

Hydrogen Sulfide Modulation of Matrix Metalloproteinases and CD147/EMMPRIN: Mechanistic Pathways and Impact on Atherosclerosis Progression

 , ,
, ,
Abstract
1. Introduction
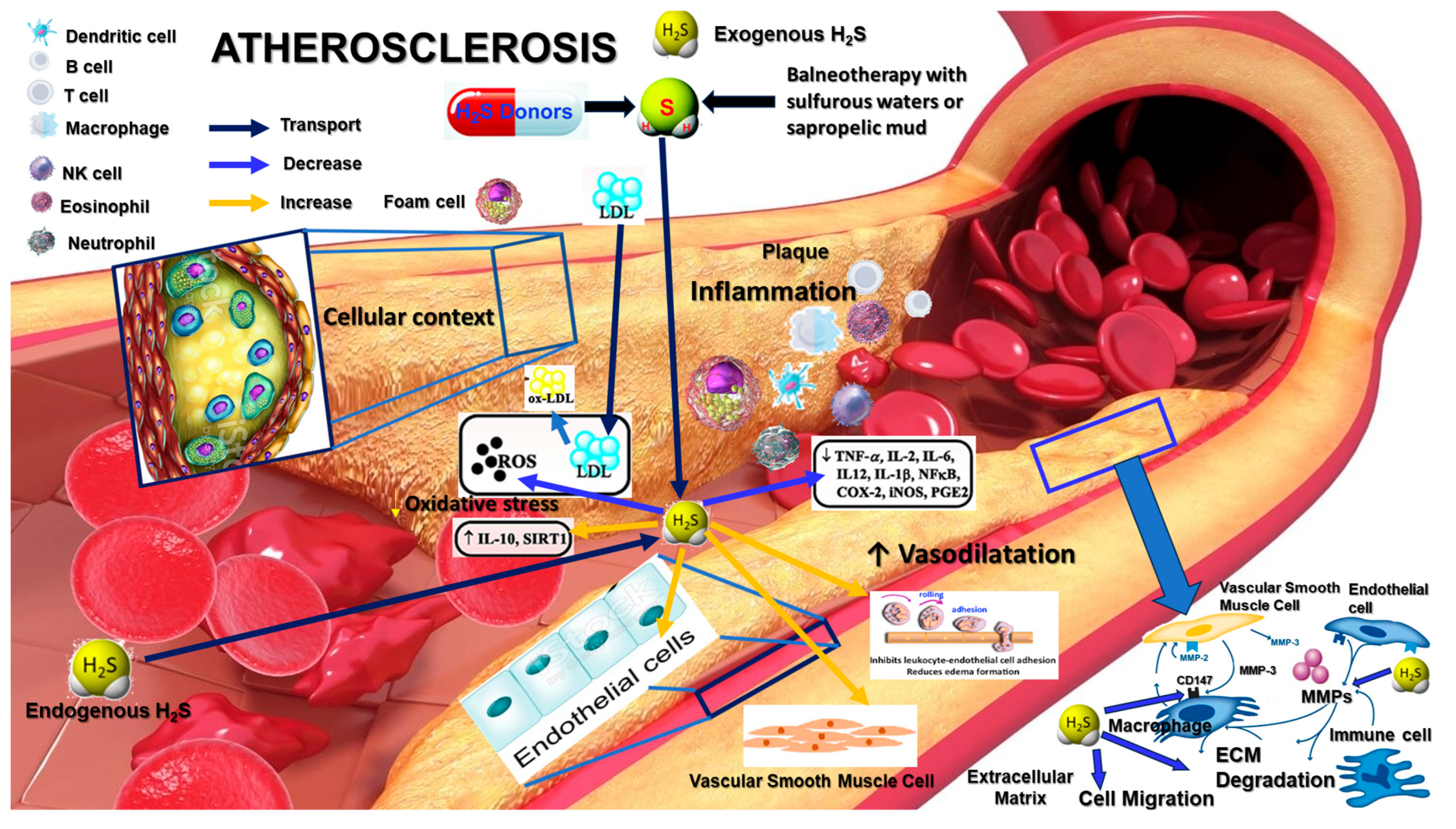
2. Role of Hydrogen Sulfide in Atherosclerosis
3. H2S Modulation of MMPs
4. CD147/EMMPRIN and Its Regulation by H2S
5. Potential Therapeutic Strategies Targeting H2S Pathways to Modulate MMP and CD147 Activity
6. Conclusions
Funding
Conflicts of Interest
References
- Rafieian-Kopaei, M.; Setorki, M.; Doudi, M.; Baradaran, A.; Nasri, H. Atherosclerosis: Process, indicators, risk factors and new hopes. Int. J. Prev. Med. 2014, 5, 927–946. [Google Scholar] [PubMed]
- Björkegren, J.L.; Lusis, A.J. Atherosclerosis: Recent developments. Cell 2022, 185, 1630–1645. [Google Scholar] [CrossRef]
- He, Z.; Luo, J.; Lv, M.; Li, Q.; Ke, W.; Niu, X.; Zhang, Z. Characteristics and evaluation of atherosclerotic plaques: An overview of state-of-the-art techniques. Front. Neurol. 2023, 14, 1159288. [Google Scholar] [CrossRef]
- Kong, P.; Cui, Z.-Y.; Huang, X.-F.; Zhang, D.-D.; Guo, R.-J.; Han, M. Inflammation and atherosclerosis: Signaling pathways and therapeutic intervention. Signal Transduct. Target. Ther. 2022, 7, 131. [Google Scholar] [CrossRef]
- Munteanu, C. Hydrogen Sulfide and Oxygen Homeostasis in Atherosclerosis: A Systematic Review from Molecular Biology to Therapeutic Perspectives. Int. J. Mol. Sci. 2023, 24, 8376. [Google Scholar] [CrossRef] [PubMed]
- Jebari-Benslaiman, S.; Galicia-García, U.; Larrea-Sebal, A.; Olaetxea, J.R.; Alloza, I.; Vandenbroeck, K.; Benito-Vicente, A.; Martín, C. Pathophysiology of Atherosclerosis. Int. J. Mol. Sci. 2022, 23, 3346. [Google Scholar] [CrossRef]
- Galkina, E.; Ley, K. Immune and inflammatory mechanisms of atherosclerosis. Annu. Rev. Immunol. 2009, 27, 165–197. [Google Scholar] [CrossRef]
- Medina-Leyte, D.J.; Zepeda-García, O.; Domínguez-Pérez, M.; González-Garrido, A.; Villarreal-Molina, T.; Jacobo-Albavera, L. Endothelial dysfunction, inflammation and coronary artery disease: Potential biomarkers and promising therapeutical ap-proaches. Int. J. Mol. Sci. 2021, 22, 3850. [Google Scholar] [CrossRef]
- Krüger-Genge, A.; Blocki, A.; Franke, R.-P.; Jung, F. Vascular endothelial cell biology: An update. Int. J. Mol. Sci. 2019, 20, 4411. [Google Scholar] [CrossRef] [PubMed]
- Drożdż, D.; Drożdż, M.; Wójcik, M. Endothelial dysfunction as a factor leading to arterial hypertension. Pediatr. Nephrol. 2023, 38, 2973–2985. [Google Scholar] [CrossRef] [PubMed]
- Rhoads, J.P.; Major, A.S. How oxidized low-density lipoprotein activates inflammatory responses. Crit. Rev. Immunol. 2018, 38, 333–342. [Google Scholar] [CrossRef]
- Khatana, C.; Saini, N.K.; Chakrabarti, S.; Saini, V.; Sharma, A.; Saini, R.V.; Saini, A.K. Mechanistic Insights into the Oxidized Low-Density Lipoprotein-Induced Atherosclerosis. Oxid. Med. Cell. Longev. 2020, 2020, 5245308. [Google Scholar] [CrossRef]
- Han, Z.; Liu, Q.; Li, H.; Zhang, M.; You, L.; Lin, Y.; Wang, K.; Gou, Q.; Wang, Z.; Zhou, S.; et al. The role of monocytes in thrombotic diseases: A review. Front. Cardiovasc. Med. 2023, 10, 1113827. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Herranz, L.; Albarrán-Juárez, J.; Bentzon, J.F. Mechanisms of fibrous cap formation in atherosclerosis. Front. Cardiovasc. Med. 2023, 10, 1254114. [Google Scholar] [CrossRef]
- Gierig, M.; Tragoudas, A.; Haverich, A.; Wriggers, P. Mechano-chemo-biological model of atherosclerosis formation based on the outside-in theory. Biomech. Model. Mechanobiol. 2023, 23, 539–552. [Google Scholar] [CrossRef]
- Jaminon, A.; Reesink, K.; Kroon, A.; Schurgers, L. The role of vascular smooth muscle cells in arterial remodeling: Focus on calcification-related processes. Int. J. Mol. Sci. 2019, 20, 5694. [Google Scholar] [CrossRef] [PubMed]
- Renna, N.F.; de las Heras, N.; Miatello, R.M. Pathophysiology of vascular remodeling in hypertension. Int. J. Hypertens. 2013, 2013, 808353. [Google Scholar] [CrossRef]
- Kowara, M.; Cudnoch-Jedrzejewska, A. Pathophysiology of Atherosclerotic Plaque Development-Contemporary Experience and New Directions in Research. Int. J. Mol. Sci. 2021, 20, 4337. [Google Scholar] [CrossRef]
- Henein, M.Y.; Vancheri, S.; Longo, G.; Vancheri, F. The Role of Inflammation in Cardiovascular Disease. Int. J. Mol. Sci. 2022, 23, 12906. [Google Scholar] [CrossRef]
- Watanabe, N.; Ikeda, U. Matrix Metalloproteinases and Atherosclerosis. Curr. Atheroscler. Rep. 2004, 6, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Cabral-Pacheco, G.A.; Garza-Veloz, I.; la Rosa, C.C.-D.; Ramirez-Acuña, J.M.; A Perez-Romero, B.; Guerrero-Rodriguez, J.F.; Martinez-Avila, N.; Martinez-Fierro, M.L. The roles of matrix metalloproteinases and their inhibitors in human diseases. Int. J. Mol. Sci. 2020, 21, 9739. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Kim, W.J. The Role of Matrix Metalloproteinase in Inflammation with a Focus on Infectious Diseases. Int. J. Mol. Sci. 2022, 23, 10546. [Google Scholar] [CrossRef]
- Lu, P.; Takai, K.; Weaver, V.M.; Werb, Z. Extracellular Matrix degradation and remodeling in development and disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a005058. [Google Scholar] [CrossRef] [PubMed]
- Hafiane, A. Vulnerable plaque, characteristics, detection, and potential therapies. J. Cardiovasc. Dev. Dis. 2019, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Tomaniak, M.; Katagiri, Y.; Modolo, R.; de Silva, R.; Khamis, R.Y.; Bourantas, C.V.; Torii, R.; Wentzel, J.J.; Gijsen, F.J.H.; van Soest, G.; et al. Vulnerable plaques and patients: State-of-the-art. Eur. Heart J. 2020, 41, 2997–3004. [Google Scholar] [CrossRef]
- Laronha, H.; Caldeira, J. Structure and function of human matrix metalloproteinases. Cells 2020, 9, 1076. [Google Scholar] [CrossRef]
- Cui, N.; Hu, M.; Khalil, R.A. Biochemical and Biological Attributes of Matrix Metalloproteinases. Prog. Mol. Biol. Transl. Sci. 2017, 147, 1–73. [Google Scholar] [CrossRef]
- Nyalali, A.M.K.; Leonard, A.U.; Xu, Y.; Li, H.; Zhou, J.; Zhang, X.; Rugambwa, T.K.; Shi, X.; Li, F. CD147: An integral and potential molecule to abrogate hallmarks of cancer. Front. Oncol. 2023, 13, 1238051. [Google Scholar] [CrossRef]
- Grass, G.D.; Toole, B.P. How, with whom and when: An overview of CD147-mediated regulatory networks influencing matrix metalloproteinase activity. Biosci. Rep. 2016, 36, e00283. [Google Scholar] [CrossRef]
- Xiong, L.; Edwards, C.K.; Zhou, L. The biological function and clinical utilization of CD147 in human diseases: A review of the current scientific literature. Int. J. Mol. Sci. 2014, 15, 17411–17441. [Google Scholar] [CrossRef]
- Iacono, K.T.; Brown, A.L.; Greene, M.I.; Saouaf, S.J. CD147 Immunoglobulin Superfamily Receptor Function and Role in Pathology. Exp. Mol. Pathol. 2007, 83, 283–295. [Google Scholar] [CrossRef]
- Muramatsu, T. Basigin (CD147), a multifunctional transmembrane glycoprotein with various binding partners. J. Biochem. 2016, 159, 481–490. [Google Scholar] [CrossRef]
- Ghandour, F.; Kassem, S.; Simanovich, E.; Rahat, M.A. Glucose Promotes EMMPRIN/CD147 and the Secretion of Pro-Angiogenic Factors in a Co-Culture System of Endothelial Cells and Monocytes. Biomedicines 2024, 12, 706. [Google Scholar] [CrossRef]
- Megha, K.; Joseph, X.; Akhil, V.; Mohanan, P. Cascade of immune mechanism and consequences of inflammatory disorders. Phytomedicine 2021, 91, 153712. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.-J.; Wang, H.; Zhang, C.; Zhang, T.-J.; Wei, H.-L.; Liu, Z.-K.; Ma, Y.-H.; Yang, Z.; He, Q.; Wang, L.-J.; et al. CD147 Sparks Atherosclerosis by Driving M1 Phenotype and Impairing Efferocytosis. Circ. Res. 2024, 134, 165–185. [Google Scholar] [CrossRef] [PubMed]
- Manicone, A.M.; McGuire, J.K. Matrix Metalloproteinases as Modulators of Inflammation. Semin. Cell Dev. Biol. 2007, 19, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Munteanu, C.; Turnea, M.A.; Rotariu, M. Hydrogen Sulfide: An Emerging Regulator of Oxidative Stress and Cellular Homeostasis—A Comprehensive One-Year Review. Antioxidants 2023, 12, 1737. [Google Scholar] [CrossRef]
- Batty, M.; Bennett, M.R.; Yu, E. The Role of Oxidative Stress in Atherosclerosis. Cells 2022, 11, 3843. [Google Scholar] [CrossRef]
- Lv, B.; Chen, S.; Tang, C.; Jin, H.; Du, J.; Huang, Y. Hydrogen sulfide and vascular regulation—An update. J. Adv. Res. 2021, 27, 85–97. [Google Scholar] [CrossRef]
- Sun, Y.; Huang, Y.; Zhang, R.; Chen, Q.; Chen, J.; Zong, Y.; Liu, J.; Feng, S.; Liu, A.D.; Holmberg, L.; et al. Hydrogen sulfide upregulates KATP channel expression in vascular smooth muscle cells of spontaneously hypertensive rats. J. Mol. Med. 2015, 93, 439–455. [Google Scholar] [CrossRef]
- Munteanu, C.; Călin, M.A.; Manea, D.; Popescu, C.; Iliescu, M.; Ionescu, E.V.; Stanciu, L.; Minea, M.; Oprea, C.; Oprea, D.; et al. Current data regarding homeostasis of tissues oxygenation in pathophysiological and therapeutic circumstances. Balneo PRM Res. J. 2023, 14, 565. [Google Scholar] [CrossRef]
- Pandey, T.; Pandey, V. Hydrogen sulfide (H2S) metabolism: Unraveling cellular regulation, disease implications, and therapeutic prospects for precision medicine. Nitric Oxide 2024, 144, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Munteanu, C.; Munteanu, D.; Onose, G. Hydrogen sulfide (H2S)—Therapeutic relevance in rehabilitation and balneotherapy Systematic literature review and meta-analysis based on the PRISMA paradig. Balneo PRM Res. J. 2021, 12, 176–195. [Google Scholar] [CrossRef]
- Munteanu, C.; Rotariu, M.; Turnea, M.; Dogaru, G.; Popescu, C.; Spînu, A.; Andone, I.; Postoiu, R.; Ionescu, E.V.; Oprea, C.; et al. Recent Advances in Molecular Research on Hydrogen Sulfide (H2S) Role in Diabetes Mellitus (DM)—A Systematic Review. Int. J. Mol. Sci. 2022, 23, 6720. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, Y.; Li, Y.; Li, L.; Xu, S.; Feng, X.; Liu, S. Hydrogen sulfide (H2S)-releasing compounds: Therapeutic potential in cardiovascular diseases. Front. Pharmacol. 2018, 9, 1066. [Google Scholar] [CrossRef]
- Andrés, C.M.C.; de la Lastra, J.M.P.; Juan, C.A.; Plou, F.J.; Pérez-Lebeña, E. Chemistry of Hydrogen Sulfide—Pathological and Physiological Functions in Mammalian Cells. Cells 2023, 12, 2684. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.; Yang, G.; Wang, R. A critical life-supporting role for cystathionine γ-lyase in the absence of dietary cysteine supply. Free. Radic. Biol. Med. 2011, 50, 1280–1287. [Google Scholar] [CrossRef]
- Ascenção, K.; Szabo, C. Emerging roles of cystathionine β-synthase in various forms of cancer. Redox Biol. 2022, 53, 102331. [Google Scholar] [CrossRef]
- Rao, S.P.; Dobariya, P.; Bellamkonda, H.; More, S.S. Role of 3-Mercaptopyruvate Sulfurtransferase (3-MST) in Physiology and Disease. Antioxidants 2023, 12, 603. [Google Scholar] [CrossRef]
- Liu, X.-Y.; Qian, L.-L.; Wang, R.-X. Hydrogen Sulfide-Induced Vasodilation: The Involvement of Vascular Potassium Channels. Front. Pharmacol. 2022, 13, 911704. [Google Scholar] [CrossRef]
- Corvino, A.; Frecentese, F.; Magli, E.; Perissutti, E.; Santagada, V.; Scognamiglio, A.; Caliendo, G.; Fiorino, F.; Severino, B. Trends in H2S-donors chemistry and their effects in cardiovascular diseases. Antioxidants 2021, 10, 429. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Vitvitsky, V.; Sethaudom, A.; Singhal, R.; Solanki, S.; Alibeckoff, S.; Hiraki, H.L.; Bell, H.N.; Andren, A.; Baker, B.M.; et al. Sulfide oxidation promotes hypoxic angiogenesis and neovascularization. Nat. Chem. Biol. 2024. [Google Scholar] [CrossRef] [PubMed]
- Gorini, F.; Del Turco, S.; Sabatino, L.; Gaggini, M.; Vassalle, C. H2S as a bridge linking inflammation, oxidative stress and endothelial biology: A possible defense in the fight against SARS-CoV-2 infection? Biomedicines 2021, 9, 1107. [Google Scholar] [CrossRef]
- He, K.; Zhang, H.; Tan, B.; Song, C.; Liang, Z.; Zhang, L.; Tian, D.; Xiao, L.; Xue, H.; Guo, Q.; et al. Hydrogen Sulfide Ameliorates Heart Aging by Downregulating Matrix Metalloproteinase-9. Cardiovasc. Drugs Ther. 2024. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, N.; Givvimani, S.; Qipshidze, N.; Kundu, S.; Kapoor, S.; Vacek, J.C.; Tyagi, S.C. Hydrogen sulfide mitigates matrix metalloproteinase-9 activity and neurovascular permeability in hyperhomocysteinemic mice. Neurochem. Int. 2010, 56, 301–307. [Google Scholar] [CrossRef]
- Wang, M.; Tang, J.; Zhang, S.; Pang, K.; Zhao, Y.; Liu, N.; Huang, J.; Kang, J.; Dong, S.; Li, H.; et al. Exogenous H2S initiating Nrf2/GPx4/GSH pathway through promoting Syvn1-Keap1 interaction in diabetic hearts. Cell Death Discov. 2023, 9, 394. [Google Scholar] [CrossRef]
- Zhang, C.-Y.; Li, X.-H.; Zhang, T.; Fu, J.; Cui, X.-D. Hydrogen sulfide suppresses the expression of MMP-8, MMP-13, and TIMP-1 in left ventricles of rats with cardiac volume overload. Acta Pharmacol. Sin. 2013, 34, 1301–1309. [Google Scholar] [CrossRef]
- Umezawa, K.; Lin, Y. Inhibition of matrix metalloproteinase expression and cellular invasion by NF-κB inhibitors of microbial origin. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2020, 1868, 140412. [Google Scholar] [CrossRef]
- Zhao, H.-L.; Wu, B.-Q.; Luo, Y.; Zhang, W.-Y.; Hao, Y.-L.; Liang, J.-J.; Fang, F.; Liu, W.; Chen, X.-H. Exogenous hydrogen sulfide ameliorates high glucose-induced myocardial injury & inflammation via the CIRP-MAPK signaling pathway in H9c2 cardiac cells. Life Sci. 2018, 208, 315–324. [Google Scholar] [CrossRef]
- Dogaru, B.G.; Munteanu, C. The Role of Hydrogen Sulfide (H2S) in Epigenetic Regulation of Neurodegenerative Diseases: A Systematic Review. Int. J. Mol. Sci. 2023, 24, 12555. [Google Scholar] [CrossRef]
- Jiang, Y.; Qian, H.-Y. Transcription factors: Key regulatory targets of vascular smooth muscle cell in atherosclerosis. Mol. Med. 2023, 29, 2. [Google Scholar] [CrossRef]
- Song, Y.; Cao, S.; Sun, X.; Chen, G. The interplay of hydrogen sulfide and microRNAs in cardiovascular diseases: Insights and future perspectives. Mamm. Genome 2024, 35, 309–323. [Google Scholar] [CrossRef] [PubMed]
- Coates-Park, S.; Lazaroff, C.; Gurung, S.; Rich, J.; Colladay, A.; O’Neill, M.; Butler, G.S.; Overall, C.M.; Stetler-Stevenson, W.G.; Peeney, D. Tissue inhibitors of metalloproteinases are proteolytic targets of matrix metalloproteinase 9. Matrix Biol. 2023, 123, 59–70. [Google Scholar] [CrossRef]
- Grootaert, M.O.J.; Bennett, M.R. Vascular smooth muscle cells in atherosclerosis: Time for a re-assessment. Cardiovasc. Res. 2021, 117, 2326–2339. [Google Scholar] [CrossRef]
- Xu, F.; Chen, H.; Zhou, C.; Zang, T.; Wang, R.; Shen, S.; Li, C.; Yu, Y.; Pei, Z.; Shen, L.; et al. Targeting deubiquitinase OTUB1 protects vascular smooth muscle cells in atherosclerosis by modulating PDGFRβ. Front. Med. 2024, 18, 465–483. [Google Scholar] [CrossRef] [PubMed]
- Blagov, A.V.; Markin, A.M.; Bogatyreva, A.I.; Tolstik, T.V.; Sukhorukov, V.N.; Orekhov, A.N. The Role of Macrophages in the Pathogenesis of Atherosclerosis. Cells 2023, 12, 522. [Google Scholar] [CrossRef]
- Moore, K.J.; Sheedy, F.J.; Fisher, E.A. Macrophages in atherosclerosis: A dynamic balance. Nat. Rev. Immunol. 2013, 13, 709–721. [Google Scholar] [CrossRef]
- Paul, B.D.; Snyder, S.H. H2S signalling through protein sulfhydration and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 499–507. [Google Scholar] [CrossRef]
- Pang, P.-P.; Zhang, H.-Y.; Zhang, D.-C.; Tang, J.-X.; Gong, Y.; Guo, Y.-C.; Zheng, C.-B. Investigating the impact of protein S-sulfhydration modification on vascular diseases: A comprehensive review. Eur. J. Pharmacol. 2024, 966, 176345. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Teng, H.; Yang, G.; Wu, L.; Wang, R. Hydrogen sulfide inhibits the translational expression of hypoxia-inducible factor-1α. Br. J. Pharmacol. 2012, 167, 1492–1505. [Google Scholar] [CrossRef]
- Guindolet, D.; Gabison, E.E. Role of CD147 (EMMPRIN/Basigin) in Tissue Remodeling. Anat. Rec. 2020, 303, 1584–1589. [Google Scholar] [CrossRef]
- Asgari, R.; Vaisi-Raygani, A.; Aleagha, M.S.E.; Mohammadi, P.; Bakhtiari, M.; Arghiani, N. CD147 and MMPs as key factors in physiological and pathological processes. Biomed. Pharmacother. 2023, 157, 113983. [Google Scholar] [CrossRef]
- Sun, J.; Hemler, M.E. Regulation of MMP-1 and MMP-2 Production through CD147/Extracellular Matrix Metalloproteinase Inducer Interactions. Cancer Res. 2001, 61, 2276–2281. Available online: http://aacrjournals.org/cancerres/article-pdf/61/5/2276/2493282/ch050102276.pdf (accessed on 23 July 2024). [PubMed]
- Wang, C.; Jin, R.; Zhu, X.; Yan, J.; Li, G. Function of CD147 in Atherosclerosis and Atherothrombosis. J. Cardiovasc. Transl. Res. 2015, 8, 59–66. [Google Scholar] [CrossRef]
- Huang, W.; Luo, W.-J.; Zhu, P.; Tang, J.; Yu, X.-L.; Cui, H.-Y.; Wang, B.; Zhang, Y.; Jiang, J.-L.; Chen, Z.-N. Modulation of CD147-induced matrix metalloproteinase activity: Role of CD147 N-glycosylation. Biochem. J. 2012, 449, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Heinzmann, D.; Noethel, M.; von Ungern-Sternberg, S.; Mitroulis, I.; Gawaz, M.; Chavakis, T.; May, A.E.; Seizer, P. CD147 is a novel interaction partner of integrin αMβ2 mediating leukocyte and platelet adhesion. Biomolecules 2020, 10, 541. [Google Scholar] [CrossRef]
- Yurchenko, V.; Constant, S.; Eisenmesser, E.; Bukrinsky, M. Cyclophilin–CD147 interactions: A new target for anti-inflammatory therapeutics. Clin. Exp. Immunol. 2010, 160, 305–317. [Google Scholar] [CrossRef]
- Landras, A.; de Moura, C.R.; Jouenne, F.; Lebbe, C.; Menashi, S.; Mourah, S. CD147 is a promising target of tumor progression and a prognostic biomarker. Cancers 2019, 11, 1803. [Google Scholar] [CrossRef]
- Kaur, S.; Schwartz, A.L.; Miller, T.W.; Roberts, D.D. CD47-Dependent regulation of H2S biosynthesis and signaling in T cells. Methods Enzymol. 2015, 555, 145–168. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Rao, D.; Jin, Q.; Lai, M.; Zhang, J.; Lai, Z.; Shen, H.; Zhong, T. Role of CD147 in the development and diagnosis of hepatocellular carcinoma. Front. Immunol. 2023, 14, 1149931. [Google Scholar] [CrossRef]
- Chen, H.-J.; Qian, L.; Li, K.; Qin, Y.-Z.; Zhou, J.-J.; Ji, X.-Y.; Wu, D.-D. Hydrogen sulfide-induced post-translational modification as a potential drug target. Genes Dis. 2023, 10, 1870–1882. [Google Scholar] [CrossRef]
- Shefa, U.; Kim, M.-S.; Jeong, N.Y.; Jung, J. Antioxidant and Cell-Signaling Functions of Hydrogen Sulfide in the Central Nervous System. Oxidative Med. Cell. Longev. 2018, 2018, 1873962. [Google Scholar] [CrossRef] [PubMed]
- Sen, N. Functional and Molecular Insights of Hydrogen Sulfide Signaling and Protein Sulfhydration. J. Mol. Biol. 2017, 429, 543–561. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Bai, Z.; Zhu, L.; Liang, Y.; Fan, X.; Li, J.; Wen, H.; Shi, T.; Zhao, Q.; Wang, Z. Hydrogen sulfide donors: Therapeutic potential in anti-atherosclerosis. Eur. J. Med. Chem. 2020, 205, 112665. [Google Scholar] [CrossRef] [PubMed]
- Cirino, G.; Szabo, C.; Papapetropoulos, A. Physiological roles of hydrogen sulfide in mammalian cells, tissues, and organs. Physiol. Rev. 2023, 103, 31–276. [Google Scholar] [CrossRef] [PubMed]
- Townsend, D.M.; Tew, K.D.; Tapiero, H. Sulfur containing amino acids and human disease. Biomed. Pharmacother. 2003, 58, 47–55. [Google Scholar] [CrossRef]
- Kolluru, G.K.; Shackelford, R.E.; Shen, X.; Dominic, P.; Kevil, C.G. Sulfide regulation of cardiovascular function in health and disease. Nat. Rev. Cardiol. 2023, 20, 109–125. [Google Scholar] [CrossRef]
- Powell, C.R.; Dillon, K.M.; Matson, J.B. A review of hydrogen sulfide (H2S) donors: Chemistry and potential therapeutic applications. Biochem. Pharmacol. 2018, 149, 110–123. [Google Scholar] [CrossRef]
- Rose, P.; Moore, P.K.; Zhu, Y.Z. H2S biosynthesis and catabolism: New insights from molecular studies. Cell. Mol. Life Sci. 2017, 74, 1391–1412. [Google Scholar] [CrossRef]
- Olas, B. Hydrogen sulfide in signaling pathways. Clin. Chim. Acta 2015, 439, 212–218. [Google Scholar] [CrossRef]
- Rong, F.; Wang, T.; Zhou, Q.; Peng, H.; Yang, J.; Fan, Q.; Li, P. Intelligent polymeric hydrogen sulfide delivery systems for therapeutic applications. Bioact. Mater. 2023, 19, 198–216. [Google Scholar] [CrossRef]
- Gemici, B.; Elsheikh, W.; Feitosa, K.B.; Costa, S.K.; Muscara, M.N.; Wallace, J.L. H2S-releasing drugs: Anti-inflammatory, cytoprotective and chemopreventative potential. Nitric Oxide 2015, 46, 25–31. [Google Scholar] [CrossRef]
- Dillon, K.M.; Carrazzone, R.J.; Matson, J.B.; Kashfi, K. The evolving landscape for cellular nitric oxide and hydrogen sulfide delivery systems: A new era of customized medications. Biochem. Pharmacol. 2020, 176, 113931. [Google Scholar] [CrossRef]
- Saigusa, D.; Matsukawa, N.; Hishinuma, E.; Koshiba, S. Identification of biomarkers to diagnose diseases and find adverse drug reactions by metabolomics. Drug Metab. Pharmacokinet. 2021, 37, 100373. [Google Scholar] [CrossRef] [PubMed]
- Qiao, X.; Gu, Y.; Yu, J.; Wang, J.; Liu, X.; Gu, M.; Ma, L.; Jia, Y.; Zhang, S. The Combination of CD147 and MMP-9 Serum Levels Is Identified as Novel Chemotherapy Response Markers of Advanced Non-Small-Cell Lung Cancer. Dis. Markers 2020, 2020, 8085053. [Google Scholar] [CrossRef]
- Wang, Y.-Z.; Ngowi, E.E.; Wang, D.; Qi, H.-W.; Jing, M.-R.; Zhang, Y.-X.; Cai, C.-B.; He, Q.-L.; Khattak, S.; Khan, N.H.; et al. The potential of hydrogen sulfide donors in treating cardiovascular diseases. Int. J. Mol. Sci. 2021, 22, 2194. [Google Scholar] [CrossRef]
- Mani, S.; Untereiner, A.; Wu, L.; Wang, R. Hydrogen sulfide and the pathogenesis of atherosclerosis. Antioxidants Redox Signal. 2014, 20, 805–817. [Google Scholar] [CrossRef]
- Wallace, J.L.; Wang, R. Hydrogen sulfide-based therapeutics: Exploiting a unique but ubiquitous gasotransmitter. Nat. Rev. Drug Discov. 2015, 14, 329–345. [Google Scholar] [CrossRef]
- Maldonado, C.S.; Weir, A.; Rumbeiha, W.K. A comprehensive review of treatments for hydrogen sulfide poisoning: Past, present, and future. Toxicol. Mech. Methods 2023, 33, 183–196. [Google Scholar] [CrossRef]
- Han, Y.; Shang, Q.; Yao, J.; Ji, Y. Hydrogen sulfide: A gaseous signaling molecule modulates tissue homeostasis: Implications in ophthalmic diseases. Cell Death Dis. 2019, 10, 293. [Google Scholar] [CrossRef]
- Al Ragib, A.; Chakma, R.; Dewan, K.; Islam, T.; Kormoker, T.; Idris, A.M. Current advanced drug delivery systems: Challenges and potentialities. J. Drug Deliv. Sci. Technol. 2022, 76, 103727. [Google Scholar] [CrossRef]
- Bordbar-Khiabani, A.; Gasik, M. Smart Hydrogels for Advanced Drug Delivery Systems. Int. J. Mol. Sci. 2022, 23, 3665. [Google Scholar] [CrossRef] [PubMed]
- Wojtasińska, A.; Frąk, W.; Lisińska, W.; Sapeda, N.; Młynarska, E.; Rysz, J.; Franczyk, B. Novel Insights into the Molecular Mechanisms of Atherosclerosis. Int. J. Mol. Sci. 2023, 24, 13434. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Mechanism | Effect on MMPs | Effect on CD147/EMMPRIN | Outcome on Atherosclerosis | Ref. No. |
|---|---|---|---|---|
| Direct Inhibition of MMP Activity | H₂S directly interacts with the catalytic zinc ion in MMPs, inhibiting their proteolytic activity. | Not directly involved. | Stabilizes plaques by preventing excessive ECM degradation. | [54] |
| Regulation of Oxidative Stress | H₂S reduces oxidative stress by scavenging reactive oxygen species (ROS), preventing ROS-mediated activation of MMPs. | Reduces oxidative stress, indirectly decreasing CD147 expression and activity. | Reduces inflammation and ECM degradation, enhancing plaque stability. | [5,37,38,53] |
| Modulation of NF-κB Signaling Pathway | H₂S inhibits NF-κB activation, leading to decreased transcription of MMP genes such as MMP-9. | Inhibits NF-κB activation, reducing CD147 expression and MMP induction. | Lowers inflammation, decreases ECM degradation, and stabilizes plaques. | [37,58] |
| S-Sulfhydration (Post-Translational Modification) | Potentially alters MMP activity by modifying cysteine residues. | Modifies CD147, potentially affecting its interaction with MMPs and inflammatory signaling molecules. | Modulates protein function and signaling, contributing to reduced ECM degradation and inflammation. | [68,69] |
| Inhibition of Smooth Muscle Cell (SMC) Migration and Proliferation | Indirectly reduces MMP activity by inhibiting SMC migration and proliferation, key processes in plaque formation. | Not directly involved. | Limits plaque growth and promotes stability by maintaining ECM integrity. | [16,40,61,64,65] |
| Interaction with Hypoxia-Inducible Factor 1-alpha (HIF-1α) | Not directly involved. | H₂S modulates HIF-1α, reducing hypoxia-induced CD147 expression. | Decreases hypoxia-related inflammation and ECM degradation. | [28,70] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Munteanu, C.; Galaction, A.I.; Poștaru, M.; Rotariu, M.; Turnea, M.; Blendea, C.D. Hydrogen Sulfide Modulation of Matrix Metalloproteinases and CD147/EMMPRIN: Mechanistic Pathways and Impact on Atherosclerosis Progression. Biomedicines 2024, 12, 1951. https://doi.org/10.3390/biomedicines12091951
Munteanu C, Galaction AI, Poștaru M, Rotariu M, Turnea M, Blendea CD. Hydrogen Sulfide Modulation of Matrix Metalloproteinases and CD147/EMMPRIN: Mechanistic Pathways and Impact on Atherosclerosis Progression. Biomedicines. 2024; 12(9):1951. https://doi.org/10.3390/biomedicines12091951
Chicago/Turabian StyleMunteanu, Constantin, Anca Irina Galaction, Mădălina Poștaru, Mariana Rotariu, Marius Turnea, and Corneliu Dan Blendea. 2024. "Hydrogen Sulfide Modulation of Matrix Metalloproteinases and CD147/EMMPRIN: Mechanistic Pathways and Impact on Atherosclerosis Progression" Biomedicines 12, no. 9: 1951. https://doi.org/10.3390/biomedicines12091951
APA StyleMunteanu, C., Galaction, A. I., Poștaru, M., Rotariu, M., Turnea, M., & Blendea, C. D. (2024). Hydrogen Sulfide Modulation of Matrix Metalloproteinases and CD147/EMMPRIN: Mechanistic Pathways and Impact on Atherosclerosis Progression. Biomedicines, 12(9), 1951. https://doi.org/10.3390/biomedicines12091951