
NCI 159456 PERK Inhibitor as a Targeted Therapy for Lung Cancer: An In Vitro Study
 , , , , , and
, , , , , and 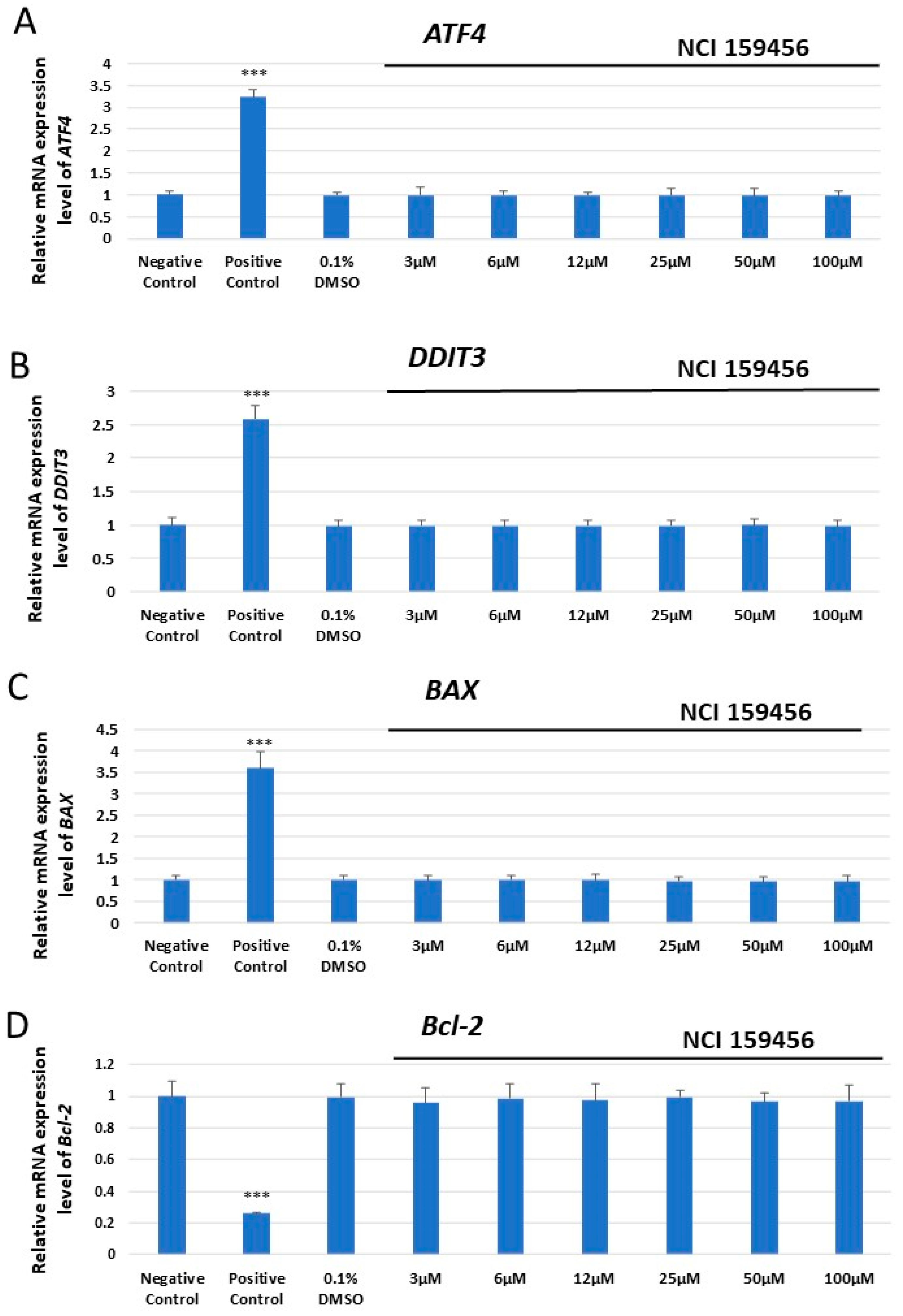{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Screening for the NCI 159456 PERK Inhibitor
2.2. Cell Cultures
2.3. Gene Expression Analysis
2.4. Cytotoxicity Analysis
2.5. Genotoxicity Analysis
2.6. Apoptosis Analysis
2.7. Evaluation of the Level of Reactive Oxygen Species (ROS)
2.8. Statistical Analysis
3. Results
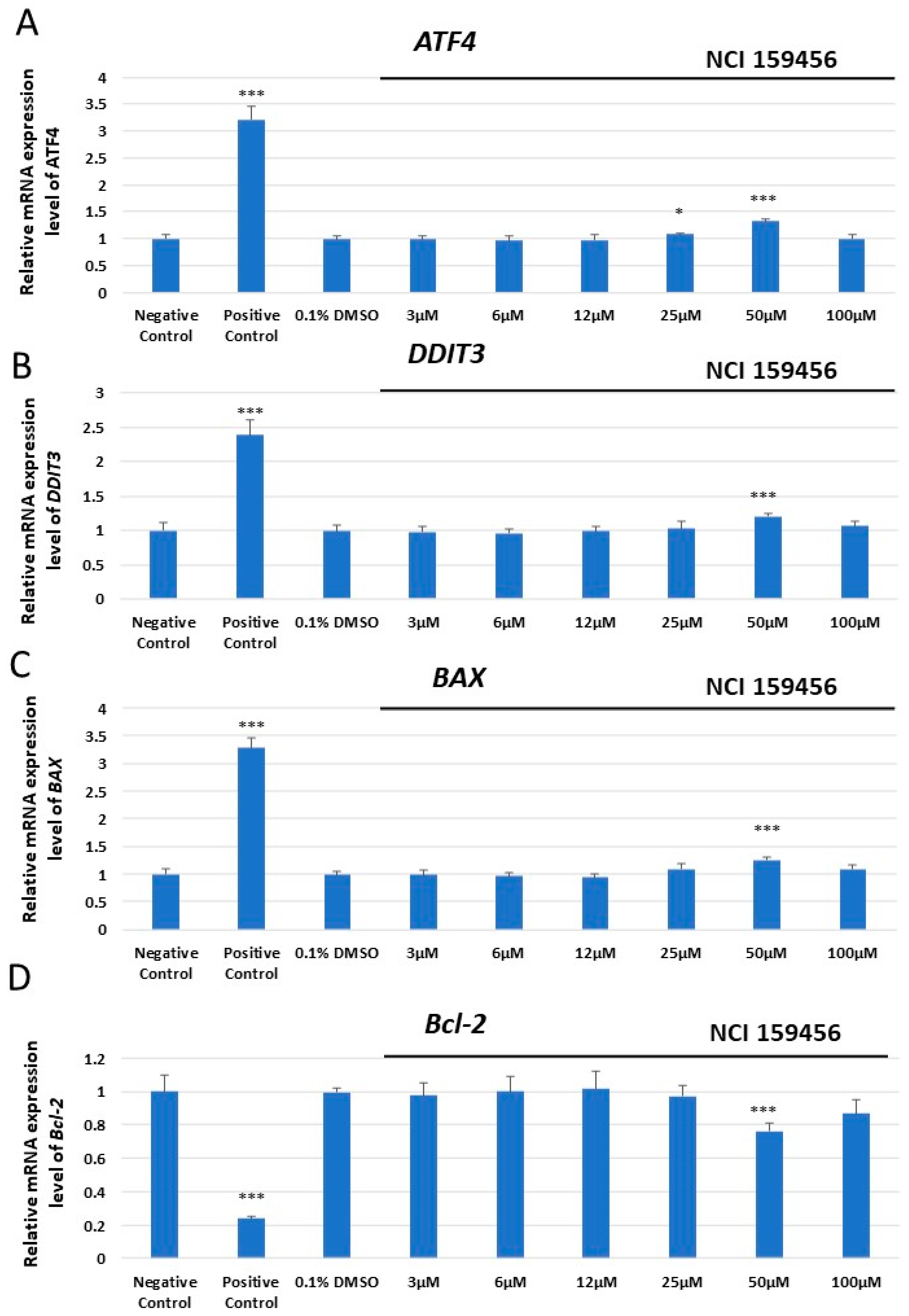
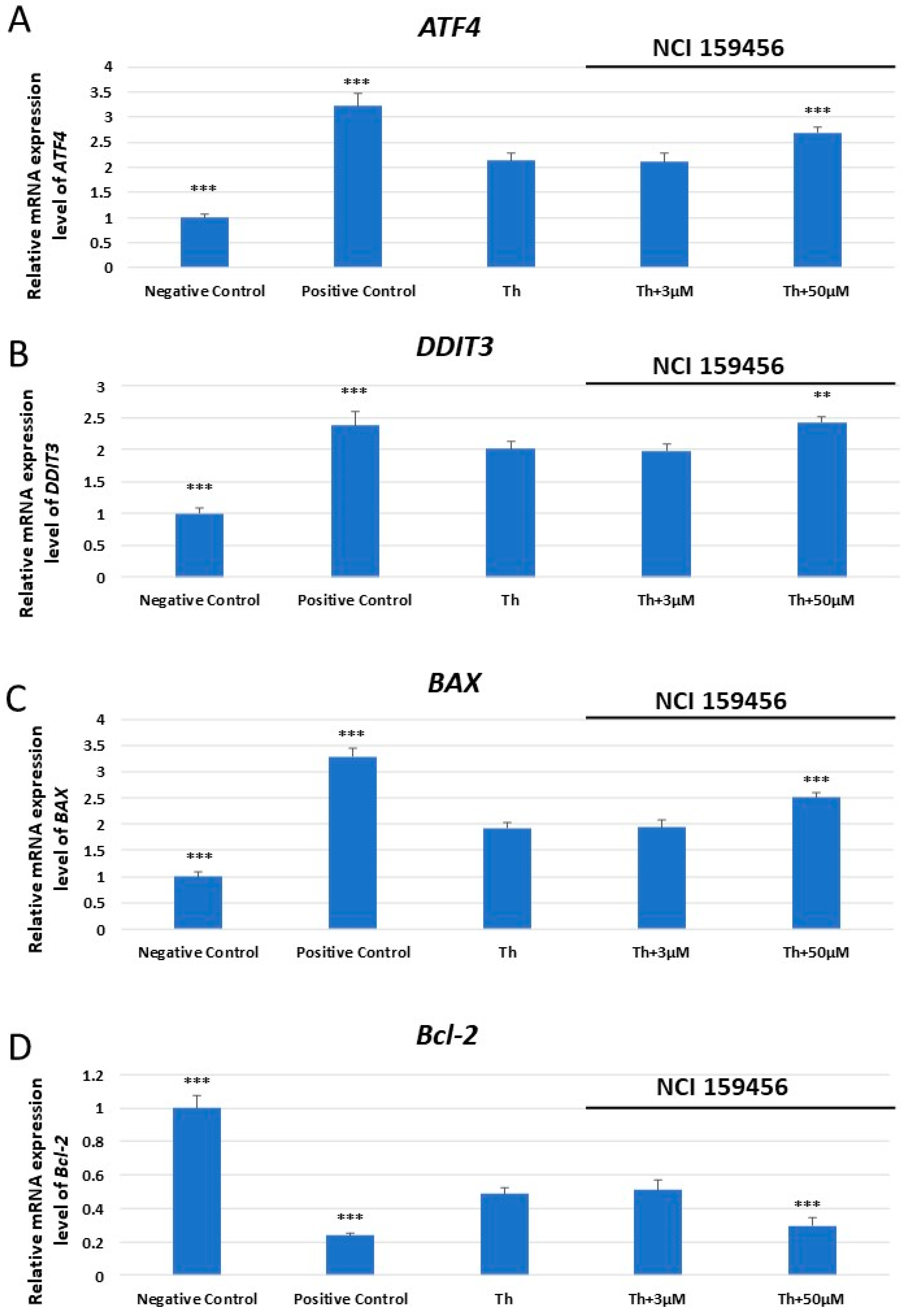
3.1. mRNA Expression Analysis of the ER Stress-Related Pro-Apoptotic Genes in A549 and HPF Cells Treated with NCI 159456 PERK Inhibitor
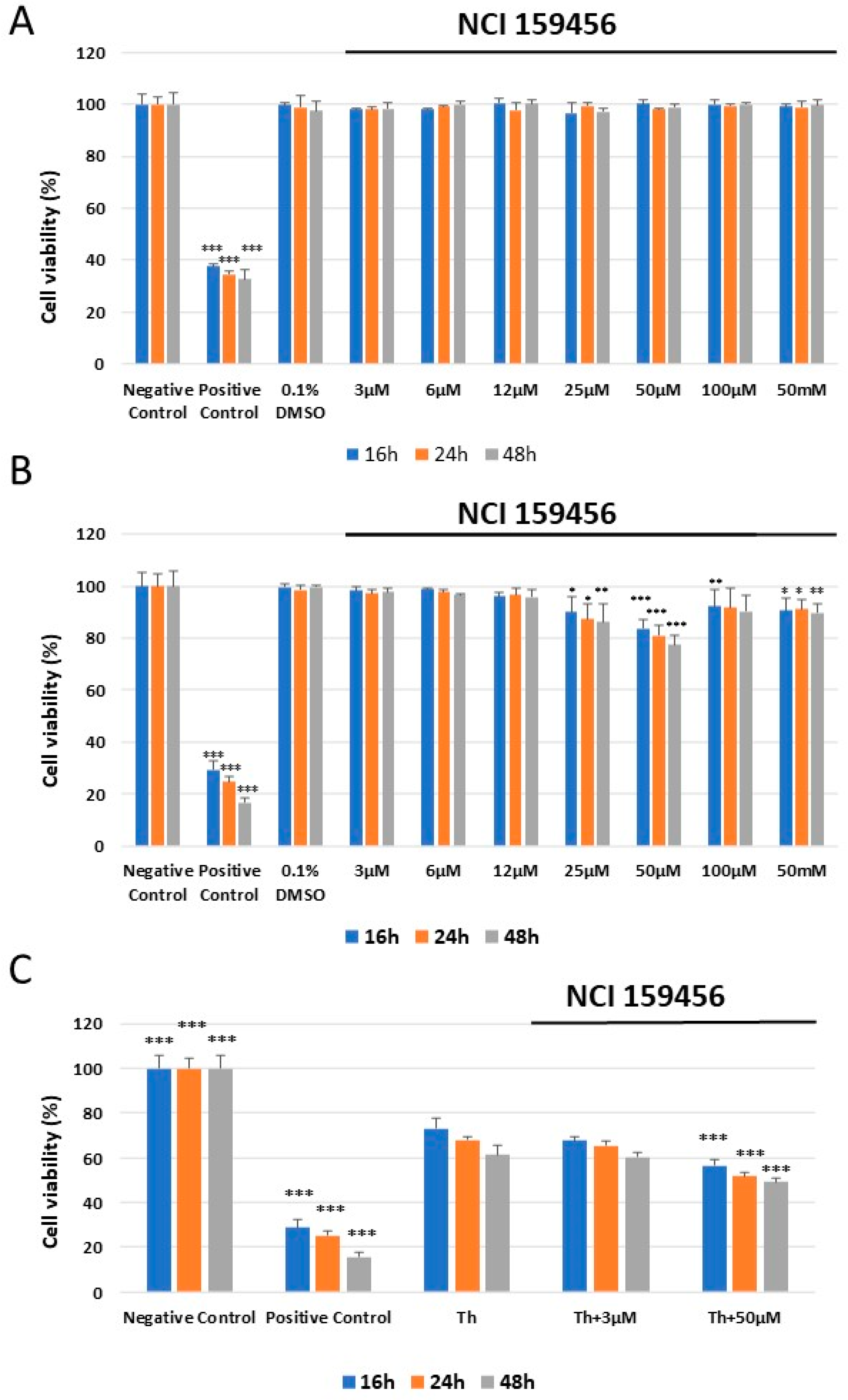
3.2. Evaluation of the Cellular Toxicity of the NCI 159456 PERK Inhibitor
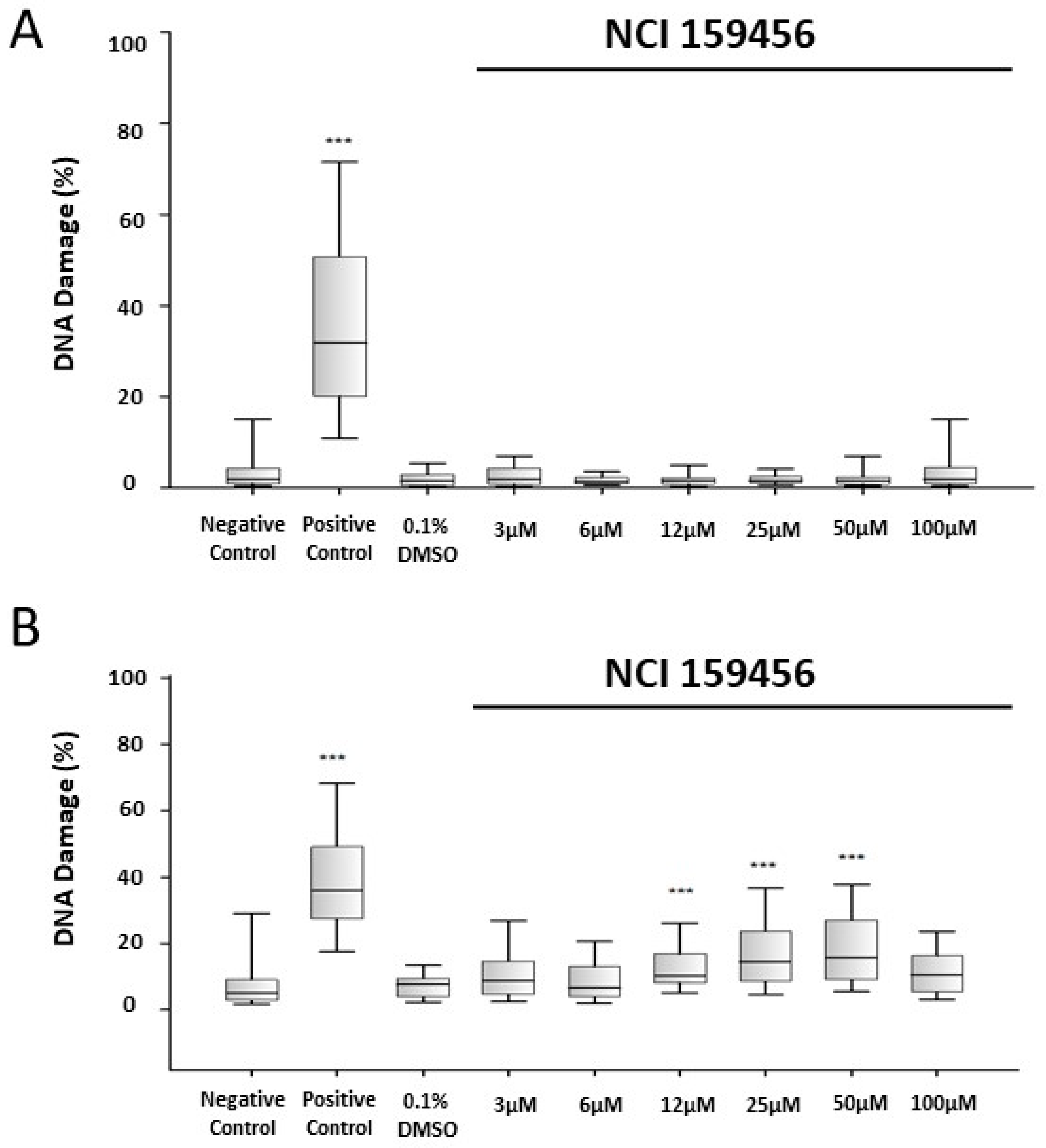
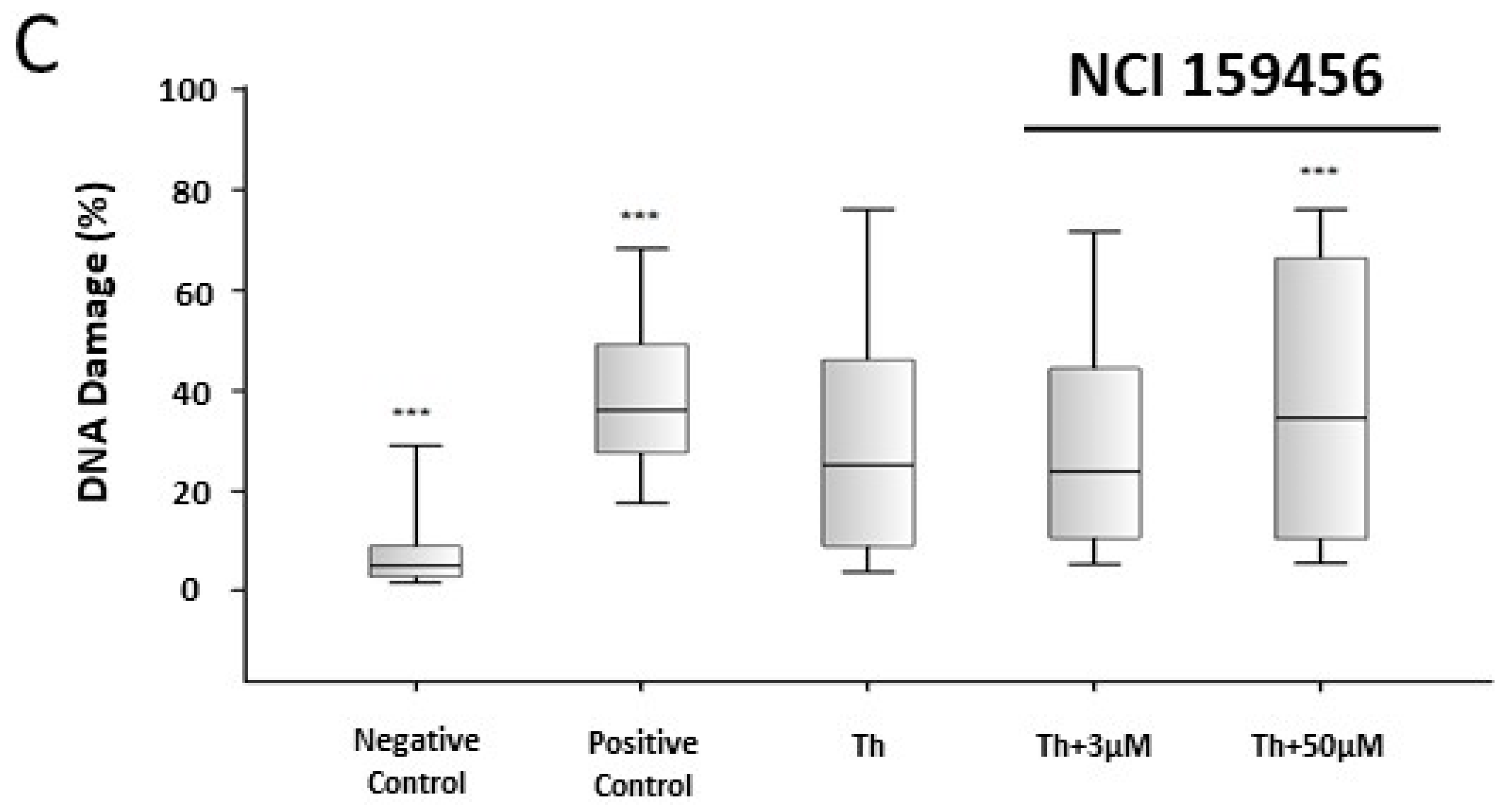
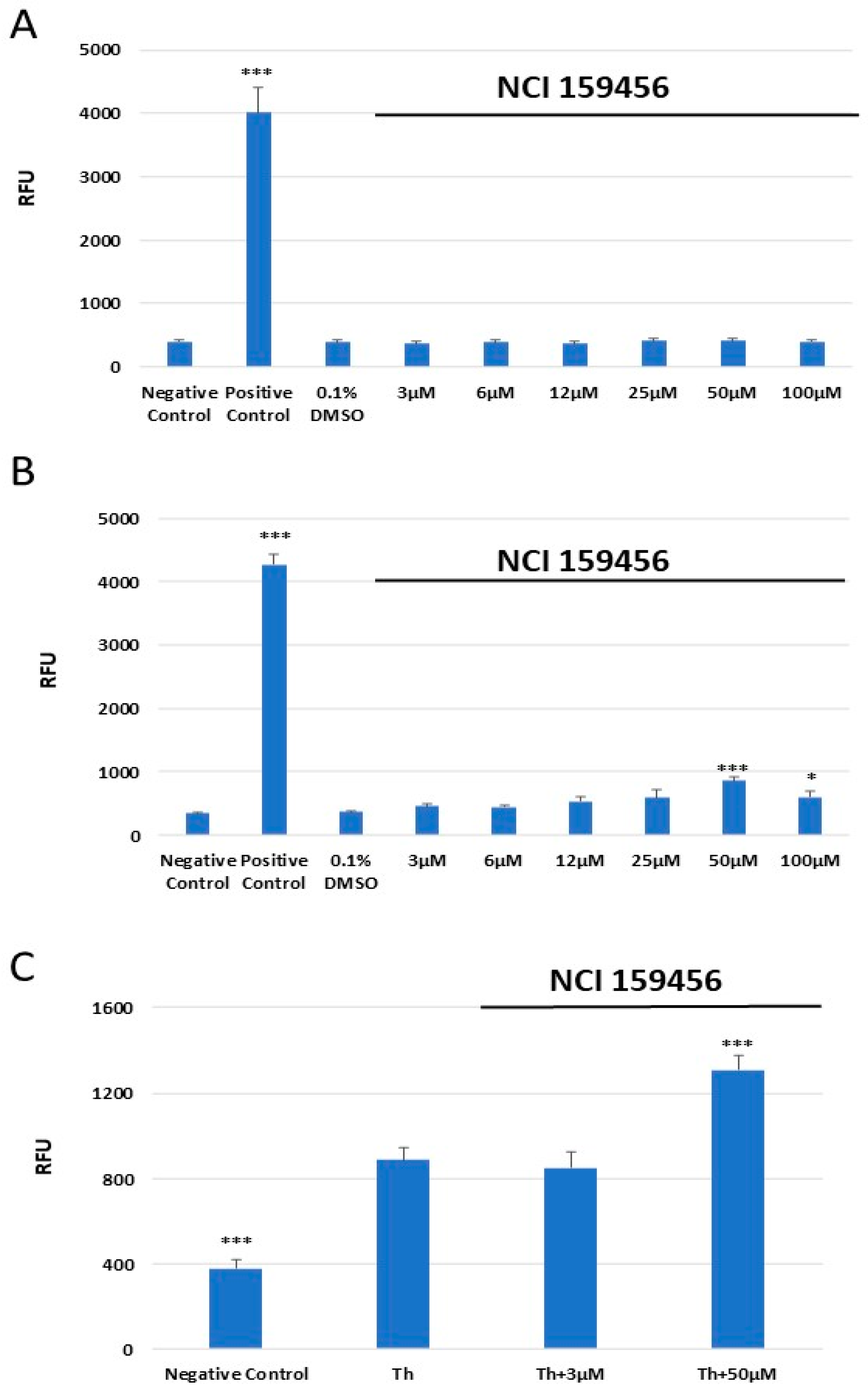
3.3. Genotoxicity Assessment of the NCI 159456 PERK Inhibitor
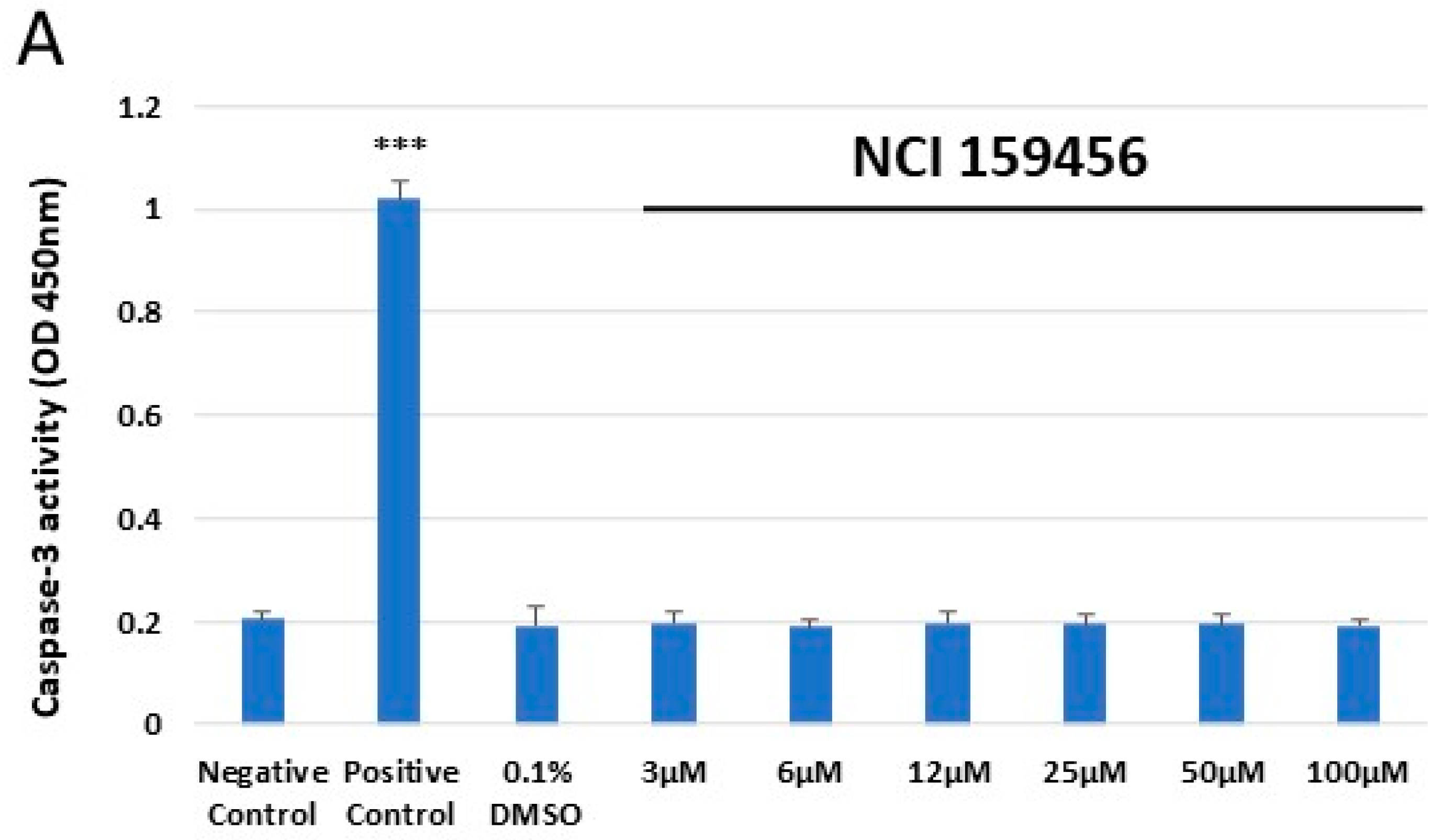
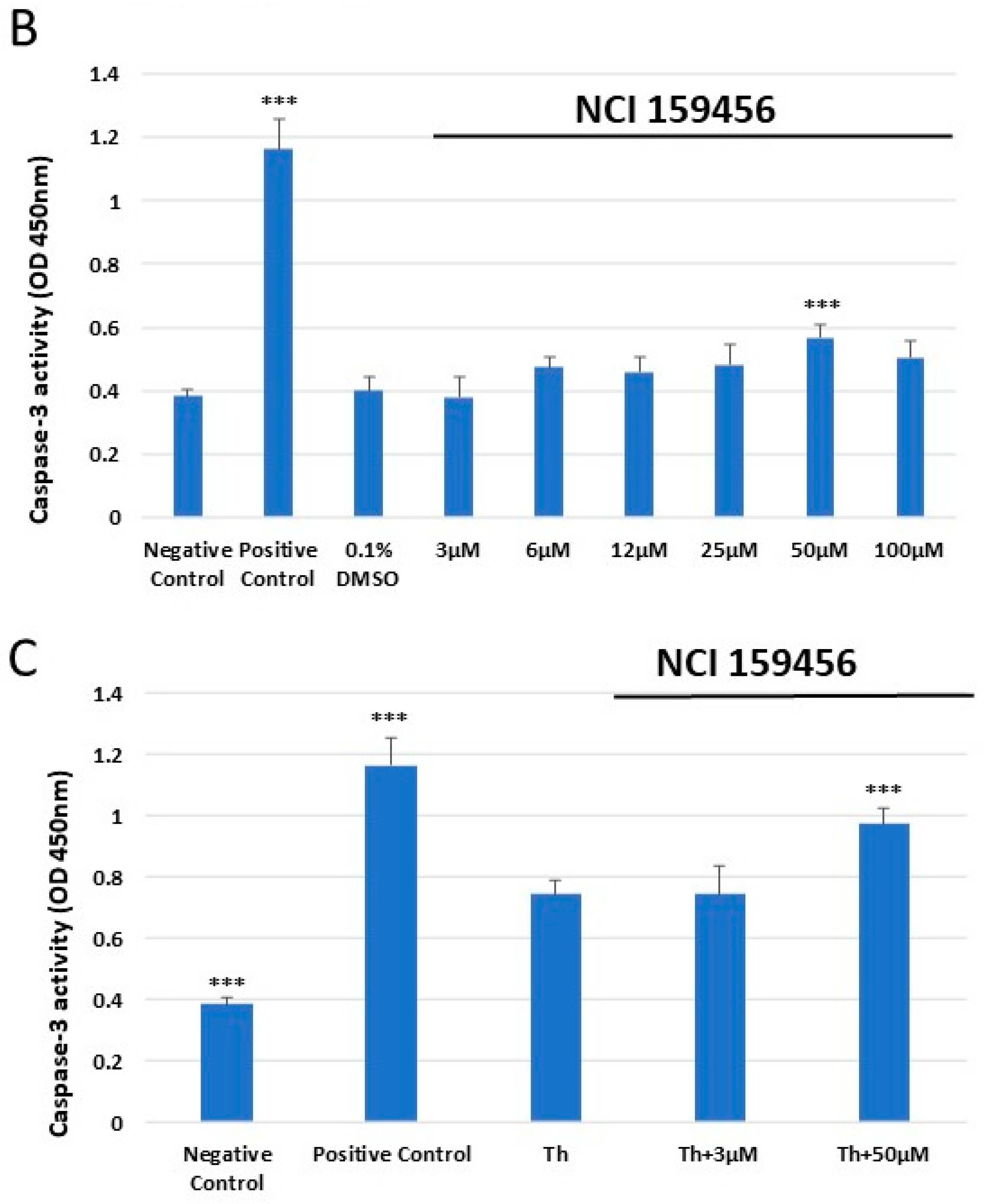
3.4. Evaluation of the Level of Apoptosis
3.5. Evaluation of the Level of Reactive Oxygen Species (ROS)
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Ganti, A.K.; Klein, A.B.; Cotarla, I.; Seal, B.; Chou, E. Update of Incidence, Prevalence, Survival, and Initial Treatment in Patients with Non–Small Cell Lung Cancer in the US. JAMA Oncol. 2021, 7, 1824. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Fillmore, C.M.; Hammerman, P.S.; Kim, C.F.; Wong, K.-K. Non-Small-Cell Lung Cancers: A Heterogeneous Set of Diseases. Nat. Rev. Cancer 2014, 14, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Cubillos-Ruiz, J.R. Endoplasmic Reticulum Stress Signals in the Tumour and Its Microenvironment. Nat. Rev. Cancer 2021, 21, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Kaufman, R.J. The Impact of the Endoplasmic Reticulum Protein-Folding Environment on Cancer Development. Nat. Rev. Cancer 2014, 14, 581–597. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, Regulation and Functions of the Unfolded Protein Response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef] [PubMed]
- Salaroglio, I.C.; Panada, E.; Moiso, E.; Buondonno, I.; Provero, P.; Rubinstein, M.; Kopecka, J.; Riganti, C. PERK induces Resistance to Cell Death Elicited by Endoplasmic Reticulum Stress and Chemotherapy. Mol. Cancer 2017, 16, 91. [Google Scholar] [CrossRef] [PubMed]
- Bu, Y.; Diehl, J.A. PERK Integrates Oncogenic Signaling and Cell Survival during Cancer Development. J. Cell. Physiol. 2016, 231, 2088–2096. [Google Scholar] [CrossRef] [PubMed]
- Cubillos-Ruiz, J.R.; Bettigole, S.E.; Glimcher, L.H. Tumorigenic and Immunosuppressive Effects of Endoplasmic Reticulum Stress in Cancer. Cell 2017, 168, 692–706. [Google Scholar] [CrossRef] [PubMed]
- Hart, L.S.; Cunningham, J.T.; Datta, T.; Dey, S.; Tameire, F.; Lehman, S.L.; Qiu, B.; Zhang, H.; Cerniglia, G.; Bi, M.; et al. ER Stress–Mediated Autophagy Promotes Myc-Dependent Transformation and Tumor Growth. J. Clin. Investig. 2012, 122, 4621–4634. [Google Scholar] [CrossRef]
- Zhang, W.; Neo, S.P.; Gunaratne, J.; Poulsen, A.; Boping, L.; Ong, E.H.; Sangthongpitag, K.; Pendharkar, V.; Hill, J.; Cohen, S.M. Feedback Regulation on PTEN/AKT Pathway by the ER Stress Kinase PERK Mediated by Interaction with the Vault Complex. Cell. Signal 2015, 27, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Liu, H.; Mao, X.; Qin, Y.; Fan, C. ATF4 Promotes Lung Cancer Cell Proliferation and Invasion Partially through Regulating Wnt/β-Catenin Signaling. Int. J. Med. Sci. 2021, 18, 1442–1448. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.-X.; Jin, D.X.; Sokol, E.S.; Reinhardt, F.; Miller, D.H.; Gupta, P.B. Cancer-Specific PERK Signaling Drives Invasion and Metastasis through CREB3L1. Nat. Commun. 2017, 8, 1079. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Yu, X.; Yuan, M.; Lv, W.; Feng, T.; Bai, R.; Zhong, H. Activation of the PERK-ATF4 Pathway Promotes Chemo-Resistance in Colon Cancer Cells. Sci. Rep. 2019, 9, 3210. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Q.; Sun, C.; Han, C.; Han, N.; Zhang, M.; Li, G. Endoplasmic Reticulum Stress Pathway PERK-EIF2α Confers Radioresistance in Oropharyngeal Carcinoma by Activating NF-ΚB. Cancer Sci. 2017, 108, 1421–1431. [Google Scholar] [CrossRef]
- Siwecka, N.; Rozpędek, W.; Pytel, D.; Wawrzynkiewicz, A.; Dziki, A.; Dziki, Ł.; Diehl, J.A.; Majsterek, I. Dual Role of Endoplasmic Reticulum Stress-Mediated Unfolded Protein Response Signaling Pathway in Carcinogenesis. Int. J. Mol. Sci. 2019, 20, 4354. [Google Scholar] [CrossRef]
- Calvo, V.; Zheng, W.; Adam-Artigues, A.; Staschke, K.A.; Huang, X.; Cheung, J.F.; Nobre, A.R.; Fujisawa, S.; Liu, D.; Fumagalli, M.; et al. A PERK-Specific Inhibitor Blocks Metastatic Progression by Limiting Integrated Stress Response–Dependent Survival of Quiescent Cancer Cells. Clin. Cancer Res. 2023, 29, 5155–5172. [Google Scholar] [CrossRef]
- Bagratuni, T.; Patseas, D.; Mavrianou-Koutsoukou, N.; Liacos, C.I.; Sklirou, A.D.; Rousakis, P.; Gavriatopoulou, M.; Terpos, E.; Tsitsilonis, O.E.; Trougakos, I.P.; et al. Characterization of a PERK Kinase Inhibitor with Anti-Myeloma Activity. Cancers 2020, 12, 2864. [Google Scholar] [CrossRef] [PubMed]
- Rozpedek-Kaminska, W.; Piotrzkowska, D.; Galita, G.; Pytel, D.; Kucharska, E.; Dziki, Ł.; Dziki, A.; Majsterek, I. Small-Molecule Inhibitors of the PERK-Mediated Unfolded Protein Response Signaling Pathway in Targeted Therapy for Colorectal Cancer. Pol. J. Surg. 2022, 94, 17–25. [Google Scholar] [CrossRef]
- Stokes, M.E.; Calvo, V.; Fujisawa, S.; Dudgeon, C.; Huang, S.; Ballal, N.; Shen, L.; Gasparek, J.; Betzenhauser, M.; Taylor, S.J.; et al. PERK Inhibition by HC-5404 Sensitizes Renal Cell Carcinoma Tumor Models to Antiangiogenic Tyrosine Kinase Inhibitors. Clin. Cancer Res. 2023, 29, 4870–4882. [Google Scholar] [CrossRef]
- Xiao, W.; Sun, Y.; Xu, J.; Zhang, N.; Dong, L. UORF-Mediated Translational Regulation of ATF4 Serves as an Evolutionarily Conserved Mechanism Contributing to Non-Small-Cell Lung Cancer (NSCLC) and Stress Response. J. Mol. Evol. 2022, 90, 375–388. [Google Scholar] [CrossRef]
- Albert, A.E.; Adua, S.J.; Cai, W.L.; Arnal-Estapé, A.; Cline, G.W.; Liu, Z.; Zhao, M.; Cao, P.D.; Mariappan, M.; Nguyen, D.X. Adaptive Protein Translation by the Integrated Stress Response Maintains the Proliferative and Migratory Capacity of Lung Adenocarcinoma Cells. Mol. Cancer Res. 2019, 17, 2343–2355. [Google Scholar] [CrossRef] [PubMed]
- Ghaddar, N.; Wang, S.; Woodvine, B.; Krishnamoorthy, J.; van Hoef, V.; Darini, C.; Kazimierczak, U.; Ah-son, N.; Popper, H.; Johnson, M.; et al. The Integrated Stress Response Is Tumorigenic and Constitutes a Therapeutic Liability in KRAS-Driven Lung Cancer. Nat. Commun. 2021, 12, 4651. [Google Scholar] [CrossRef]
- Jorgensen, E.; Stinson, A.; Shan, L.; Yang, J.; Gietl, D.; Albino, A.P. Cigarette Smoke Induces Endoplasmic Reticulum Stress and the Unfolded Protein Response in Normal and Malignant Human Lung Cells. BMC Cancer 2008, 8, 229. [Google Scholar] [CrossRef] [PubMed]
- Emanuelli, G.; Nassehzadeh-Tabriz, N.; Morrell, N.W.; Marciniak, S.J. The Integrated Stress Response in Pulmonary Disease. Eur. Respir. Rev. 2020, 29, 200184. [Google Scholar] [CrossRef]
- Golubovskaya, V.M.; Palma, N.L.; Zheng, M.; Ho, B.; Magis, A.; Ostrov, D.; Cance, W.G. A Small-Molecule Inhibitor, 5′-O-Tritylthymidine, Targets FAK and Mdm-2 Interaction, and Blocks Breast and Colon Tumorigenesis in vivo. Anticancer. Agents Med. Chem. 2013, 13, 532–545. [Google Scholar] [CrossRef]
- Xu, Y.; Chen, Z.; Zhang, G.; Xi, Y.; Sun, R.; Wang, X.; Wang, W.; Chai, F.; Li, X. HSP90B1 Overexpression Predicts Poor Prognosis in NSCLC Patients. Tumor Biol. 2016, 37, 14321–14328. [Google Scholar] [CrossRef] [PubMed]
- Del Vecchio, C.A.; Feng, Y.; Sokol, E.S.; Tillman, E.J.; Sanduja, S.; Reinhardt, F.; Gupta, P.B. De-Differentiation Confers Multidrug Resistance Via Noncanonical PERK-Nrf2 Signaling. PLoS Biol. 2014, 12, e1001945. [Google Scholar] [CrossRef] [PubMed]
- Peñaranda-Fajardo, N.M.; Meijer, C.; Liang, Y.; Dijkstra, B.M.; Aguirre-Gamboa, R.; den Dunnen, W.F.A.; Kruyt, F.A.E. ER Stress and UPR Activation in Glioblastoma: Identification of a Noncanonical PERK Mechanism Regulating GBM Stem Cells through SOX2 Modulation. Cell Death Dis. 2019, 10, 690. [Google Scholar] [CrossRef]
- Wang, Y.; Alam, G.N.; Ning, Y.; Visioli, F.; Dong, Z.; Nör, J.E.; Polverini, P.J. The Unfolded Protein Response Induces the Angiogenic Switch in Human Tumor Cells through the PERK/ATF4 Pathway. Cancer Res. 2012, 72, 5396–5406. [Google Scholar] [CrossRef]
- Feng, Y.; Sokol, E.S.; Del Vecchio, C.A.; Sanduja, S.; Claessen, J.H.L.; Proia, T.A.; Jin, D.X.; Reinhardt, F.; Ploegh, H.L.; Wang, Q.; et al. Epithelial-to-Mesenchymal Transition Activates PERK–EIF2α and Sensitizes Cells to Endoplasmic Reticulum Stress. Cancer Discov. 2014, 4, 702–715. [Google Scholar] [CrossRef]
- Dey, S.; Sayers, C.M.; Verginadis, I.I.; Lehman, S.L.; Cheng, Y.; Cerniglia, G.J.; Tuttle, S.W.; Feldman, M.D.; Zhang, P.J.L.; Fuchs, S.Y.; et al. ATF4-Dependent Induction of Heme Oxygenase 1 Prevents Anoikis and Promotes Metastasis. J. Clin. Investig. 2015, 125, 2592–2608. [Google Scholar] [CrossRef]
- Wang, L.; Wen, J.; Sun, Y.; Yang, X.; Ma, Y.; Tian, X. Knockdown of NUPR1 Inhibits Angiogenesis in Lung Cancer through IRE1/XBP1 and PERK/EIF2α/ATF4 Signaling Pathways. Open Med. 2023, 18, 20230796. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, M.; Izumi, H.; Ise, T.; Higuchi, S.; Yamori, T.; Yasumoto, K.; Kohno, K. Activating Transcription Factor 4 Increases the Cisplatin Resistance of Human Cancer Cell Lines. Cancer Res. 2003, 63, 8592–8595. [Google Scholar] [PubMed]
- Seong, Y.-A.; Shin, P.-G.; Yoon, J.-S.; Yadunandam, A.K.; Kim, G.-D. Induction of the Endoplasmic Reticulum Stress and Autophagy in Human Lung Carcinoma A549 Cells by Anacardic Acid. Cell Biochem. Biophys. 2014, 68, 369–377. [Google Scholar] [CrossRef]
- Xie, W.-Y.; Zhou, X.-D.; Li, Q.; Chen, L.-X.; Ran, D.-H. Acid-Induced Autophagy Protects Human Lung Cancer Cells from Apoptosis by Activating ER Stress. Exp. Cell Res. 2015, 339, 270–279. [Google Scholar] [CrossRef]
- Gwinn, D.M.; Lee, A.G.; Briones-Martin-del-Campo, M.; Conn, C.S.; Simpson, D.R.; Scott, A.I.; Le, A.; Cowan, T.M.; Ruggero, D.; Sweet-Cordero, E.A. Oncogenic KRAS Regulates Amino Acid Homeostasis and Asparagine Biosynthesis via ATF4 and Alters Sensitivity to L-Asparaginase. Cancer Cell 2018, 33, 91–107.e6. [Google Scholar] [CrossRef]
- Yang, H.; Liang, S.-Q.; Xu, D.; Yang, Z.; Marti, T.M.; Gao, Y.; Kocher, G.J.; Zhao, H.; Schmid, R.A.; Peng, R.-W. HSP90/AXL/EIF4E-Regulated Unfolded Protein Response as an Acquired Vulnerability in Drug-Resistant KRAS-Mutant Lung Cancer. Oncogenesis 2019, 8, 45. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Tan, P.; Yan, B.; Gao, R.; Zhao, J.; Wang, J.; Guo, J.; Li, N.; Ma, Z. ER Stress and Autophagy Are Involved in the Apoptosis Induced by Cisplatin in Human Lung Cancer Cells. Oncol. Rep. 2016, 35, 2606–2614. [Google Scholar] [CrossRef]
- Li, Y.; Chen, C.; Liu, H.; Li, C.; Zhang, Z.; Wang, C. Pazopanib Restricts Small Cell Lung Cancer Proliferation via Reactive Oxygen species-mediated Endoplasmic Reticulum Stress. Thorac. Cancer 2022, 13, 2421–2428. [Google Scholar] [CrossRef]
- O’Brien, M.E.R.; Gaafar, R.; Hasan, B.; Menis, J.; Cufer, T.; Popat, S.; Woll, P.J.; Surmont, V.; Georgoulias, V.; Montes, A.; et al. Maintenance Pazopanib versus Placebo in Non-Small Cell Lung Cancer Patients Non-Progressive after First Line Chemotherapy: A Double Blind Randomised Phase III Study of the Lung Cancer Group, EORTC 08092 (EudraCT: 2010-018566-23, NCT01208064). Eur. J. Cancer 2015, 51, 1511–1528. [Google Scholar] [CrossRef]
- Li, D.; Liu, L.; Li, F.; Ma, C.; Ge, K. Nifuroxazide Induces the Apoptosis of Human Non-small Cell Lung Cancer Cells through the Endoplasmic Reticulum Stress PERK Signaling Pathway. Oncol. Lett. 2023, 25, 248. [Google Scholar] [CrossRef]
- Di, S.; Fan, C.; Ma, Z.; Li, M.; Guo, K.; Han, D.; Li, X.; Mu, D.; Yan, X. PERK/EIF-2α/CHOP Pathway Dependent ROS Generation Mediates Butein-Induced Non-Small-Cell Lung Cancer Apoptosis and G2/M Phase Arrest. Int. J. Biol. Sci. 2019, 15, 1637–1653. [Google Scholar] [CrossRef]
- Della Corte, C.M.; Ciaramella, V.; Di Mauro, C.; Castellone, M.D.; Papaccio, F.; Fasano, M.; Sasso, F.C.; Martinelli, E.; Troiani, T.; De Vita, F.; et al. Metformin Increases Antitumor Activity of MEK Inhibitors through GLI1 Downregulation in LKB1 Positive Human NSCLC Cancer Cells. Oncotarget 2016, 7, 4265–4278. [Google Scholar] [CrossRef]
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The Biology and Management of Non-Small Cell Lung Cancer. Nature 2018, 553, 446–454. [Google Scholar] [CrossRef]
- Araghi, M.; Mannani, R.; Heidarnejad Maleki, A.; Hamidi, A.; Rostami, S.; Safa, S.H.; Faramarzi, F.; Khorasani, S.; Alimohammadi, M.; Tahmasebi, S.; et al. Recent Advances in Non-Small Cell Lung Cancer Targeted Therapy; an Update Review. Cancer Cell Int. 2023, 23, 162. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rozpędek-Kamińska, W.; Galita, G.; Siwecka, N.; Granek, Z.; Barczuk, J.; Saramowicz, K.; Majsterek, I. NCI 159456 PERK Inhibitor as a Targeted Therapy for Lung Cancer: An In Vitro Study. Biomedicines 2024, 12, 889. https://doi.org/10.3390/biomedicines12040889
Rozpędek-Kamińska W, Galita G, Siwecka N, Granek Z, Barczuk J, Saramowicz K, Majsterek I. NCI 159456 PERK Inhibitor as a Targeted Therapy for Lung Cancer: An In Vitro Study. Biomedicines. 2024; 12(4):889. https://doi.org/10.3390/biomedicines12040889
Chicago/Turabian StyleRozpędek-Kamińska, Wioletta, Grzegorz Galita, Natalia Siwecka, Zuzanna Granek, Julia Barczuk, Kamil Saramowicz, and Ireneusz Majsterek. 2024. "NCI 159456 PERK Inhibitor as a Targeted Therapy for Lung Cancer: An In Vitro Study" Biomedicines 12, no. 4: 889. https://doi.org/10.3390/biomedicines12040889