
PSEN2 Mutations May Mimic Frontotemporal Dementia: Two New Case Reports and a Review

 ,
, 
 , , ,
, , ,  and
and
Abstract
1. Introduction
2. Detailed Case Description
2.1. Materials and Methods
2.1.1. Clinical and Neuropsychological Workup
2.1.2. Structural and Functional Neuroimaging Tests
2.1.3. Laboratory Study (Including AD Biomarkers)
2.1.4. Genetic Tests
2.2. Case A
2.2.1. Clinical Course
2.2.2. Neuroimaging Tests
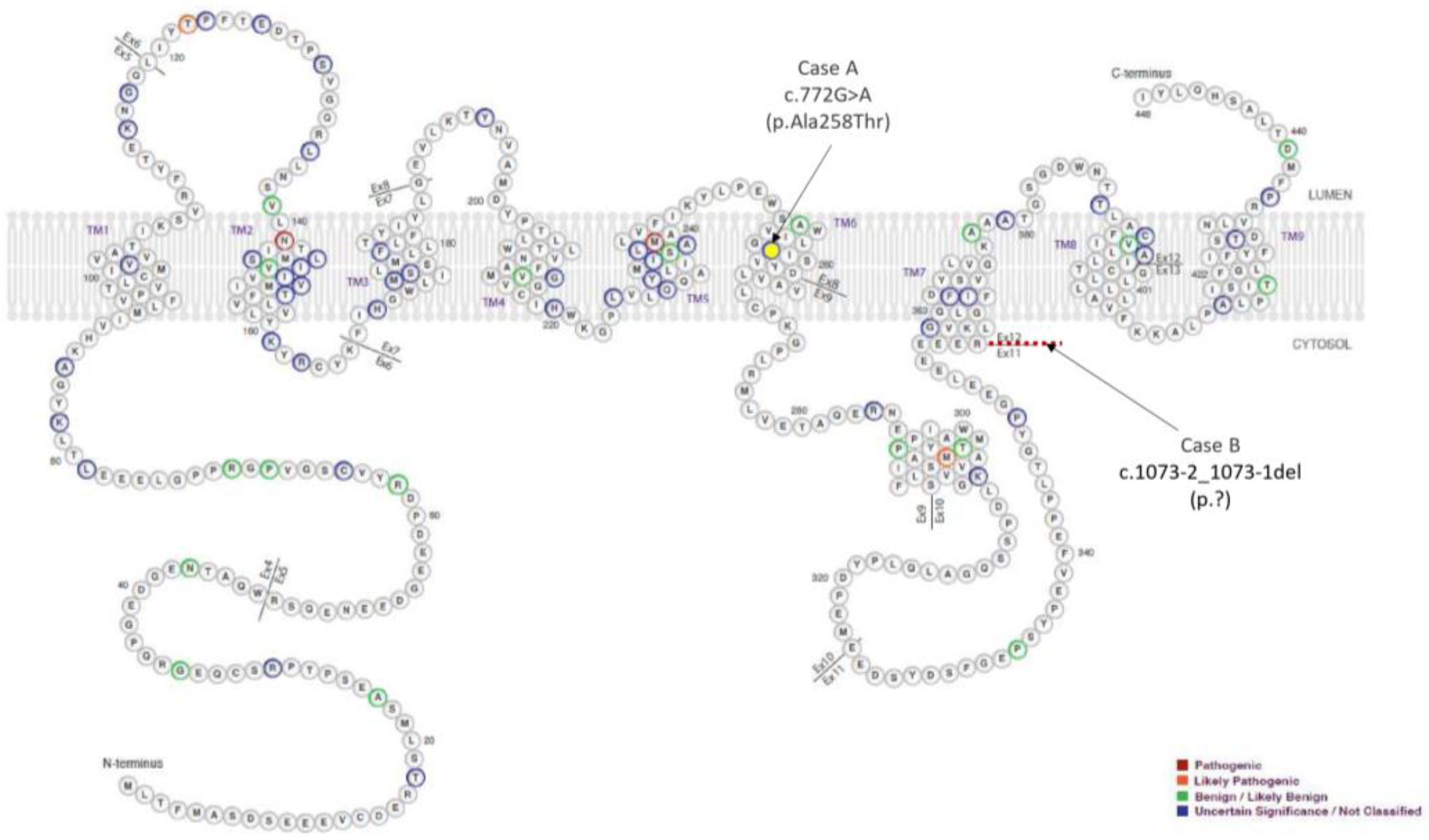
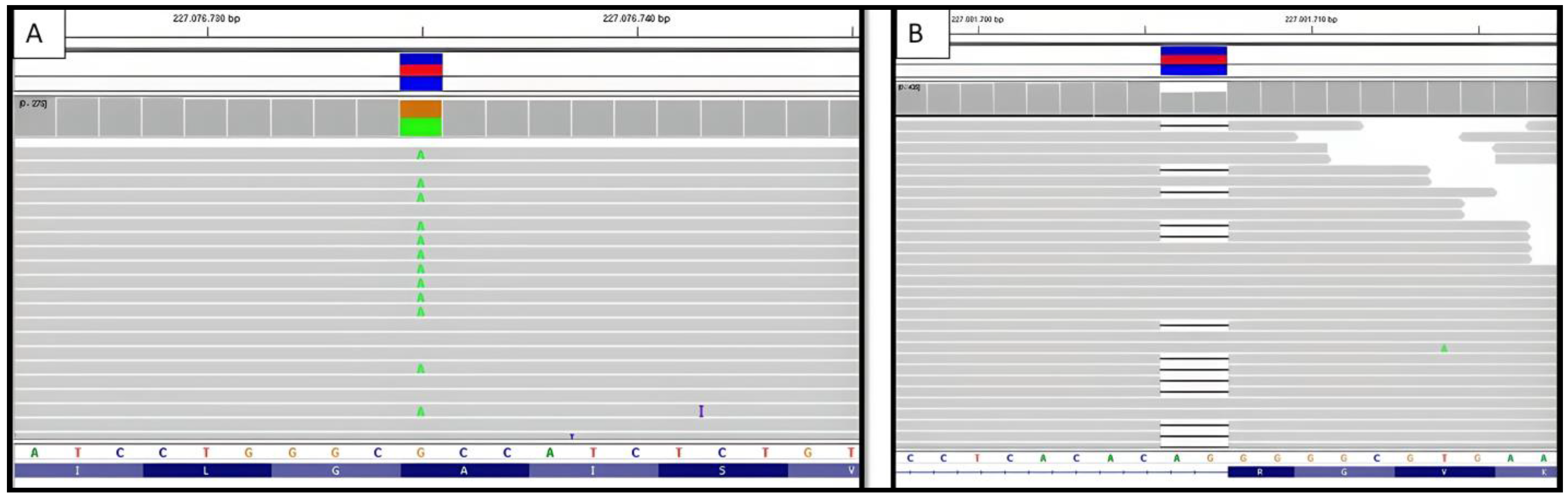
2.2.3. Genetic Testing
2.3. Case B
2.3.1. Clinical Course
2.3.2. Neuroimaging Tests
2.3.3. Genetic Testing
2.4. AD Blood and CSF Biomarkers
3. Discussion
4. Conclusions
Limitations and Strengths of the Study
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Orphanet. Copyright, INSERM 1999. Available online: http://www.orpha.net (accessed on 23 April 2023).
- Sociedad Española de Neurología. Guía Oficial de Práctica Clínica en Demencias; Sociedad Española de Neurología: Madrid, Spain, 2018. [Google Scholar]
- Goate, A.; Chartier-Harlin, M.-C.; Mullan, M.; Brown, J.; Crawford, F.; Fidani, L.; Giuffra, L.; Haynes, A.; Irving, N.; James, L.; et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 1991, 349, 704–706. [Google Scholar] [CrossRef]
- Sherrington, R.; Rogaev, E.I.; Liang, Y.; Rogaeva, E.A.; Levesque, G.; Ikeda, M.; Chi, H.; Lin, C.; Li, G.; Holman, K.; et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 1995, 375, 754–760. [Google Scholar] [CrossRef] [PubMed]
- Levy-Lahad, E.; Wasco, W.; Poorkaj, P.; Romano, D.M.; Oshima, J.; Pettingell, W.H.; Yu, C.-E.; Jondro, P.D.; Schmidt, S.D.; Wang, K.; et al. Candidate gene for the chromosome 1 familial Alzheimer’ disease locus. Science 1995, 269, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Chen, M. The maze of APP processing in Alzheimer’s disease: Where did we go wrong in reasoning? Front. Cell. Neurosci. 2015, 9, 186. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; An, S.S.A.; Kim, S. Mutations in presenilin 2 and its implications in Alzheimer’s disease and other dementia-associated disorders. Clin. Interv. Aging 2015, 10, 1163–1172. [Google Scholar] [PubMed]
- Canevelli, M.; Piscopo, P.; Talarico, G.; Vanacore, N.; Blasimme, A.; Crestini, A.; Tosto, G.; Troili, F.; Lenzi, G.L.; Confaloni, A.; et al. Familial Alzheimer’s disease sustained by presenilin 2 mutations: Systematic review of literature and genotype-phenotype correlation. Neurosci. Biobehav. Rev. 2014, 42, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Shea, Y.F.; Chu, L.W.; Chan, A.O.K.; Ha, J.; Li, Y.; Song, Y.Q. A systematic review of familial Alzheimer’s disease: Differences in presentation of clinical features among three mutated genes and potential ethnic differences. J. Formos. Med. Assoc. 2016, 115, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Folstein, M.F.; Fostein, S.E.; McHugh, P.R. MiniMental State. A practical method for grading the cognitive state of patients for the clinician. J. Psychiat Res. 1975, 12, 189–198. [Google Scholar] [CrossRef]
- Morris, J.C. The Clinical Dementia Rating (CDR): Current version and scoring rules. Neurology 1993, 43, 2412–2414. [Google Scholar] [CrossRef]
- Goodglass, H.; Kaplan, E.; Barresi, B. Aphasia Severity Subscale of the Boston Diagnostic Aphasia Examination, 3rd ed.; Spanish Ad-Aptation by García-Albea, J.E.; Editorial Médica Panamericana: Buenos Aires, Argentina; Madrid, Spain, 2005. [Google Scholar]
- Peña-Casanova, J.; Gramunt, N.F.; Gich, J.F. Neuropsychological Tests. Fundamentals for Evidence-Based Clinical Psychology; Masson: Barcelona, Spain, 2004. [Google Scholar]
- Buschke, H.; Kuslansky, G.; Katz, M.; Stewart, W.F.; Sliwinski, M.J.; Eckholdt, H.M.; Lipton, R.B. Screening for dementia with the Memory Impairment Screen. Neurology 1999, 52, 231–238. [Google Scholar] [CrossRef]
- Cummings, J.L.; Mega, M.; Gray, K.; Rosenberg-Thompson, S.; Carusi, D.A.; Gornbein, J. The neuropsychiatric inventory. Comprehensive assessment of psychopathology in dementia. Neurology 1994, 44, 2308. [Google Scholar] [CrossRef] [PubMed]
- Lawton, M.P.; Brody, E.M. Assessment of older people: Self-Maintaining and Instrumental Activities of Daily Living. Gerontologist 1969, 9, 179–186. [Google Scholar] [CrossRef]
- Global Biomarker Standardization Consortium (GBSC)|Alzheimer’s Association [Internet]. Available online: https://www.alz.org/research/for_researchers/partnerships/gbsc (accessed on 27 December 2021).
- Carrillo, M.C.; Blennow, K.; Soares, H.; Lewczuk, P.; Mattsson, N.; Oberoi, P.; Umek, R.; Vandijck, M.; Salamone, S.; Bittner, T.; et al. Global standardization measurement of cerebral spinal fluid for Alzheimer’s disease: An update from the Alzheimer’s Association Global Biomarkers Consortium. Alzheimer’s Dement. 2013, 9, 137–140. [Google Scholar] [CrossRef]
- Delaby, C.; Muñoz, L.; Torres, S.; Nadal, A.; Le Bastard, N.; Lehmann, S.; Lleó, A.; Alcolea, D. Impact of CSF storage volume on the analysis of Alzheimer’s disease biomarkers on an automated platform. Clin. Chim. Acta 2019, 490, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Greaves, C.V.; Rohrer, J.D. An update on genetic frontotemporal dementia. J. Neurol. 2019, 266, 2075–2086. [Google Scholar] [CrossRef] [PubMed]
- Perrone, F.; Cacace, R.; Van Mossevelde, S.; Van den Bossche, T.; De Deyn, P.P.; Cras, P.; Engelborghs, S.; van der Zee, J.; Van Broeckhoven, C. Genetic screening in early-onset dementia patients with unclear phenotype: Relevance for clinical diagnosis. Neurobiol. Aging 2018, 69, 292. [Google Scholar] [CrossRef] [PubMed]
- Yagi, R.; Miyamoto, R.; Morino, H.; Izumi, Y.; Kuramochi, M.; Kurashige, T.; Maruyama, H.; Mizuno, N.; Kurihara, H.; Kawakami, H. Detecting gene mutations in Japanese Alzheimer’s patients by semiconductor sequencing. Neurobiol. Aging 2014, 35, 1780.e1–1780.e5. [Google Scholar] [CrossRef]
- Farrer, L.A.; Cupples, L.A.; Haines, J.L.; Hyman, B.; Kukull, W.A.; Mayeux, R.; Myers, R.H.; Pericak-Vance, M.A.; Risch, N.; van Duijn, C.M. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. JAMA 1997, 278, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Mattsson, N.; Zetterberg, H.; Janelidze, S.; Insel, P.S.; Andreasson, U.; Stomrud, E.; Palmqvist, S.; Baker, D.; Tan Hehir, C.A.; Jeromin, A.; et al. DNI Investigators. Plasma tau in Alzheimer disease. Neurology 2016, 87, 1827–1835. [Google Scholar] [CrossRef] [PubMed]
- Janelidze, S.; Berron, D.; Smith, R.; Strandberg, O.; Proctor, N.K.; Dage, J.L.; Stomrud, E.; Palmqvist, S.; Mattsson-Carlgren, N.; Hansson, O. Associations of Plasma Phospho-Tau217 Levels with Tau Positron Emission Tomography in Early Alzheimer Disease. JAMA Neurol. 2021, 78, 149–156. [Google Scholar] [CrossRef]
- Illán-Gala, I.; Lleo, A.; Karydas, A.; Staffaroni, A.M.; Zetterberg, H.; Sivasankaran, R.; Grinberg, L.T.; Spina, S.; Kramer, J.H.; Ramos, E.M.; et al. Plasma Tau and Neurofilament Light in Frontotemporal Lobar Degeneration and Alzheimer Disease. Neurology 2021, 96, e671–e683. [Google Scholar] [CrossRef]
- Dumurgier, J.; Schraen, S.; Gabelle, A.; Vercruysse, O.; Bombois, S.; Laplanche, J.-L.; Peoc’h, K.; Sablonnière, B.; Kastanenka, K.V.; Delaby, C.; et al. Cerebrospinal fluid amyloid-beta 42/40 ratio in clinical setting of memory centers: A multicentric study. Alzheimer’s Res. Ther. 2015, 7, 30. [Google Scholar] [CrossRef]
- Lewczuk, P.; Lelental, N.; Spitzer, P.; Maler, J.M.; Kornhuber, J. Amyloid-beta 42/40 cerebrospinal fluid concentration ratio in the diagnostics of Alzheimer’s disease: Validation of two novel assays. J. Alzheimer’s Dis. 2015, 43, 183–191. [Google Scholar] [CrossRef]
- Scheuner, D.; Eckman, C.; Jensen, M.; Song, X.; Citron, M.; Suzuki, N.; Bird, T.D.; Hardy, J.; Hutton, M.; Kukull, W.; et al. Secreted amyloid β-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat. Med. 1996, 2, 864–870. [Google Scholar] [CrossRef]
- Shen, J.; Kelleher, R.J. The presenilin hypothesis of Alzheimer’s disease: Evidence for a loss-of-function pathogenic mechanism. Proc. Natl. Acad. Sci. USA 2007, 104, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Jayadev, S.; Leverenz, J.B.; Steinbart, E.; Stahl, J.; Klunk, W.; Yu, C.E.; Bird, T.D. Alzheimer’s disease phenotypes and genotypes associated with mutations in presenilin. Brain 2010, 133, 1143–1154. [Google Scholar] [CrossRef] [PubMed]
- Zarea, A.; Charbonnier, C.; Rovelet-Lecrux, A.; Nicolas, G.; Rousseau, S.; Borden, A.; Pariente, J.; Le Ber, I.; Pasquier, F.; Formaglio, M.; et al. Seizures in dominantly inherited Alzheimer disease. Neurology 2016, 87, 912–919. [Google Scholar] [CrossRef] [PubMed]
- Lleó, A.; Blesa, R.; Queralt, R.; Ezquerra, M.; Molinuevo, J.L.; Pena-Casanova, J.; Rojo, A.; Oliva, R. Frequency of mutations in the presenilin and amyloid precursor protein genes in earlyonset Alzheimer disease in Spain. Arch. Neurol. 2002, 59, 1759–1763. [Google Scholar] [CrossRef] [PubMed]
- Tabaee Damavandi, P.; Storti, B.; Fabin, N.; Bianchi, E.; Ferrarese, C.; DiFrancesco, J.C. Epilepsy in cerebral amyloid angiopathy: An observational retrospective study of a large population. Epilepsia 2023, 64, 500–510. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, G.; Collinge, J.; Fox, N.C.; Lashley, T.; Mead, S.; Schott, J.M.; Werring, D.J.; Ryan, N.S. Clinical considerations in early-onset cerebral amyloid angiopathy. Brain 2023, 146, 3991–4014. [Google Scholar] [CrossRef] [PubMed]
- Moscoso, A.; Rey-Bretal, D.; Silva-Rodríguez, J.; Aldrey, J.M.; Cortés, J.; Pías-Peleteiro, J.; Ruibal, Á.; Aguiar, P.; Alzheimer’s Disease Neuroimaging Initiative. White matter hyperintensities are associated with subthreshold amyloid accumulation. Neuroimage 2020, 218, 116944. [Google Scholar] [CrossRef] [PubMed]
- Moscoso, A.; Silva-Rodríguez, J.; Aldrey, J.M.; Cortés, J.; Pías-Peleteiro, J.M.; Ruibal, Á.; Aguiar, P.; Alzheimer’s Disease Neuroimaging Initiative. 18F-florbetapir PET as a marker of myelin integrity across the Alzheimer’s disease spectrum. Eur. J. Nucl. Med. Mol. Imaging 2022, 49, 1242–1253. [Google Scholar] [CrossRef] [PubMed]
- Kapasi, A.; DeCarli, C.; Schneider, J.A. Impact of multiple pathologies on the threshold for clinically overt dementia. Acta Neuropathol. 2017, 134, 171–186. [Google Scholar] [CrossRef]
- Chang Wong, E.; Chang Chui, H. Vascular Cognitive Impairment and Dementia. Continuum 2022, 28, 750–780. [Google Scholar] [CrossRef] [PubMed]
- Marcon, G.; Di Fede, G.; Giaccone, G.; Rossi, G.; Giovagnoli, A.R.; Maccagnano, E.; Tagliavini, F. A novel Italian presenilin 2 gene mutation with prevalent behavioral phenotype. J. Alzheimer’s Dis. 2009, 16, 509–511. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case B | Case A | |||||
|---|---|---|---|---|---|---|
| CDR_ | 26 April 2017 | 4 December 2018 | 4 June 2020 | 18 June 2014 | 22 June 2016 | 22 June 2018 |
| CDR_MEMORY | 0.5 | 2 | 3 | 1 | NV * | NV |
| CDR_ORIENTATION | 1 | 1 | 3 | 1 | 2 | 3 |
| CDR_REASONING AND PROBLEM SOLVING | 0 | 2 | 3 | 1 | NV | 3 |
| CDR_OUT-OF-HOME ACTIVITIES | 0.5 | 2 | 3 | 1 | 3 | 3 |
| CDR_DOMESTIC ACTIVITIES AND HOBBIES | 0 | 2 | 3 | 1 | 2 | 3 |
| CDR_SELF-CARE | 0 | 2 | 3 | 1 | 2 | 3 |
| CDR_TOTAL | 0.5 | 2 | 3 | 1 | 2 | 3 |
| MMSE | 21 | 8 | 0 | 8 | NV | NV |
| NPI_TOTAL | 25 | 34 | 46 | 12 | 16 | 25 |
| NPI_DELUSIONS (frequencyXgravity) | ×1 | 0 | 0 | 0 | 0 | 0 |
| NPI_HALLUCINATIONS (frequencyXgravity) | 0 | 0 | 0 | 0 | 0 | 0 |
| NPI_AGITATION (frequencyXgravity) | 0 | 0 | 4 × 2 | 0 | 0 | 1 × 1 |
| NPI_DEPRESSION-DYSPHORIA (frequencyXgravity) | 4 × 2 | 4 × 1 | 0 | 4 × 1 | 4 × 2 | 4 × 2 |
| NPI_ANXIETY (frequencyXgravity) | 4 × 2 | 4 × 1 | 4 × 2 | 4 × 1 | 0 | 1 × 1 |
| NPI_EUPHORIA (frequencyXgravity) | 0 | 4 × 2 | 4 × 2 | 0 | 0 | 4 × 3 |
| NPI_APATHY (frequencyXgravity) | 4 × 2 | 4 × 2 | 4 × 2 | 0 | 4 × 2 | 1 × 1 |
| NPI_DISINHIBITION (frequencyXgravity) | 0 | 4 × 2 | 4 × 2 | 2 × 2 | 0 | 1 × 1 |
| NPI_IRRITABILITY (frequencyXgravity) | 0 | 2 × 2 | 3 × 2 | 2 × 2 (hygiene) | 0 | 1 × 1 |
| NPI_ABERRANT MOTOR BEHAVIOR (frequencyXgravity) | 0 | 0 | 0 | 0 | 0 | 0 |
| NPI_CAREGIVER STRESS | 6 | 8 | 10 | 3 | 3 | 3 |
| APETITE | OK | HYPERPHAGIA | HYPERPHAGIA | HYPERPHAGIA | HYPERPHAGIA | OK |
| SLEEP | OK | OK with medication | OK with medication | OK | Fragmentated | Fragmentated |
| BARTHEL | 100 | 90 | 40 | 100 | 50 | 20 |
| AIVD | 8 | 2 | 2 | 3 | 0 | 0 |
| MIS | 2/8 | 0/8 | 0 | 2/8 | NV | NV |
| SEMANTIC FLUENCY | -- | 2 | 0 | 2 | 0 | 0 |
| PHONETIC FLUENCY | -- | 0 | 0 | 0 | 0 | 0 |
| VISOCONSTRUCTIVE PX (pentagons) | 0 (JUST ONE) | 0 | 0 | |||
| ORIENTATION | 5/10 | 1/10 | 0/10 | 2/10 | NV | NV |
| Boston Aphasia Severity Scale | 4/5 | 3/5 | 1/5 | 2/5 | 1/5 | 0/5 |
| Cut-Off Points | None Proposed | None Proposed | <0.083 | >2 pg/mL |
|---|---|---|---|---|
| ID | Aβ1–42 | Aβ1–40 | β-amyloid ratio (1–42/1–40) | p-tau |
| Case A | 18.31 | 318.71 | 0.057 | 2.39 |
| Case B | 2.96 | 265.42 | 0.011 | 2.91 |
| Cut-Off Points | <638 pg/mL | None Proposed | <0.069 | >56.5 pg/mL | >404 pg/mL |
|---|---|---|---|---|---|
| ID | Aβ1–42 | Aβ1–40 | β-amyloid ratio (1–42/1–40) | p-tau | t-tau |
| Case A | 988 | 10,693 | 0.092 | 122.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Minguillón Pereiro, A.M.; Quintáns Castro, B.; Ouro Villasante, A.; Aldrey Vázquez, J.M.; Cortés Hernández, J.; Aramburu-Núñez, M.; Arias Gómez, M.; Jiménez Martín, I.; Sobrino, T.; Pías-Peleteiro, J.M. PSEN2 Mutations May Mimic Frontotemporal Dementia: Two New Case Reports and a Review. Biomedicines 2024, 12, 1881. https://doi.org/10.3390/biomedicines12081881
Minguillón Pereiro AM, Quintáns Castro B, Ouro Villasante A, Aldrey Vázquez JM, Cortés Hernández J, Aramburu-Núñez M, Arias Gómez M, Jiménez Martín I, Sobrino T, Pías-Peleteiro JM. PSEN2 Mutations May Mimic Frontotemporal Dementia: Two New Case Reports and a Review. Biomedicines. 2024; 12(8):1881. https://doi.org/10.3390/biomedicines12081881
Chicago/Turabian StyleMinguillón Pereiro, Anxo Manuel, Beatriz Quintáns Castro, Alberto Ouro Villasante, José Manuel Aldrey Vázquez, Julia Cortés Hernández, Marta Aramburu-Núñez, Manuel Arias Gómez, Isabel Jiménez Martín, Tomás Sobrino, and Juan Manuel Pías-Peleteiro. 2024. "PSEN2 Mutations May Mimic Frontotemporal Dementia: Two New Case Reports and a Review" Biomedicines 12, no. 8: 1881. https://doi.org/10.3390/biomedicines12081881
APA StyleMinguillón Pereiro, A. M., Quintáns Castro, B., Ouro Villasante, A., Aldrey Vázquez, J. M., Cortés Hernández, J., Aramburu-Núñez, M., Arias Gómez, M., Jiménez Martín, I., Sobrino, T., & Pías-Peleteiro, J. M. (2024). PSEN2 Mutations May Mimic Frontotemporal Dementia: Two New Case Reports and a Review. Biomedicines, 12(8), 1881. https://doi.org/10.3390/biomedicines12081881