perez-Otano, I.; Larsen, R.S.; Wesseling, J.F. Emerging roles of GluN3-containing NMDA receptors in the CNS. Nat. Rev. Neurosci. 2016, 17, 623–635. [Google Scholar] [CrossRef]
- Stroebel, D.; Mony, L.; Paoletti, P. Glycine agonism in ionotropic glutamate receptors. Neuropharmacology 2021, 193, 108631. [Google Scholar] [CrossRef] [PubMed]
- Pachernegg, S.; Strutz-Seebohm, N.; Hollmann, M. GluN3 subunit-containing NMDA receptors: Not just one-trick ponies. Trends Neurosci. 2012, 35, 240–249. [Google Scholar] [CrossRef]
- Ma, H.; Khaled, H.G.; Wang, X.; Mandelberg, N.J.; Cohen, S.M.; He, X.; Tsien, R.W. Excitation-transcription coupling, neuronal gene expression and synaptic plasticity. Nat. Rev. Neurosci. 2023, 24, 672–692. [Google Scholar] [CrossRef]
- Montes de Oca Balderas, P.; Aguilera, P. A Metabotropic-Like Flux-Independent NMDA Receptor Regulates Ca2+ Exit from Endoplasmic Reticulum and Mitochondrial Membrane Potential in Cultured Astrocytes. PLoS ONE 2015, 10, e0126314. [Google Scholar] [CrossRef]
- Montes de Oca Balderas, P.; Matus Nunez, M.; Picones, A.; Hernandez-Cruz, A. NMDAR in cultured astrocytes: Flux-independent pH sensor and flux-dependent regulator of mitochondria and plasma membrane-mitochondria bridging. FASEB J. 2020, 34, 16622–16644. [Google Scholar] [CrossRef]
- Negri, S.; Faris, P.; Maniezzi, C.; Pellavio, G.; Spaiardi, P.; Botta, L.; Laforenza, U.; Biella, G.; Moccia, D.F. NMDA receptors elicit flux-independent intracellular Ca2+ signals via metabotropic glutamate receptors and flux-dependent nitric oxide release in human brain microvascular endothelial cells. Cell Calcium 2021, 99, 102454. [Google Scholar] [CrossRef] [PubMed]
- Mehra, A.; Guerit, S.; Macrez, R.; Gosselet, F.; Sevin, E.; Lebas, H.; Maubert, E.; De Vries, H.E.; Bardou, I.; Vivien, D.; et al. Non-ionotropic action of endothelial NMDA receptors on blood-brain barrier permeability via Rho/ROCK mediated phosphorylation of myosin. J. Neurosci. 2020, 40, 1778–1787. [Google Scholar] [CrossRef]
- Vissel, B.; Krupp, J.J.; Heinemann, S.F.; Westbrook, G.L. A use-dependent tyrosine dephosphorylation of NMDA receptors is independent of ion flux. Nat. Neurosci. 2001, 4, 587–596. [Google Scholar] [CrossRef]
- Wang, Y.T.; Yu, X.M.; Salter, M.W. Ca2+-independent reduction of N-methyl-D-aspartate channel activity by protein tyrosine phosphatase. Proc. Natl. Acad. Sci. USA 1996, 93, 1721–1725. [Google Scholar] [CrossRef] [PubMed]
- Nong, Y.; Huang, Y.Q.; Ju, W.; Kalia, L.V.; Ahmadian, G.; Wang, Y.T.; Salter, M.W. Glycine binding primes NMDA receptor internalization. Nature 2003, 422, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Campanucci, V.A.; Cooke, J.; Salter, M.W. Identification of a single amino acid in GluN1 that is critical for glycine-primed internalization of NMDA receptors. Mol. Brain 2013, 6, 36. [Google Scholar] [CrossRef]
- Li, H.; Rajani, V.; Han, L.; Chung, D.; Cooke, J.E.; Sengar, A.S.; Salter, M.W. Alternative splicing of GluN1 gates glycine site-dependent nonionotropic signaling by NMDAR receptors. Proc. Natl. Acad. Sci. USA 2021, 118, e2026411118. [Google Scholar] [CrossRef] [PubMed]
- Barria, A.; Malinow, R. Subunit-specific NMDA receptor trafficking to synapses. Neuron 2002, 35, 345–353. [Google Scholar] [CrossRef]
- Yashiro, K.; Philpot, B.D. Regulation of NMDA receptor subunit expression and its implications for LTD, LTP, and metaplasticity. Neuropharmacology 2008, 55, 1081–1094. [Google Scholar] [CrossRef]
- Mayford, M.; Wang, J.; Kandel, E.R.; O’Dell, T.J. CaMKII regulates the frequency-response function of hippocampal synapses for the production of both LTD and LTP. Cell 1995, 81, 891–904. [Google Scholar] [CrossRef]
- Nabavi, S.; Kessels, H.W.; Alfonso, S.; Aow, J.; Fox, R.; Malinow, R. Metabotropic NMDA receptor function is required for NMDA receptor-dependent long-term depression. Proc. Natl. Acad. Sci. USA 2013, 110, 4027–4032. [Google Scholar] [CrossRef]
- Stein, I.S.; Gray, J.A.; Zito, K. Non-Ionotropic NMDA Receptor Signaling Drives Activity-Induced Dendritic Spine Shrinkage. J. Neurosci. 2015, 35, 12303–12308. [Google Scholar] [CrossRef]
- Dore, K.; Aow, J.; Malinow, R. Agonist binding to the NMDA receptor drives movement of its cytoplasmic domain without ion flow. Proc. Natl. Acad. Sci. USA 2015, 112, 14705–14710. [Google Scholar] [CrossRef]
- Wong, J.M.; Gray, J.A. Long-Term Depression Is Independent of GluN2 Subunit Composition. J. Neurosci. 2018, 38, 4462–4470. [Google Scholar] [CrossRef] [PubMed]
- Stein, I.S.; Park, D.K.; Flores, J.C.; Jahncke, J.N.; Zito, K. Molecular Mechanisms of Non-ionotropic NMDA Receptor Signaling in Dendritic Spine Shrinkage. J. Neurosci. 2020, 40, 3741–3750. [Google Scholar] [CrossRef] [PubMed]
- Li, L.L.; Ginet, V.; Liu, X.; Vergun, O.; Tuittila, M.; Mathieu, M.; Bonny, C.; Puyal, J.; Truttmann, A.C.; Courtney, M.J. The nNOS-p38MAPK pathway is mediated by NOS1AP during neuronal death. J. Neurosci. 2013, 33, 8185–8201. [Google Scholar] [CrossRef] [PubMed]
- Eales, K.L.; Palygin, O.; O’Loughlin, T.; Rasooli-Nejad, S.; Gaestel, M.; Muller, J.; Collins, D.R.; Pankratov, Y.; Correa, S.A. The MK2/3 cascade regulates AMPAR trafficking and cognitive flexibility. Nat. Commun. 2014, 5, 4701. [Google Scholar] [CrossRef] [PubMed]
- Bayer, K.U.; Schulman, H. CaM Kinase: Still Inspiring at 40. Neuron 2019, 103, 380–394. [Google Scholar] [CrossRef]
- Aow, J.; Dore, K.; Malinow, R. Conformational signaling required for synaptic plasticity by the NMDA receptor complex. Proc. Natl. Acad. Sci. USA 2015, 112, 14711–14716. [Google Scholar] [CrossRef] [PubMed]
- Coultrap, S.J.; Freund, R.K.; O’Leary, H.; Sanderson, J.L.; Roche, K.W.; Dell’Acqua, M.L.; Bayer, K.U. Autonomous CaMKII mediates both LTP and LTD using a mechanism for differential substrate site selection. Cell Rep. 2014, 6, 431–437. [Google Scholar] [CrossRef]
- Dore, K.; Aow, J.; Malinow, R. The Emergence of NMDA Receptor Metabotropic Function: Insights from Imaging. Front. Synaptic Neurosci. 2016, 8, 20. [Google Scholar] [CrossRef]
- Mapelli, L.; Gagliano, G.; Soda, T.; Laforenza, U.; Moccia, F.; D’Angelo, E.U. Granular Layer Neurons Control Cerebellar Neurovascular Coupling Through an NMDA Receptor/NO-Dependent System. J. Neurosci. 2017, 37, 1340–1351. [Google Scholar] [CrossRef]
- Sanderson, J.L.; Gorski, J.A.; Dell’Acqua, M.L. NMDA Receptor-Dependent LTD Requires Transient Synaptic Incorporation of Ca2+-Permeable AMPARs Mediated by AKAP150-Anchored PKA and Calcineurin. Neuron 2016, 89, 1000–1015. [Google Scholar] [CrossRef]
- Dore, K.; Malinow, R. Elevated PSD-95 Blocks Ion-flux Independent LTD: A Potential New Role for PSD-95 in Synaptic Plasticity. Neuroscience 2021, 456, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Stein, V.; House, D.R.; Bredt, D.S.; Nicoll, R.A. Postsynaptic density-95 mimics and occludes hippocampal long-term potentiation and enhances long-term depression. J. Neurosci. 2003, 23, 5503–5506. [Google Scholar] [CrossRef] [PubMed]
- Shiotani, H.; Maruo, T.; Sakakibara, S.; Miyata, M.; Mandai, K.; Mochizuki, H.; Takai, Y. Aging-dependent expression of synapse-related proteins in the mouse brain. Genes Cells 2017, 22, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Li, L.J.; Hu, R.; Lujan, B.; Chen, J.; Zhang, J.J.; Nakano, Y.; Cui, T.Y.; Liao, M.X.; Chen, J.C.; Man, H.Y.; et al. Glycine Potentiates AMPA Receptor Function through Metabotropic Activation of GluN2A-Containing NMDA Receptors. Front. Mol. Neurosci. 2016, 9, 102. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Mao, L.; Tang, Q.; Samdani, S.; Liu, Z.; Wang, J.Q. A novel Ca2+-independent signaling pathway to extracellular signal-regulated protein kinase by coactivation of NMDA receptors and metabotropic glutamate receptor 5 in neurons. J. Neurosci. 2004, 24, 10846–10857. [Google Scholar] [CrossRef] [PubMed]
- Stein, I.S.; Zito, K. Dendritic Spine Elimination: Molecular Mechanisms and Implications. Neuroscientist 2019, 25, 27–47. [Google Scholar] [CrossRef]
- Thomazeau, A.; Bosch, M.; Essayan-Perez, S.; Barnes, S.A.; De Jesus-Cortes, H.; Bear, M.F. Dissociation of functional and structural plasticity of dendritic spines during NMDAR and mGluR-dependent long-term synaptic depression in wild-type and fragile X model mice. Mol. Psychiatry 2021, 26, 4652–4669. [Google Scholar] [CrossRef] [PubMed]
- Goo, M.S.; Sancho, L.; Slepak, N.; Boassa, D.; Deerinck, T.J.; Ellisman, M.H.; Bloodgood, B.L.; Patrick, G.N. Activity-dependent trafficking of lysosomes in dendrites and dendritic spines. J. Cell Biol. 2017, 216, 2499–2513. [Google Scholar] [CrossRef] [PubMed]
- Foster, W.J.; Taylor, H.B.C.; Padamsey, Z.; Jeans, A.F.; Galione, A.; Emptage, N.J. Hippocampal mGluR1-dependent long-term potentiation requires NAADP-mediated acidic store Ca2+ signaling. Sci. Signal. 2018, 11, eaat9093. [Google Scholar] [CrossRef]
- Cang, C.; Zhou, Y.; Navarro, B.; Seo, Y.J.; Aranda, K.; Shi, L.; Battaglia-Hsu, S.; Nissim, I.; Clapham, D.E.; Ren, D. mTOR regulates lysosomal ATP-sensitive two-pore Na+ channels to adapt to metabolic state. Cell 2013, 152, 778–790. [Google Scholar] [CrossRef]
- Faris, P.; Shekha, M.; Montagna, D.; Guerra, G.; Moccia, F. Endolysosomal Ca2+ Signalling and Cancer Hallmarks: Two-Pore Channels on the Move, TRPML1 Lags Behind! Cancers 2018, 11, 27. [Google Scholar] [CrossRef] [PubMed]
- Stein, I.S.; Park, D.K.; Claiborne, N.; Zito, K. Non-ionotropic NMDA receptor signaling gates bidirectional structural plasticity of dendritic spines. Cell Rep. 2021, 34, 108664. [Google Scholar] [CrossRef] [PubMed]
- Bialecki, J.; Werner, A.; Weilinger, N.L.; Tucker, C.M.; Vecchiarelli, H.A.; Egana, J.; Mendizabal-Zubiaga, J.; Grandes, P.; Hill, M.N.; Thompson, R.J. Suppression of Presynaptic Glutamate Release by Postsynaptic Metabotropic NMDA Receptor Signalling to Pannexin-1. J. Neurosci. 2020, 40, 729–742. [Google Scholar] [CrossRef] [PubMed]
- Abrahamsson, T.; Chou, C.Y.C.; Li, S.Y.; Mancino, A.; Costa, R.P.; Brock, J.A.; Nuro, E.; Buchanan, K.A.; Elgar, D.; Blackman, A.V.; et al. Differential Regulation of Evoked and Spontaneous Release by Presynaptic NMDA Receptors. Neuron 2017, 96, 839–855.e835. [Google Scholar] [CrossRef] [PubMed]
- Negri, S.; Faris, P.; Rosti, V.; Antognazza, M.R.; Lodola, F.; Moccia, F. Endothelial TRPV1 as an Emerging Molecular Target to Promote Therapeutic Angiogenesis. Cells 2020, 9, 1341. [Google Scholar] [CrossRef] [PubMed]
- Negri, S.; Faris, P.; Tullii, G.; Vismara, M.; Pellegata, A.F.; Lodola, F.; Guidetti, G.; Rosti, V.; Antognazza, M.R.; Moccia, F. Conjugated polymers mediate intracellular Ca2+ signals in circulating endothelial colony forming cells through the reactive oxygen species-dependent activation of Transient Receptor Potential Vanilloid 1 (TRPV1). Cell Calcium 2022, 101, 102502. [Google Scholar] [CrossRef] [PubMed]
- Kaeser, P.S.; Sudhof, T.C. RIM function in short- and long-term synaptic plasticity. Biochem. Soc. Trans. 2005, 33, 1345–1349. [Google Scholar] [CrossRef] [PubMed]
- Babiec, W.E.; Guglietta, R.; Jami, S.A.; Morishita, W.; Malenka, R.C.; O’Dell, T.J. Ionotropic NMDA receptor signaling is required for the induction of long-term depression in the mouse hippocampal CA1 region. J. Neurosci. 2014, 34, 5285–5290. [Google Scholar] [CrossRef] [PubMed]
- Sgritta, M.; Locatelli, F.; Soda, T.; Prestori, F.; D’Angelo, E.U. Hebbian Spike-Timing Dependent Plasticity at the Cerebellar Input Stage. J. Neurosci. 2017, 37, 2809–2823. [Google Scholar] [CrossRef] [PubMed]
- Malenka, R.C.; Bear, M.F. LTP and LTD: An embarrassment of riches. Neuron 2004, 44, 5–21. [Google Scholar] [CrossRef]
- Iadecola, C. The Neurovascular Unit Coming of Age: A Journey through Neurovascular Coupling in Health and Disease. Neuron 2017, 96, 17–42. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, S.; Iadecola, C. Revisiting the neurovascular unit. Nat. Neurosci. 2021, 24, 1198–1209. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Chvatal, A. NMDA Receptors in Astrocytes. Neurochem. Res. 2020, 45, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Ruan, J.; Peng, S.; Li, J.; Hu, X.; Zhang, Y.; Zhang, T.; Ge, Y.; Zhu, Z.; Xiao, X.; et al. Synaptic-like transmission between neural axons and arteriolar smooth muscle cells drives cerebral neurovascular coupling. Nat. Neurosci. 2024, 27, 232–248. [Google Scholar] [CrossRef]
- Letellier, M.; Park, Y.K.; Chater, T.E.; Chipman, P.H.; Gautam, S.G.; Oshima-Takago, T.; Goda, Y. Astrocytes regulate heterogeneity of presynaptic strengths in hippocampal networks. Proc. Natl. Acad. Sci. USA 2016, 113, E2685–E2694. [Google Scholar] [CrossRef] [PubMed]
- Hogan-Cann, A.D.; Lu, P.; Anderson, C.M. Endothelial NMDA receptors mediate activity-dependent brain hemodynamic responses in mice. Proc. Natl. Acad. Sci. USA 2019, 116, 10229–10231. [Google Scholar] [CrossRef]
- Lalo, U.; Pankratov, Y.; Parpura, V.; Verkhratsky, A. Ionotropic receptors in neuronal-astroglial signalling: What is the role of "excitable" molecules in non-excitable cells. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2011, 1813, 992–1002. [Google Scholar] [CrossRef]
- Skowronska, K.; Obara-Michlewska, M.; Zielinska, M.; Albrecht, J. NMDA Receptors in Astrocytes: In Search for Roles in Neurotransmission and Astrocytic Homeostasis. Int. J. Mol. Sci. 2019, 20, 309. [Google Scholar] [CrossRef]
- Gerard, F.; Hansson, E. Inflammatory activation enhances NMDA-triggered Ca2+ signalling and IL-1beta secretion in primary cultures of rat astrocytes. Brain Res. 2012, 1473, 1–8. [Google Scholar] [CrossRef]
- Schulte, A.; Bieniussa, L.; Gupta, R.; Samtleben, S.; Bischler, T.; Doering, K.; Sodmann, P.; Rittner, H.; Blum, R. Homeostatic calcium fluxes, ER calcium release, SOCE, and calcium oscillations in cultured astrocytes are interlinked by a small calcium toolkit. Cell Calcium 2022, 101, 102515. [Google Scholar] [CrossRef]
- Torres, R.; Hidalgo, C. Subcellular localization and transcriptional regulation of brain ryanodine receptors. Functional implications. Cell Calcium 2023, 116, 102821. [Google Scholar] [CrossRef]
- Lim, D.; Mapelli, L.; Canonico, P.L.; Moccia, F.; Genazzani, A.A. Neuronal Activity-Dependent Activation of Astroglial Calcineurin in Mouse Primary Hippocampal Cultures. Int. J. Mol. Sci. 2018, 19, 2997. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.; Semyanov, A.; Genazzani, A.; Verkhratsky, A. Calcium signaling in neuroglia. Int. Rev. Cell Mol. Biol. 2021, 362, 1–53. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Blasco, D.; Santofimia-Castano, P.; Gonzalez, A.; Almeida, A.; Bolanos, J.P. Astrocyte NMDA receptors’ activity sustains neuronal survival through a Cdk5-Nrf2 pathway. Cell Death Differ. 2015, 22, 1877–1889. [Google Scholar] [CrossRef]
- Martucci, L.L.; Cancela, J.M. Neurophysiological functions and pharmacological tools of acidic and non-acidic Ca2+ stores. Cell Calcium 2022, 104, 102582. [Google Scholar] [CrossRef] [PubMed]
- Gruszczynska-Biegala, J.; Strucinska, K.; Maciag, F.; Majewski, L.; Sladowska, M.; Kuznicki, J. STIM Protein-NMDA2 Receptor Interaction Decreases NMDA-Dependent Calcium Levels in Cortical Neurons. Cells 2020, 9, 160. [Google Scholar] [CrossRef] [PubMed]
- Serwach, K.; Nurowska, E.; Klukowska, M.; Zablocka, B.; Gruszczynska-Biegala, J. STIM2 regulates NMDA receptor endocytosis that is induced by short-term NMDA receptor overactivation in cortical neurons. Cell. Mol. Life Sci. 2023, 80, 368. [Google Scholar] [CrossRef] [PubMed]
- Negri, S.; Faris, P.; Moccia, F. Reactive Oxygen Species and Endothelial Ca2+ Signaling: Brothers in Arms or Partners in Crime? Int. J. Mol. Sci. 2021, 22, 9821. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef]
- Moccia, F.; Negri, S.; Faris, P.; Angelone, T. Targeting endothelial ion signalling to rescue cerebral blood flow in cerebral disorders. Vasc. Pharmacol. 2022, 145, 106997. [Google Scholar] [CrossRef]
- Stobart, J.L.; Lu, L.; Anderson, H.D.; Mori, H.; Anderson, C.M. Astrocyte-induced cortical vasodilation is mediated by D-serine and endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 2013, 110, 3149–3154. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Hogan-Cann, A.D.; Globa, A.K.; Lu, P.; Nagy, J.I.; Bamji, S.X.; Anderson, C.M. Astrocytes drive cortical vasodilatory signaling by activating endothelial NMDA receptors. J. Cereb. Blood Flow Metab. 2019, 39, 481–496. [Google Scholar] [CrossRef]
- Vazana, U.; Veksler, R.; Pell, G.S.; Prager, O.; Fassler, M.; Chassidim, Y.; Roth, Y.; Shahar, H.; Zangen, A.; Raccah, R.; et al. Glutamate-Mediated Blood-Brain Barrier Opening: Implications for Neuroprotection and Drug Delivery. J. Neurosci. 2016, 36, 7727–7739. [Google Scholar] [CrossRef]
- Kim, K.S.; Jeon, M.T.; Kim, E.S.; Lee, C.H.; Kim, D.G. Activation of NMDA receptors in brain endothelial cells increases transcellular permeability. Fluids Barriers CNS 2022, 19, 70. [Google Scholar] [CrossRef] [PubMed]
- Avemary, J.; Salvamoser, J.D.; Peraud, A.; Remi, J.; Noachtar, S.; Fricker, G.; Potschka, H. Dynamic regulation of P-glycoprotein in human brain capillaries. Mol. Pharm. 2013, 10, 3333–3341. [Google Scholar] [CrossRef]
- Qi, D.; Lin, H.; Hu, B.; Wei, Y. A review on in vitro model of the blood-brain barrier (BBB) based on hCMEC/D3 cells. J. Control. Release 2023, 358, 78–97. [Google Scholar] [CrossRef] [PubMed]
- Weksler, B.; Romero, I.A.; Couraud, P.O. The hCMEC/D3 cell line as a model of the human blood brain barrier. Fluids Barriers CNS 2013, 10, 16. [Google Scholar] [CrossRef]
- Hogan-Cann, A.D.; Anderson, C.M. Physiological Roles of Non-Neuronal NMDA Receptors. Trends Pharmacol. Sci. 2016, 37, 750–767. [Google Scholar] [CrossRef]
- Moccia, F.; Zuccolo, E.; Di Nezza, F.; Pellavio, G.; Faris, P.S.; Negri, S.; De Luca, A.; Laforenza, U.; Ambrosone, L.; Rosti, V.; et al. Nicotinic acid adenine dinucleotide phosphate activates two-pore channel TPC1 to mediate lysosomal Ca2+ release in endothelial colony-forming cells. J. Cell. Physiol. 2021, 236, 688–705. [Google Scholar] [CrossRef]
- Moccia, F.; Negri, S.; Faris, P.; Perna, A.; De Luca, A.; Soda, T.; Romani, R.B.; Guerra, G. Targeting Endolysosomal Two-Pore Channels to Treat Cardiovascular Disorders in the Novel COronaVIrus Disease 2019. Front. Physiol 2021, 12, 629119. [Google Scholar] [CrossRef]
- Negri, S.; Faris, P.; Moccia, F. Endolysosomal Ca2+ signaling in cardiovascular health and disease. Int. Rev. Cell Mol. Biol. 2021, 363, 203–269. [Google Scholar] [CrossRef] [PubMed]
- Gu, F.; Kruger, A.; Roggenkamp, H.G.; Alpers, R.; Lodygin, D.; Jaquet, V.; Mockl, F.; Hernandez, C.L.; Winterberg, K.; Bauche, A.; et al. Dual NADPH oxidases DUOX1 and DUOX2 synthesize NAADP and are necessary for Ca2+ signaling during T cell activation. Sci. Signal. 2021, 14, eabe3800. [Google Scholar] [CrossRef]
- Challiss, R.A.; Mistry, R.; Gray, D.W.; Nahorski, S.R. Modulatory effects of NMDA on phosphoinositide responses evoked by the metabotropic glutamate receptor agonist 1S,3R-ACPD in neonatal rat cerebral cortex. Br. J. Pharmacol. 1994, 112, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Moutin, E.; Raynaud, F.; Roger, J.; Pellegrino, E.; Homburger, V.; Bertaso, F.; Ollendorff, V.; Bockaert, J.; Fagni, L.; Perroy, J. Dynamic remodeling of scaffold interactions in dendritic spines controls synaptic excitability. J. Cell Biol. 2012, 198, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Hoiland, R.L.; Caldwell, H.G.; Howe, C.A.; Nowak-Fluck, D.; Stacey, B.S.; Bailey, D.M.; Paton, J.F.R.; Green, D.J.; Sekhon, M.S.; Macleod, D.B.; et al. Nitric oxide is fundamental to neurovascular coupling in humans. J. Physiol. 2020, 598, 4927–4939. [Google Scholar] [CrossRef] [PubMed]
- O’Gallagher, K.; Puledda, F.; O’Daly, O.; Ryan, M.; Dancy, L.; Chowienczyk, P.J.; Zelaya, F.; Goadsby, P.J.; Shah, A.M. Neuronal nitric oxide synthase regulates regional brain perfusion in healthy humans. Cardiovasc. Res. 2022, 118, 1321–1329. [Google Scholar] [CrossRef] [PubMed]
- Louet, E.R.; Glavan, M.; Orset, C.; Parcq, J.; Hanley, D.F.; Vivien, D. tPA-NMDAR Signaling Blockade Reduces the Incidence of Intracerebral Aneurysms. Transl. Stroke Res. 2022, 13, 1005–1016. [Google Scholar] [CrossRef] [PubMed]
- De Bock, M.; Wang, N.; Decrock, E.; Bol, M.; Gadicherla, A.K.; Culot, M.; Cecchelli, R.; Bultynck, G.; Leybaert, L. Endothelial calcium dynamics, connexin channels and blood-brain barrier function. Prog. Neurobiol. 2013, 108, 1–20. [Google Scholar] [CrossRef]
- Helms, H.C.; Abbott, N.J.; Burek, M.; Cecchelli, R.; Couraud, P.O.; Deli, M.A.; Forster, C.; Galla, H.J.; Romero, I.A.; Shusta, E.V.; et al. In vitro models of the blood-brain barrier: An overview of commonly used brain endothelial cell culture models and guidelines for their use. J. Cereb. Blood Flow Metab. 2016, 36, 862–890. [Google Scholar] [CrossRef]
- Longden, T.A.; Dabertrand, F.; Koide, M.; Gonzales, A.L.; Tykocki, N.R.; Brayden, J.E.; Hill-Eubanks, D.; Nelson, M.T. Capillary K+-sensing initiates retrograde hyperpolarization to increase local cerebral blood flow. Nat. Neurosci. 2017, 20, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Sancho, M.; Gao, Y.; Hald, B.O.; Yin, H.; Boulton, M.; Steven, D.A.; MacDougall, K.W.; Parrent, A.G.; Pickering, J.G.; Welsh, D.G. An assessment of KIR channel function in human cerebral arteries. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H794–H800. [Google Scholar] [CrossRef] [PubMed]
- Berra-Romani, R.; Brunetti, V.; Pellavio, G.; Soda, T.; Laforenza, U.; Scarpellino, G.; Moccia, F. Allyl Isothiocianate Induces Ca2+ Signals and Nitric Oxide Release by Inducing Reactive Oxygen Species Production in the Human Cerebrovascular Endothelial Cell Line hCMEC/D3. Cells 2023, 12, 1732. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.P.; Jiang, M.Q.; Shim, S.S.; Pourkhodadad, S.; Wei, L. Extrasynaptic NMDA receptors in acute and chronic excitotoxicity: Implications for preventive treatments of ischemic stroke and late-onset Alzheimer’s disease. Mol. Neurodegener. 2023, 18, 43. [Google Scholar] [CrossRef]
- Belov Kirdajova, D.; Kriska, J.; Tureckova, J.; Anderova, M. Ischemia-Triggered Glutamate Excitotoxicity From the Perspective of Glial Cells. Front. Cell. Neurosci. 2020, 14, 51. [Google Scholar] [CrossRef] [PubMed]
- Gauchy, C.; Nairn, A.C.; Glowinski, J.; Premont, J. N-Methyl-D-aspartate receptor activation inhibits protein synthesis in cortical neurons independently of its ionic permeability properties. Neuroscience 2002, 114, 859–867. [Google Scholar] [CrossRef]
- Weilinger, N.L.; Lohman, A.W.; Rakai, B.D.; Ma, E.M.; Bialecki, J.; Maslieieva, V.; Rilea, T.; Bandet, M.V.; Ikuta, N.T.; Scott, L.; et al. Metabotropic NMDA receptor signaling couples Src family kinases to pannexin-1 during excitotoxicity. Nat. Neurosci. 2016, 19, 432–442. [Google Scholar] [CrossRef]
- Minnella, A.M.; Zhao, J.X.; Jiang, X.; Jakobsen, E.; Lu, F.; Wu, L.; El-Benna, J.; Gray, J.A.; Swanson, R.A. Excitotoxic superoxide production and neuronal death require both ionotropic and non-ionotropic NMDA receptor signaling. Sci. Rep. 2018, 8, 17522. [Google Scholar] [CrossRef]
- Brennan-Minnella, A.M.; Shen, Y.; El-Benna, J.; Swanson, R.A. Phosphoinositide 3-kinase couples NMDA receptors to superoxide release in excitotoxic neuronal death. Cell Death Dis. 2013, 4, e580. [Google Scholar] [CrossRef]
- Cheng, Y.D.; Al-Khoury, L.; Zivin, J.A. Neuroprotection for ischemic stroke: Two decades of success and failure. NeuroRx 2004, 1, 36–45. [Google Scholar] [CrossRef]
- Saver, J.L.; Starkman, S.; Eckstein, M.; Stratton, S.J.; Pratt, F.D.; Hamilton, S.; Conwit, R.; Liebeskind, D.S.; Sung, G.; Kramer, I.; et al. Prehospital use of magnesium sulfate as neuroprotection in acute stroke. N. Engl. J. Med. 2015, 372, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Jackson, J.; Jambrina, E.; Li, J.; Marston, H.; Menzies, F.; Phillips, K.; Gilmour, G. Targeting the Synapse in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 735. [Google Scholar] [CrossRef] [PubMed]
- Tamburri, A.; Dudilot, A.; Licea, S.; Bourgeois, C.; Boehm, J. NMDA-receptor activation but not ion flux is required for amyloid-beta induced synaptic depression. PLoS ONE 2013, 8, e65350. [Google Scholar] [CrossRef] [PubMed]
- Kessels, H.W.; Nabavi, S.; Malinow, R. Metabotropic NMDA receptor function is required for beta-amyloid-induced synaptic depression. Proc. Natl. Acad. Sci. USA 2013, 110, 4033–4038. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, J.H.; Bali, J.; Rajendran, L.; Nitsch, R.M.; Tackenberg, C. Calcium flux-independent NMDA receptor activity is required for Abeta oligomer-induced synaptic loss. Cell Death Dis. 2015, 6, e1791. [Google Scholar] [CrossRef] [PubMed]
- Dore, K.; Carrico, Z.; Alfonso, S.; Marino, M.; Koymans, K.; Kessels, H.W.; Malinow, R. PSD-95 protects synapses from beta-amyloid. Cell Rep. 2021, 35, 109194. [Google Scholar] [CrossRef]
- Park, D.K.; Petshow, S.; Anisimova, M.; Barragan, E.V.; Gray, J.A.; Stein, I.S.; Zito, K. Reduced d-serine levels drive enhanced non-ionotropic NMDA receptor signaling and destabilization of dendritic spines in a mouse model for studying schizophrenia. Neurobiol. Dis. 2022, 170, 105772. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brunetti, V.; Soda, T.; Berra-Romani, R.; De Sarro, G.; Guerra, G.; Scarpellino, G.; Moccia, F. Two Signaling Modes Are Better than One: Flux-Independent Signaling by Ionotropic Glutamate Receptors Is Coming of Age. Biomedicines 2024, 12, 880. https://doi.org/10.3390/biomedicines12040880
Brunetti V, Soda T, Berra-Romani R, De Sarro G, Guerra G, Scarpellino G, Moccia F. Two Signaling Modes Are Better than One: Flux-Independent Signaling by Ionotropic Glutamate Receptors Is Coming of Age. Biomedicines. 2024; 12(4):880. https://doi.org/10.3390/biomedicines12040880
Chicago/Turabian StyleBrunetti, Valentina, Teresa Soda, Roberto Berra-Romani, Giovambattista De Sarro, Germano Guerra, Giorgia Scarpellino, and Francesco Moccia. 2024. "Two Signaling Modes Are Better than One: Flux-Independent Signaling by Ionotropic Glutamate Receptors Is Coming of Age" Biomedicines 12, no. 4: 880. https://doi.org/10.3390/biomedicines12040880
APA StyleBrunetti, V., Soda, T., Berra-Romani, R., De Sarro, G., Guerra, G., Scarpellino, G., & Moccia, F. (2024). Two Signaling Modes Are Better than One: Flux-Independent Signaling by Ionotropic Glutamate Receptors Is Coming of Age. Biomedicines, 12(4), 880. https://doi.org/10.3390/biomedicines12040880