

ALCAT1-Mediated Pathological Cardiolipin Remodeling and PLSCR3-Mediated Cardiolipin Transferring Contribute to LPS-Induced Myocardial Injury
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Viability
2.3. Measurement of Oxidative Stress
2.4. Immunofluorescent Staining
2.5. Electron Microscopy
2.6. Western Blot Analysis
2.7. Statistical Analysis
3. Results
3.1. The Effect of LPS on Cellular Injury and Oxidative Stress in the HL-1 Cell Line
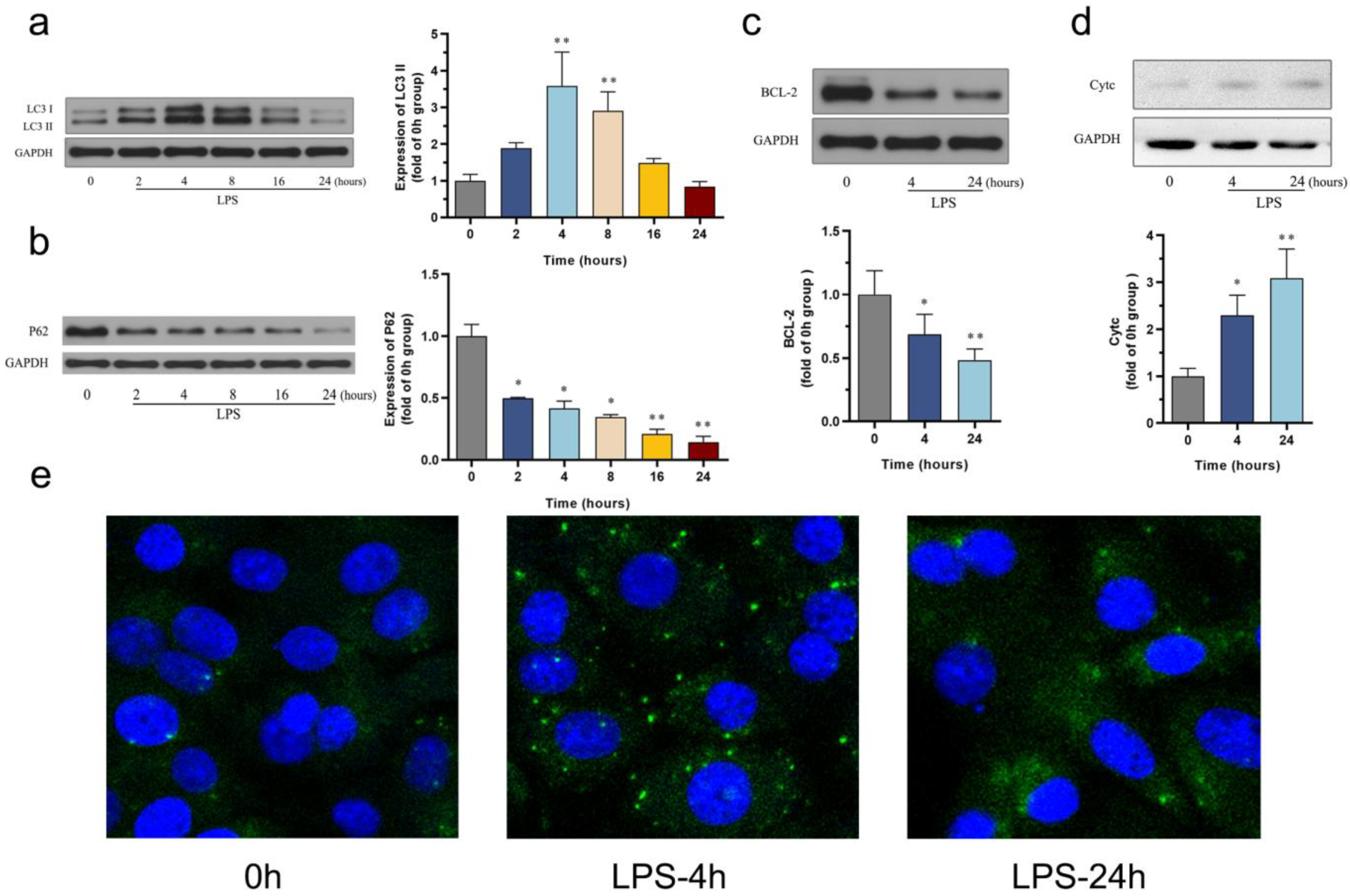
3.2. The Effect of LPS on Autophagy and Apoptosis in the HL-1 Cell Line
3.3. The Effect of LPS on Mitophagy in the HL-1 Cell Line
3.4. The Effect of LPS on CL Remodeling and CL Externalization in the HL-1 Cell Line
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.-D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). J. Am. Med. Assoc. 2016, 315, 801–810. [Google Scholar] [CrossRef]
- Hollenberg, S.M.; Singer, M. Pathophysiology of sepsis-induced cardiomyopathy. Nat. Rev. Cardiol. 2021, 18, 424–434. [Google Scholar] [CrossRef] [PubMed]
- Parker, M.M.; Shelhamer, J.H.; Bacharach, S.L.; Green, M.V.; Natanson, C.; Frederick, T.M.; Damske, B.A.; Parrillo, J.E. Profound but reversible myocardial depression in patients with septic shock. Ann. Intern. Med. 1984, 100, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Stanzani, G.; Duchen, M.R.; Singer, M. The role of mitochondria in sepsis-induced cardiomyopathy. Biochim. Biophys. Acta Mol. Basis. Dis. 2019, 1865, 759–773. [Google Scholar] [CrossRef]
- Chen, T.; Ye, L.; Zhu, J.; Tan, B.; Yi, Q.; Sun, Y.; Xie, Q.; Xiang, H.; Wang, R.; Tian, J.; et al. Inhibition of pyruvate dehydrogenase kinase 4 attenuates myocardial and mitochondrial injury in sepsis-induced cardiomyopathy. J. Infect. Dis. 2023, 229, 1178–1188. [Google Scholar] [CrossRef]
- Lin, Y.; Xu, Y.; Zhang, Z. Sepsis-Induced Myocardial Dysfunction (SIMD): The Pathophysiological Mechanisms and Therapeutic Strategies Targeting Mitochondria. Inflammation 2020, 43, 1184–1200. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Long, F.; Kang, R.; Klionsky, D.J.; Yang, M.; Tang, D. The lipid basis of cell death and autophagy. Autophagy 2024, 20, 469–488. [Google Scholar] [CrossRef]
- Oemer, G.; Koch, J.; Wohlfarter, Y.; Alam, M.T.; Lackner, K.; Sailer, S.; Neumann, L.; Lindner, H.H.; Watschinger, K.; Haltmeier, M.; et al. Phospholipid Acyl Chain Diversity Controls the Tissue-Specific Assembly of Mitochondrial Cardiolipins. Cell Rep. 2020, 30, 4281–4291.e4. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Shi, Y. An upstream open reading frame (5′-uORF) links oxidative stress to translational control of ALCAT1 through phosphorylation of eIF2α. Free. Radic. Biol. Med. 2024, 214, 129–136. [Google Scholar] [CrossRef]
- Liu, X.; Ye, B.; Miller, S.; Yuan, H.; Zhang, H.; Tian, L.; Nie, J.; Imae, R.; Arai, H.; Li, Y.; et al. Ablation of ALCAT1 mitigates hypertrophic cardiomyopathy through effects on oxidative stress and mitophagy. Mol. Cell. Biol. 2012, 32, 4493–4504. [Google Scholar] [CrossRef]
- Jia, D.; Zhang, J.; Nie, J.; Andersen, J.-P.; Rendon, S.; Zheng, Y.; Liu, X.; Tian, Z.; Shi, Y. Cardiolipin remodeling by ALCAT1 links hypoxia to coronary artery disease by promoting mitochondrial dysfunction. Mol. Ther. 2021, 29, 3498–3511. [Google Scholar] [CrossRef]
- Zhang, J.; Shi, Y. In Search of the Holy Grail: Toward a Unified Hypothesis on Mitochondrial Dysfunction in Age-Related Diseases. Cells 2022, 11, 1906. [Google Scholar] [CrossRef]
- Ren, W.; Xu, Z.; Pan, S.; Ma, Y.; Li, H.; Wu, F.; Bo, W.; Cai, M.; Tian, Z. Irisin and ALCAT1 mediated aerobic exercise-alleviated oxidative stress and apoptosis in skeletal muscle of mice with myocardial infarction. Free. Radic. Biol. Med. 2022, 193 Pt 2, 526–537. [Google Scholar] [CrossRef]
- Rieger, B.; Krajčová, A.; Duwe, P.; Busch, K.B. ALCAT1 Overexpression Affects Supercomplex Formation and Increases ROS in Respiring Mitochondria. Oxid. Med. Cell. Longev. 2019, 2019, 9186469. [Google Scholar] [CrossRef] [PubMed]
- Palanirajan, S.K.; Gummadi, S.N. Heavy-Metals-Mediated Phospholipids Scrambling by Human Phospholipid Scramblase 3: A Probable Role in Mitochondrial Apoptosis. Chem. Res. Toxicol. 2020, 33, 553–564. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, M.; Bharambe, N.; Shang, Y.; Lu, B.; Mandal, A.; Mohan, P.M.; Wang, R.; Boatz, J.C.; Galvez, J.M.M.; Shnyrova, A.V.; et al. NMR identification of a conserved Drp1 cardiolipin-binding motif essential for stress-induced mitochondrial fission. Proc. Natl. Acad. Sci. USA 2021, 118, e2023079118. [Google Scholar] [CrossRef] [PubMed]
- Iriondo, M.N.; Etxaniz, A.; Varela, Y.R.; Ballesteros, U.; Hervás, J.H.; Montes, L.R.; Goñi, F.M.; Alonso, A. LC3 subfamily in cardiolipin-mediated mitophagy: A comparison of the LC3A, LC3B and LC3C homologs. Autophagy 2022, 18, 2985–3003. [Google Scholar] [CrossRef]
- Liu, Y.J.; McIntyre, R.L.; Janssens, G.E.; Houtkooper, R.H. Mitochondrial fission and fusion: A dynamic role in aging and potential target for age-related disease. Mech. Ageing Dev. 2020, 186, 111212. [Google Scholar] [CrossRef]
- Wang, C.; Yuan, J.; Du, J. Resveratrol alleviates acute lung injury through regulating PLSCR-3-mediated mitochondrial dysfunction and mitophagy in a cecal ligation and puncture model. Eur. J. Pharmacol. 2021, 913, 174643. [Google Scholar] [CrossRef]
- Palanirajan, S.K.; Gummadi, S.N. Phospholipid scramblase 3: A latent mediator connecting mitochondria and heavy metal apoptosis. Cell Biochem. Biophys. 2023, 81, 443–458. [Google Scholar] [CrossRef]
- Zhou, H.; Toan, S.; Zhu, P.; Wang, J.; Ren, J.; Zhang, Y. DNA-PKcs promotes cardiac ischemia reperfusion injury through mitigating BI-1-governed mitochondrial homeostasis. Basic Res. Cardiol. 2020, 115, 11. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Shi, C.; Hu, S.; Zhu, H.; Ren, J.; Chen, Y. BI1 is associated with microvascular protection in cardiac ischemia reperfusion injury via repressing Syk-Nox2-Drp1-mitochondrial fission pathways. Angiogenesis 2018, 21, 599–615. [Google Scholar] [CrossRef] [PubMed]
- Eleftheriadis, T.; Pissas, G.; Liakopoulos, V.; Stefanidis, I. Cytochrome c as a Potentially Clinical Useful Marker of Mitochondrial and Cellular Damage. Front. Immunol. 2016, 7, 279. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Dai, Z.; Li, J.; Wang, J.; Zhu, H.; Chang, X.; Wang, Y. TMBIM6 prevents VDAC1 multimerization and improves mitochondrial quality control to reduce sepsis-related myocardial injury. Metabolism 2023, 140, 155383. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Cai, Y.; Zang, Q.S. Cardiac Autophagy in Sepsis. Cells 2019, 8, 141. [Google Scholar] [CrossRef]
- Ikeda, S.; Zablocki, D.; Sadoshima, J. The role of autophagy in death of cardiomyocytes. J. Mol. Cell. Cardiol. 2022, 165, 1–8. [Google Scholar] [CrossRef]
- Yang, H.; Zhang, Z. Sepsis-induced myocardial dysfunction: The role of mitochondrial dysfunction. Inflamm. Res. 2021, 70, 379–387. [Google Scholar] [CrossRef]
- Harrington, J.S.; Ryter, S.W.; Plataki, M.; Price, D.R.; Choi, A. Mitochondria in health, disease, and aging. Physiol. Rev. 2023, 103, 2349–2422. [Google Scholar] [CrossRef]
- Cao, J.; Liu, Y.; Lockwood, J.; Burn, P.; Shi, Y. A novel cardiolipin-remodeling pathway revealed by a gene encoding an endoplasmic reticulum-associated acyl-CoA:lysocardiolipin acyltransferase (ALCAT1) in mouse. J. Biol. Chem. 2004, 279, 31727–31734. [Google Scholar] [CrossRef]
- Liu, N.; Zhu, Y.; Song, W.; Ren, W.; Tian, Z. Cardioprotection Attributed to Aerobic Exercise-Mediated Inhibition of ALCAT1 and Oxidative Stress-Induced Apoptosis in MI Rats. Biomedicines 2022, 10, 2250. [Google Scholar] [CrossRef]
- Li, J.; Romestaing, C.; Han, X.; Li, Y.; Hao, X.; Wu, Y.; Sun, C.; Liu, X.; Jefferson, L.S.; Xiong, J.; et al. Cardiolipin remodeling by ALCAT1 links oxidative stress and mitochondrial dysfunction to obesity. Cell Metab. 2010, 12, 154–165. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Yang, J.; Yang, K.; Zhongdan, Z.; Abendschein, D.R.; Gross, R.W. Alterations in myocardial cardiolipin content and composition occur at the very earliest stages of diabetes: A shotgun lipidomics study. Biochemistry 2007, 46, 6417–6428. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, X.; Nie, J.; Zhang, J.; Kimball, S.R.; Zhang, H.; Zhang, W.J.; Jefferson, L.S.; Cheng, Z.; Ji, Q.; et al. ALCAT1 controls mitochondrial etiology of fatty liver diseases, linking defective mitophagy to steatosis. Hepatology 2015, 61, 486–496. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Zhang, J.; Qi, S.; Liu, Z.; Zhang, X.; Zheng, Y.; Andersen, J.; Zhang, W.; Strong, R.; Martinez, P.A.; et al. Cardiolipin remodeling by ALCAT1 links mitochondrial dysfunction to Parkinson’s diseases. Aging Cell 2019, 18, e12941. [Google Scholar] [CrossRef]
- Liu, J.; Dai, Q.; Chen, J.; Durrant, D.; Freeman, A.; Liu, T.; Grossman, D.; Lee, R.M. Phospholipid scramblase 3 controls mitochondrial structure, function, and apoptotic response. Mol. Cancer Res. 2003, 1, 892–902. [Google Scholar]
- Ndebele, K.; Gona, P.; Jin, T.-G.; Benhaga, N.; Chalah, A.; Degli-Esposti, M.; Khosravi-Far, R. Tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) induced mitochondrial pathway to apoptosis and caspase activation is potentiated by phospholipid scramblase-3. Apoptosis 2008, 13, 845–856. [Google Scholar] [CrossRef]
- Vendrov, A.E.; Xiao, H.; Lozhkin, A.; Hayami, T.; Hu, G.; Brody, M.J.; Sadoshima, J.; Zhang, Y.-Y.; Runge, M.S.; Madamanchi, N.R. Cardiomyocyte NOX4 regulates resident macrophage-mediated inflammation and diastolic dysfunction in stress cardiomyopathy. Redox Biol. 2023, 67, 102937. [Google Scholar] [CrossRef]
- Münzel, T.; Templin, C.; Cammann, V.L.; Hahad, O. Takotsubo Syndrome: Impact of endothelial dysfunction and oxidative stress. Free. Radic. Biol. Med. 2021, 169, 216–223. [Google Scholar] [CrossRef]
- Wang, T.; Xiong, T.; Yang, Y.; Zuo, B.; Chen, X.; Wang, D. Metabolic remodeling in takotsubo syndrome. Front. Cardiovasc. Med. 2022, 9, 1060070. [Google Scholar] [CrossRef]
- Miyamoto, S. Hippo signaling pathway and mitochondrial dysfunction in Takotsubo syndrome. Am. J. Physiol. Heart Circ. Physiol. 2023, 324, H525–H527. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, D.; Wang, C.; Feng, X.; Hu, L.; Wang, B.; Hu, X.; Wu, J. ALCAT1-Mediated Pathological Cardiolipin Remodeling and PLSCR3-Mediated Cardiolipin Transferring Contribute to LPS-Induced Myocardial Injury. Biomedicines 2024, 12, 2013. https://doi.org/10.3390/biomedicines12092013
Han D, Wang C, Feng X, Hu L, Wang B, Hu X, Wu J. ALCAT1-Mediated Pathological Cardiolipin Remodeling and PLSCR3-Mediated Cardiolipin Transferring Contribute to LPS-Induced Myocardial Injury. Biomedicines. 2024; 12(9):2013. https://doi.org/10.3390/biomedicines12092013
Chicago/Turabian StyleHan, Dong, Chenyang Wang, Xiaojing Feng, Li Hu, Beibei Wang, Xinyue Hu, and Jing Wu. 2024. "ALCAT1-Mediated Pathological Cardiolipin Remodeling and PLSCR3-Mediated Cardiolipin Transferring Contribute to LPS-Induced Myocardial Injury" Biomedicines 12, no. 9: 2013. https://doi.org/10.3390/biomedicines12092013