

Digging Through the Complexities of Immunological Approaches in Emerging Osteosarcoma Therapeutics: A Comprehensive Narrative Review with Updated Clinical Trials

 ,
,  , , and
, , and
Abstract
:1. Introduction
2. Tumor Microenvironment and Tumor-Associated Macrophages (TAMs)
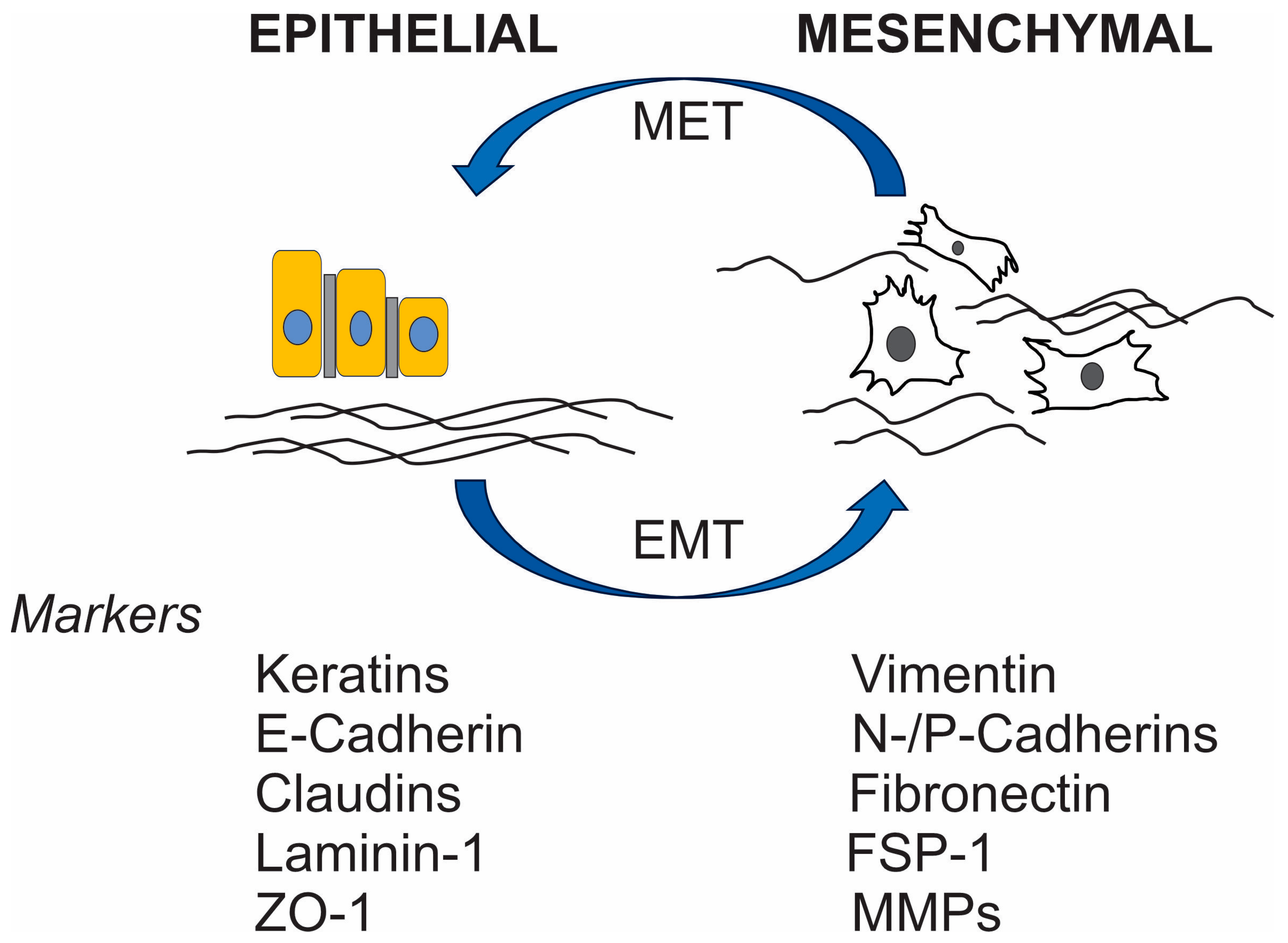
Myeloid-Derived Suppressor Cells (MDSCs) and EMT
3. Tumor-Infiltrating Lymphocytes (TILs)
4. Immune-Related Cells
5. Immune Checkpoint Inhibitors (ICIs)
6. CAR T and TCR T Therapies
7. Vaccine Approaches
8. Conclusive Remarks
9. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhou, H.; He, S.; Zhang, D.; Wang, J.; Yang, X.; Jiao, J.; Xu, W.; Yang, J.; Xiao, J. Prognostic factors of sarcomas occurring in bone and joint: A SEER based study. Medicine 2023, 102, e34231. [Google Scholar] [CrossRef] [PubMed]
- Karimi, A.; Ebrahimpour, A.; Sadighi, M.; Chehrassan, M.; Biglari, F.; Jafari Kafiabadi, M.; Akbari, M.E.; Azizmohammad Looha, M. Descriptive Epidemiology and Survival Rate of Osteosarcoma: The First National Population-Based Study in the Middle East (2008–2014). Arch. Bone Jt. Surg. 2023, 11, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Lupo, P.J.; Marcotte, E.L.; Scheurer, M.E.; Poynter, J.N.; Spector, L.G.; COG Epidemiology Committee. Children’s Oncology Group’s 2023 blueprint for research: Epidemiology. Pediatr. Blood Cancer 2023, 70 (Suppl. S6), e30566. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Cui, L.; He, Z.; Chen, Z.; Tao, H.; Yang, J. Epidemiology and nomogram of pediatric and young adulthood osteosarcoma patients with synchronous lung metastasis: A SEER analysis. PLoS ONE 2023, 18, e0288492. [Google Scholar] [CrossRef] [PubMed]
- Trama, A.; Stark, D.; Bozovic-Spasojevic, I.; Gaspar, N.; Peccatori, F.; Toss, A.; Bernasconi, A.; Quarello, P.; Scheinemann, K.; Jezdic, S.; et al. Cancer burden in adolescents and young adults in Europe. ESMO Open 2023, 8, 100744. [Google Scholar] [CrossRef]
- Gaspar, N.; Occean, B.V.; Pacquement, H.; Bompas, E.; Bouvier, C.; Brisse, H.J.; Castex, M.P.; Cheurfa, N.; Corradini, N.; Delaye, J.; et al. Results of methotrexate-etoposide-ifosfamide based regimen (M-EI) in osteosarcoma patients included in the French OS2006/sarcome-09 study. Eur. J. Cancer 2018, 88, 57–66. [Google Scholar] [CrossRef]
- Hinshaw, D.C.; Shevde, L.A. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef]
- Wu, C.; Tan, J.; Shen, H.; Deng, C.; Kleber, C.; Osterhoff, G.; Schopow, N. Exploring the relationship between metabolism and immune microenvironment in osteosarcoma based on metabolic pathways. J. Biomed. Sci. 2024, 31, 4. [Google Scholar] [CrossRef]
- Li, S.; Zhang, H.; Shang, G. Current status and future challenges of CAR-T cell therapy for osteosarcoma. Front. Immunol. 2023, 14, 1290762. [Google Scholar] [CrossRef]
- Park, J.A.; Cheung, N.V. Promise and Challenges of T Cell Immunotherapy for Osteosarcoma. Int. J. Mol. Sci. 2023, 24, 12520. [Google Scholar] [CrossRef]
- Panez-Toro, I.; Munoz-Garcia, J.; Vargas-Franco, J.W.; Renodon-Corniere, A.; Heymann, M.F.; Lezot, F.; Heymann, D. Advances in Osteosarcoma. Curr. Osteoporos. Rep. 2023, 21, 330–343. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Liao, Y.; Sun, P.; Tu, J.; Zou, Y.; Fang, J.; Chen, Z.; Li, H.; Chen, J.; Peng, Y.; et al. Construction of an ER stress-related prognostic signature for predicting prognosis and screening the effective anti-tumor drug in osteosarcoma. J. Transl. Med. 2024, 22, 66. [Google Scholar] [CrossRef]
- Zhu, J.; Fan, J.; Xia, Y.; Wang, H.; Li, Y.; Feng, Z.; Fu, C. Potential targets and applications of nanodrug targeting myeloid cells in osteosarcoma for the enhancement of immunotherapy. Front. Pharmacol. 2023, 14, 1271321. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Zhou, J.; Wu, Q.; Luo, G.; Zhao, M.; Zhong, G.; Zheng, Y.; Meng, X.; Cheng, S.; Zhang, Y. Multifunctional 3D-printed scaffolds eradiate orthotopic osteosarcoma and promote osteogenesis via microwave thermo-chemotherapy combined with immunotherapy. Biomaterials 2023, 301, 122236. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Song, F.; Jin, J.; Zou, B.; Dai, P.; Sun, M.; Xu, W.; Wang, L.; Kang, Y. Prognostic and immunological significance of an M1 macrophage-related gene signature in osteosarcoma. Front. Immunol. 2023, 14, 1202725. [Google Scholar] [CrossRef]
- Wu, C.; Gong, S.; Duan, Y.; Deng, C.; Kallendrusch, S.; Berninghausen, L.; Osterhoff, G.; Schopow, N. A tumor microenvironment-based prognostic index for osteosarcoma. J. Biomed. Sci. 2023, 30, 23. [Google Scholar] [CrossRef]
- Hu, C.; Liu, C.; Tian, S.; Wang, Y.; Shen, R.; Rao, H.; Li, J.; Yang, X.; Chen, B.; Ye, L. Comprehensive analysis of prognostic tumor microenvironment-related genes in osteosarcoma patients. BMC Cancer 2020, 20, 814. [Google Scholar] [CrossRef]
- Zhang, C.; Zheng, J.H.; Lin, Z.H.; Lv, H.Y.; Ye, Z.M.; Chen, Y.P.; Zhang, X.Y. Profiles of immune cell infiltration and immune-related genes in the tumor microenvironment of osteosarcoma. Aging 2020, 12, 3486–3501. [Google Scholar] [CrossRef]
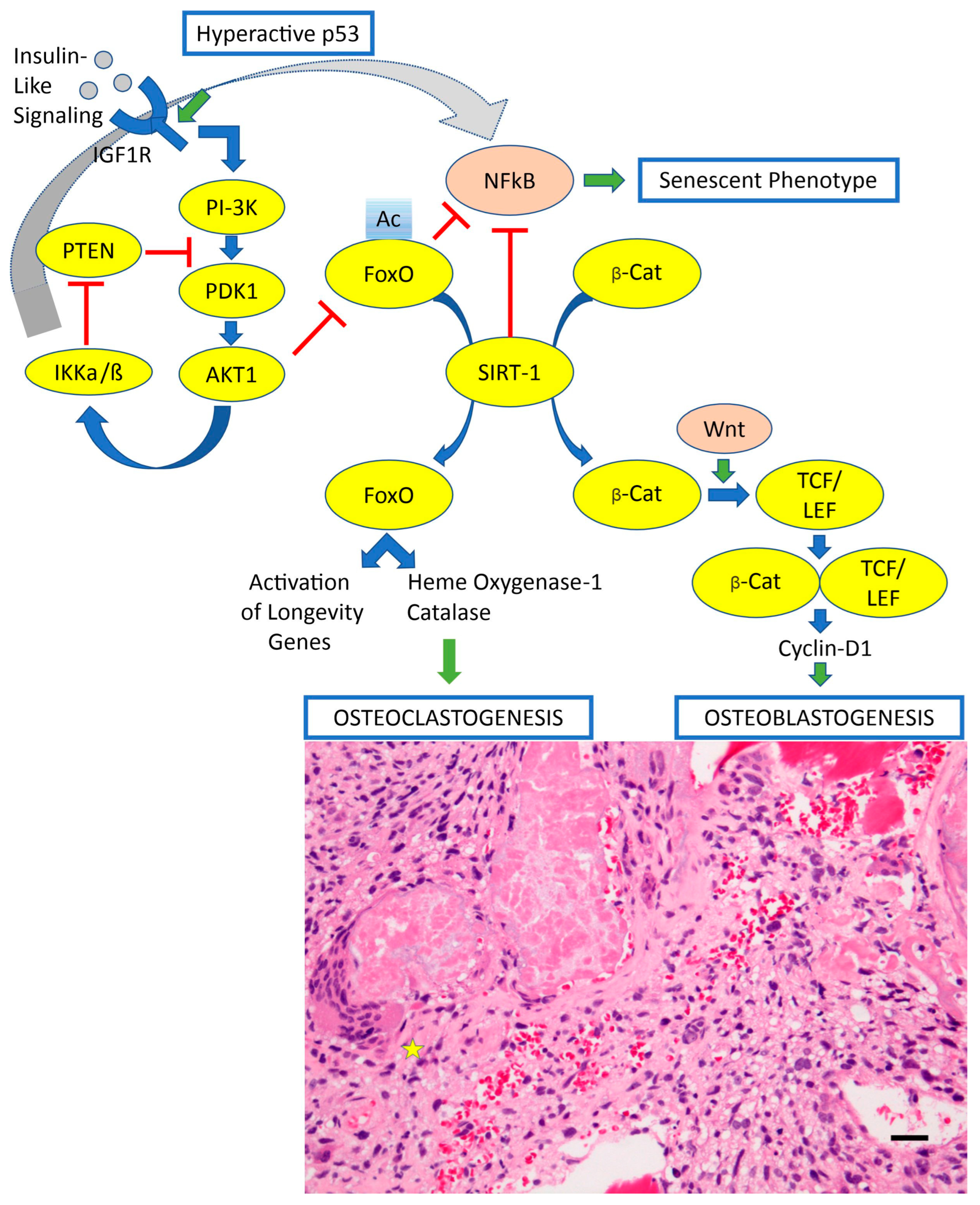
- Sergi, C.; Shen, F.; Liu, S.M. Insulin/IGF-1R, SIRT1, and FOXOs Pathways-An Intriguing Interaction Platform for Bone and Osteosarcoma. Front. Endocrinol. 2019, 10, 93. [Google Scholar] [CrossRef]
- Gao, Y.M.; Pei, Y.; Zhao, F.F.; Wang, L. Osteoclasts in Osteosarcoma: Mechanisms, Interactions, and Therapeutic Prospects. Cancer Manag. Res. 2023, 15, 1323–1337. [Google Scholar] [CrossRef]
- Huang, H.; Wang, X.; Zhang, S.; Bai, X.; Griffin, N.; Shan, Y.; Shan, F. In vitro and in vivo killing effects of methionine enkephalin on osteosarcoma. Int. Immunopharmacol. 2023, 125 Pt B, 111226. [Google Scholar] [CrossRef]
- Liao, S.; Li, J.; Gao, S.; Han, Y.; Han, X.; Wu, Y.; Bi, J.; Xu, M.; Bi, W. Sulfatinib, a novel multi-targeted tyrosine kinase inhibitor of FGFR1, CSF1R, and VEGFR1-3, suppresses osteosarcoma proliferation and invasion via dual role in tumor cells and tumor microenvironment. Front. Oncol. 2023, 13, 1158857. [Google Scholar] [CrossRef]
- Ammons, D.T.; Harris, R.A.; Hopkins, L.S.; Kurihara, J.; Weishaar, K.; Dow, S. A single-cell RNA sequencing atlas of circulating leukocytes from healthy and osteosarcoma affected dogs. Front. Immunol. 2023, 14, 1162700. [Google Scholar] [CrossRef]
- Xia, Y.; Wang, D.; Piao, Y.; Chen, M.; Wang, D.; Jiang, Z.; Liu, B. Modulation of immunosuppressive cells and noncoding RNAs as immunotherapy in osteosarcoma. Front. Immunol. 2022, 13, 1025532. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Tong, C.; Xu, M.; He, H.; Chen, C. Bioinformatics Analysis Reveals an Association between Autophagy, Prognosis, Tumor Microenvironment, and Immunotherapy in Osteosarcoma. J. Oncol. 2022, 2022, 4220331. [Google Scholar] [CrossRef]
- Thakur, M.D.; Franz, C.J.; Brennan, L.; Brouwer-Visser, J.; Tam, R.; Korski, K.; Koeppen, H.; Ziai, J.; Babitzki, G.; Ranchere-Vince, D.; et al. Immune contexture of paediatric cancers. Eur. J. Cancer 2022, 170, 179–193. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; Zuo, J.; Tian, H.; Huang, C.; Nice, E.C.; Shi, Z.; Kong, Q. Nanoengineering a metal-organic framework for osteosarcoma chemo-immunotherapy by modulating indoleamine-2,3-dioxygenase and myeloid-derived suppressor cells. J. Exp. Clin. Cancer Res. 2022, 41, 162. [Google Scholar] [CrossRef]
- Ling, Z.; Yang, C.; Tan, J.; Dou, C.; Chen, Y. Beyond immunosuppressive effects: Dual roles of myeloid-derived suppressor cells in bone-related diseases. Cell. Mol. Life Sci. CMLS 2021, 78, 7161–7183. [Google Scholar] [CrossRef] [PubMed]
- Ligon, J.A.; Choi, W.; Cojocaru, G.; Fu, W.; Hsiue, E.H.; Oke, T.F.; Siegel, N.; Fong, M.H.; Ladle, B.; Pratilas, C.A.; et al. Pathways of immune exclusion in metastatic osteosarcoma are associated with inferior patient outcomes. J. Immunother. Cancer 2021, 9, e001772. [Google Scholar] [CrossRef]
- Deng, C.; Xu, Y.; Fu, J.; Zhu, X.; Chen, H.; Xu, H.; Wang, G.; Song, Y.; Song, G.; Lu, J.; et al. Reprograming the tumor immunologic microenvironment using neoadjuvant chemotherapy in osteosarcoma. Cancer Sci. 2020, 111, 1899–1909. [Google Scholar] [CrossRef]
- Jin, Y.; Huang, Y.; Ren, H.; Huang, H.; Lai, C.; Wang, W.; Tong, Z.; Zhang, H.; Wu, W.; Liu, C.; et al. Nano-enhanced immunotherapy: Targeting the immunosuppressive tumor microenvironment. Biomaterials 2024, 305, 122463. [Google Scholar] [CrossRef]
- Lasser, S.A.; Ozbay Kurt, F.G.; Arkhypov, I.; Utikal, J.; Umansky, V. Myeloid-derived suppressor cells in cancer and cancer therapy. Nat. Rev. Clin. Oncol. 2024, 21, 147–164. [Google Scholar] [CrossRef] [PubMed]
- Chamani, F.K.; Etebari, A.; Hajivalili, M.; Mosaffa, N.; Jalali, S.A. Hypoxia and programmed cell death-ligand 1 expression in the tumor microenvironment: A review of the effects of hypoxia-induced factor-1 on immunotherapy. Mol. Biol. Rep. 2024, 51, 88. [Google Scholar] [CrossRef]
- Xu, M.; Zhao, X.; Wen, T.; Qu, X. Unveiling the role of KRAS in tumor immune microenvironment. Biomed. Pharmacother. Biomed. Pharmacother. 2024, 171, 116058. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Zhang, X.; Liu, X.; Cai, X.; Shen, T.; Pan, D.; Liang, R.; Ding, R.; Hu, R.; Dong, J.; et al. Single-cell sequencing reveals the immune microenvironment landscape related to anti-PD-1 resistance in metastatic colorectal cancer with high microsatellite instability. BMC Med. 2023, 21, 161. [Google Scholar] [CrossRef]
- Liu, A.; Gammon, S.T.; Pisaneschi, F.; Boda, A.; Ager, C.R.; Piwnica-Worms, D.; Hong, D.S.; Curran, M.A. Hypoxia-activated prodrug and antiangiogenic therapies cooperatively treat pancreatic cancer but elicit immunosuppressive G-MDSC infiltration. JCI Insight 2024, 9, e169150. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Z.; Chan, S.L.; Zhou, J.; Vong, J.S.L.; Kwong, T.T.; Zeng, X.; Wu, H.; Cao, J.; Tu, Y.; Feng, Y.; et al. Targeting PPAR-gamma counteracts tumour adaptation to immune-checkpoint blockade in hepatocellular carcinoma. Gut 2023, 72, 1758–1773. [Google Scholar] [CrossRef]
- Yang, H.; Kang, B.; Ha, Y.; Lee, S.H.; Kim, I.; Kim, H.; Lee, W.S.; Kim, G.; Jung, S.; Rha, S.Y.; et al. High serum IL-6 correlates with reduced clinical benefit of atezolizumab and bevacizumab in unresectable hepatocellular carcinoma. JHEP Rep. 2023, 5, 100672. [Google Scholar] [CrossRef]
- Wang, C.; Zheng, X.; Zhang, J.; Jiang, X.; Wang, J.; Li, Y.; Li, X.; Shen, G.; Peng, J.; Zheng, P.; et al. CD300ld on neutrophils is required for tumour-driven immune suppression. Nature 2023, 621, 830–839. [Google Scholar] [CrossRef]
- Yu, Y.; Zhang, H.; Ren, T.; Huang, Y.; Liang, X.; Wang, W.; Niu, J.; Han, Y.; Guo, W. Development of a prognostic gene signature based on an immunogenomic infiltration analysis of osteosarcoma. J. Cell. Mol. Med. 2020, 24, 11230–11242. [Google Scholar] [CrossRef]
- Liu, J.; Lu, J.; Wu, L.; Zhang, T.; Wu, J.; Li, L.; Tai, Z.; Chen, Z.; Zhu, Q. Targeting tumor-associated macrophages: Novel insights into immunotherapy of skin cancer. J. Adv. Res. 2024, 67, 231–252. [Google Scholar] [CrossRef] [PubMed]
- Cersosimo, F.; Lonardi, S.; Bernardini, G.; Telfer, B.; Mandelli, G.E.; Santucci, A.; Vermi, W.; Giurisato, E. Tumor-Associated Macrophages in Osteosarcoma: From Mechanisms to Therapy. Int. J. Mol. Sci. 2020, 21, 5207. [Google Scholar] [CrossRef] [PubMed]
- Mirabello, L.; Zhu, B.; Koster, R.; Karlins, E.; Dean, M.; Yeager, M.; Gianferante, M.; Spector, L.G.; Morton, L.M.; Karyadi, D.; et al. Frequency of Pathogenic Germline Variants in Cancer-Susceptibility Genes in Patients with Osteosarcoma. JAMA Oncol. 2020, 6, 724–734. [Google Scholar] [CrossRef] [PubMed]
- Wolf-Dennen, K.; Gordon, N.; Kleinerman, E.S. Exosomal communication by metastatic osteosarcoma cells modulates alveolar macrophages to an M2 tumor-promoting phenotype and inhibits tumoricidal functions. Oncoimmunology 2020, 9, 1747677. [Google Scholar] [CrossRef] [PubMed]
- Dhupkar, P.; Gordon, N.; Stewart, J.; Kleinerman, E.S. Anti-PD-1 therapy redirects macrophages from an M2 to an M1 phenotype inducing regression of OS lung metastases. Cancer Med. 2018, 7, 2654–2664. [Google Scholar] [CrossRef]
- Heymann, M.F.; Lezot, F.; Heymann, D. The contribution of immune infiltrates and the local microenvironment in the pathogenesis of osteosarcoma. Cell. Immunol. 2019, 343, 103711. [Google Scholar] [CrossRef] [PubMed]
- Buddingh, E.P.; Kuijjer, M.L.; Duim, R.A.; Burger, H.; Agelopoulos, K.; Myklebost, O.; Serra, M.; Mertens, F.; Hogendoorn, P.C.; Lankester, A.C.; et al. Tumor-infiltrating macrophages are associated with metastasis suppression in high-grade osteosarcoma: A rationale for treatment with macrophage activating agents. Clin. Cancer Res. An. Off. J. Am. Assoc. Cancer Res. 2011, 17, 2110–2119. [Google Scholar] [CrossRef]
- Mudry, P.; Kyr, M.; Rohleder, O.; Mahdal, M.; Staniczkova Zambo, I.; Jezova, M.; Tomas, T.; Sterba, J. Improved osteosarcoma survival with addition of mifamurtide to conventional chemotherapy—Observational prospective single institution analysis. J. Bone Oncol. 2021, 28, 100362. [Google Scholar] [CrossRef]
- Nastasi, N.; Pasha, A.; Bruno, G.; Subbiani, A.; Pietrovito, L.; Leo, A.; Scala, L.; de Simone, L.; Casazza, G.; Lunardi, F.; et al. Blockade of IL-10 Signaling Ensures Mifamurtide Efficacy in Metastatic Osteosarcoma. Cancers 2023, 15, 4744. [Google Scholar] [CrossRef]
- Ataseven, E.; Goktepe, S.O.; Kaya, H.; Tamsel, I.; Kececi, B.; Argin, M.; Doganavsargil, B.; Sabah, D.; Burak, Z.; Kantar, M. What has changed in the last 25 years in osteosarcoma treatment? A single center experience. Turk. J. Pediatr. 2023, 65, 54–63. [Google Scholar] [CrossRef]
- Kokkali, S.; Kotsantis, I.; Magou, E.; Sophia, T.; Kormas, T.; Diakoumis, G.; Spathas, N.; Psyrri, A.; Ardavanis, A. The addition of the immunomodulator mifamurtide to adjuvant chemotherapy for early osteosarcoma: A retrospective analysis. Investig. New Drugs 2022, 40, 668–675. [Google Scholar] [CrossRef] [PubMed]
- Punzo, F.; Bellini, G.; Tortora, C.; Pinto, D.D.; Argenziano, M.; Pota, E.; Paola, A.D.; Martino, M.D.; Rossi, F. Mifamurtide and TAM-like macrophages: Effect on proliferation, migration and differentiation of osteosarcoma cells. Oncotarget 2020, 11, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Di Bella, G.; Di Bella, L.; Borghetto, V.; Moscato, I.; Costanzo, E. A retrospective observational study on cases of osteosarcomas treated with a multitherapy: The rationale and effectiveness. Neuro Endocrinol. Lett. 2022, 43, 173–179. Available online: https://www.ncbi.nlm.nih.gov/pubmed/36179729 (accessed on 30 November 2024). [PubMed]
- Paukovcekova, S.; Valik, D.; Sterba, J.; Veselska, R. Enhanced Antiproliferative Effect of Combined Treatment with Calcitriol and All-Trans Retinoic Acid in Relation to Vitamin D Receptor and Retinoic Acid Receptor alpha Expression in Osteosarcoma Cell Lines. Int. J. Mol. Sci. 2020, 21, 6591. [Google Scholar] [CrossRef]
- Shenouda, S.; Kulkarni, K.; Abuetabh, Y.; Sergi, C. Cancer Stem Cells and their Management in Cancer Therapy. Recent Pat. Anti-Cancer Drug Discov. 2020, 15, 212–227. [Google Scholar] [CrossRef] [PubMed]
- Li, H.B.; Huang, G.; Tu, J.; Lv, D.M.; Jin, Q.L.; Chen, J.K.; Zou, Y.T.; Lee, D.F.; Shen, J.N.; Xie, X.B. METTL14-mediated epitranscriptome modification of MN1 mRNA promote tumorigenicity and all-trans-retinoic acid resistance in osteosarcoma. EBioMedicine 2022, 82, 104142. [Google Scholar] [CrossRef]
- Nyffeler, J.; Willis, C.; Harris, F.R.; Taylor, L.W.; Judson, R.; Everett, L.J.; Harrill, J.A. Combining phenotypic profiling and targeted RNA-Seq reveals linkages between transcriptional perturbations and chemical effects on cell morphology: Retinoic acid as an example. Toxicol. Appl. Pharmacol. 2022, 444, 116032. [Google Scholar] [CrossRef]
- Fukuda, H.; Nakamura, S.; Chisaki, Y.; Takada, T.; Toda, Y.; Murata, H.; Itoh, K.; Yano, Y.; Takata, K.; Ashihara, E. Daphnetin inhibits invasion and migration of LM8 murine osteosarcoma cells by decreasing RhoA and Cdc42 expression. Biochem. Biophys. Res. Commun. 2016, 471, 63–67. [Google Scholar] [CrossRef]
- Lv, J.; Chen, F.K.; Liu, C.; Liu, P.J.; Feng, Z.P.; Jia, L.; Yang, Z.X.; Hou, F.; Deng, Z.Y. Zoledronic acid inhibits thyroid cancer stemness and metastasis by repressing M2-like tumor-associated macrophages induced Wnt/beta-catenin pathway. Life Sci. 2020, 256, 117925. [Google Scholar] [CrossRef]
- Gomez-Brouchet, A.; Illac, C.; Gilhodes, J.; Bouvier, C.; Aubert, S.; Guinebretiere, J.M.; Marie, B.; Larousserie, F.; Entz-Werle, N.; de Pinieux, G.; et al. CD163-positive tumor-associated macrophages and CD8-positive cytotoxic lymphocytes are powerful diagnostic markers for the therapeutic stratification of osteosarcoma patients: An immunohistochemical analysis of the biopsies fromthe French OS2006 phase 3 trial. Oncoimmunology 2017, 6, e1331193. [Google Scholar] [CrossRef]
- Gomez-Brouchet, A.; Gilhodes, J.; Acker, N.V.; Brion, R.; Bouvier, C.; Assemat, P.; Gaspar, N.; Aubert, S.; Guinebretiere, J.M.; Marie, B.; et al. Characterization of Macrophages and Osteoclasts in the Osteosarcoma Tumor Microenvironment at Diagnosis: New Perspective for Osteosarcoma Treatment? Cancers 2021, 13, 423. [Google Scholar] [CrossRef]
- Barros, M.H.; Hauck, F.; Dreyer, J.H.; Kempkes, B.; Niedobitek, G. Macrophage polarisation: An immunohistochemical approach for identifying M1 and M2 macrophages. PLoS ONE 2013, 8, e80908. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Li, H.; Zhao, X.; Yang, S.; Zhan, J.; Liu, H.; Jiang, Y.; Shi, L.; Song, Y.; Lei, Y.; et al. Expression of PD-1 mitigates phagocytic activities TAM in osteosarcoma. Heliyon 2024, 10, e23498. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; He, R.; Cao, W.; Li, D.; Zheng, Q.; Zhang, Y. Oncogenic and immunological values of RBM34 in osteosarcoma and its pan-cancer analysis. Am. J. Cancer Res. 2023, 13, 5094–5121. Available online: https://www.ncbi.nlm.nih.gov/pubmed/38058813 (accessed on 30 November 2024).
- Chen, F.; Liu, J.; Yang, T.; Sun, J.; He, X.; Fu, X.; Qiao, S.; An, J.; Yang, J. Analysis of intercellular communication in the osteosarcoma microenvironment based on single cell sequencing data. J. Bone Oncol. 2023, 41, 100493. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Liu, S.; Luo, X.; Sun, Y.; Li, X.; Luo, K.; Liao, S.; Li, F.; Liang, J.; Zhan, X.; et al. A novel molecular signature for predicting prognosis and immunotherapy response in osteosarcoma based on tumor-infiltrating cell marker genes. Front. Immunol. 2023, 14, 1150588. [Google Scholar] [CrossRef]
- Park, J.A.; Espinosa-Cotton, M.; Guo, H.F.; Monette, S.; Cheung, N.V. Targeting tumor vasculature to improve antitumor activity of T cells armed ex vivo with T cell engaging bispecific antibody. J. Immunother. Cancer 2023, 11, e006680. [Google Scholar] [CrossRef]
- Bertin, H.; Peries, S.; Amiaud, J.; Van Acker, N.; Perrot, B.; Bouvier, C.; Aubert, S.; Marie, B.; Larousserie, F.; De Pinieux, G.; et al. Characterization of the Tumor Microenvironment in Jaw Osteosarcomas, towards Prognostic Markers and New Therapeutic Targets. Cancers 2023, 15, 1004. [Google Scholar] [CrossRef]
- Xu, N.; Wang, X.; Wang, L.; Song, Y.; Zheng, X.; Hu, H. Comprehensive analysis of potential cellular communication networks in advanced osteosarcoma using single-cell RNA sequencing data. Front. Genet. 2022, 13, 1013737. [Google Scholar] [CrossRef]
- Ruiz, D.; Haynes, C.; Marable, J.; Pundkar, C.; Nance, R.L.; Bedi, D.; Agarwal, P.; Suryawanshi, A.S.; Mishra, A.; Smith, B.F.; et al. Development of OX40 agonists for canine cancer immunotherapy. iScience 2022, 25, 105158. [Google Scholar] [CrossRef]
- Wu, R.; Dou, X.; Li, H.; Sun, Z.; Li, H.; Shen, Y.; Weng, W.; Min, J. Identification of Cell Subpopulations and Interactive Signaling Pathways from a Single-Cell RNA Sequencing Dataset in Osteosarcoma: A Comprehensive Bioinformatics Analysis. Front. Oncol. 2022, 12, 853979. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Zheng, D.; Guo, W. Comprehensive Analysis of a Zinc Finger Protein Gene-Based Signature with Regard to Prognosis and Tumor Immune Microenvironment in Osteosarcoma. Front. Genet. 2022, 13, 835014. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.Y.; Park, J.A.; Long, A.; Guo, H.F.; Cheung, N.V. Novel potent anti-STEAP1 bispecific antibody to redirect T cells for cancer immunotherapy. J. Immunother. Cancer 2021, 9, e003114. [Google Scholar] [CrossRef]
- Wunder, J.S.; Lee, M.J.; Nam, J.; Lau, B.Y.; Dickson, B.C.; Pinnaduwage, D.; Bull, S.B.; Ferguson, P.C.; Seto, A.; Gokgoz, N.; et al. Osteosarcoma and soft-tissue sarcomas with an immune infiltrate express PD-L1: Relation to clinical outcome and Th1 pathway activation. Oncoimmunology 2020, 9, 1737385. [Google Scholar] [CrossRef]
- Judge, S.J.; Darrow, M.A.; Thorpe, S.W.; Gingrich, A.A.; O’Donnell, E.F.; Bellini, A.R.; Sturgill, I.R.; Vick, L.V.; Dunai, C.; Stoffel, K.M.; et al. Analysis of tumor-infiltrating NK and T cells highlights IL-15 stimulation and TIGIT blockade as a combination immunotherapy strategy for soft tissue sarcomas. J. Immunother. Cancer 2020, 8, e001355. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, M.; Wei, R.; Wu, J. Adoptive transfer of TILs plus anti-PD1 therapy: An alternative combination therapy for treating metastatic osteosarcoma. J. Bone Oncol. 2020, 25, 100332. [Google Scholar] [CrossRef]
- Shi, J.; Li, M.; Yang, R. Tumor-infiltrating lymphocytes as a feasible adjuvant immunotherapy for osteosarcoma with a poor response to neoadjuvant chemotherapy. Immunotherapy 2020, 12, 641–652. [Google Scholar] [CrossRef]
- He, Y.; Huang, X.; Ma, Y.; Yang, G.; Cui, Y.; Lv, X.; Zhao, R.; Jin, H.; Tong, Y.; Zhang, X.; et al. A novel aging-associated lncRNA signature for predicting prognosis in osteosarcoma. Sci. Rep. 2024, 14, 1386. [Google Scholar] [CrossRef]
- Hu, J.; Lazar, A.J.; Ingram, D.; Wang, W.L.; Zhang, W.; Jia, Z.; Ragoonanan, D.; Wang, J.; Xia, X.; Mahadeo, K.; et al. Cell membrane-anchored and tumor-targeted IL-12 T-cell therapy destroys cancer-associated fibroblasts and disrupts extracellular matrix in heterogenous osteosarcoma xenograft models. J. Immunother. Cancer 2024, 12, e006991. [Google Scholar] [CrossRef]
- Wu, H.; Wang, R.; Li, S.; Chen, S.; Liu, S.; Li, X.; Yang, X.; Zeng, Q.; Zhou, Y.; Zhu, X.; et al. Aspect ratio-dependent dual-regulation of the tumor immune microenvironment against osteosarcoma by hydroxyapatite nanoparticles. Acta Biomater. 2023, 170, 427–441. [Google Scholar] [CrossRef]
- Cascini, C.; Ratti, C.; Botti, L.; Parma, B.; Cancila, V.; Salvaggio, A.; Meazza, C.; Tripodo, C.; Colombo, M.P.; Chiodoni, C. Rewiring innate and adaptive immunity with TLR9 agonist to treat osteosarcoma. J. Exp. Clin. Cancer Res. 2023, 42, 154. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Cardona, X.E.; Luo, W.; Mohammed, S.I. Advances and challenges of CAR T therapy and suitability of animal models (Review). Mol. Clin. Oncol. 2022, 17, 134. [Google Scholar] [CrossRef] [PubMed]
- Le, T.; Su, S.; Shahriyari, L. Investigating Optimal Chemotherapy Options for Osteosarcoma Patients through a Mathematical Model. Cells 2021, 10, 2009. [Google Scholar] [CrossRef] [PubMed]
- Spreafico, M.; Ieva, F.; Arlati, F.; Capello, F.; Fatone, F.; Fedeli, F.; Genalti, G.; Anninga, J.; Gelderblom, H.; Fiocco, M. Novel longitudinal Multiple Overall Toxicity (MOTox) score to quantify adverse events experienced by patients during chemotherapy treatment: A retrospective analysis of the MRC BO06 trial in osteosarcoma. BMJ Open 2021, 11, e053456. [Google Scholar] [CrossRef]
- Le, T.; Su, S.; Kirshtein, A.; Shahriyari, L. Data-Driven Mathematical Model of Osteosarcoma. Cancers 2021, 13, 2367. [Google Scholar] [CrossRef]
- Fritzsching, B.; Fellenberg, J.; Moskovszky, L.; Sapi, Z.; Krenacs, T.; Machado, I.; Poeschl, J.; Lehner, B.; Szendroi, M.; Bosch, A.L.; et al. CD8+/FOXP3+-ratio in osteosarcoma microenvironment separates survivors from non-survivors: A multicenter validated retrospective study. Oncoimmunology 2015, 4, e990800. [Google Scholar] [CrossRef]
- Kunz, P.; Fellenberg, J.; Moskovszky, L.; Sapi, Z.; Krenacs, T.; Machado, I.; Poeschl, J.; Lehner, B.; Szendroi, M.; Ruef, P.; et al. Improved survival in osteosarcoma patients with atypical low vascularization. Ann. Surg. Oncol. 2015, 22, 489–496. [Google Scholar] [CrossRef]
- Kunz, P.; Fellenberg, J.; Moskovszky, L.; Sapi, Z.; Krenacs, T.; Poeschl, J.; Lehner, B.; Szendroi, M.; Ewerbeck, V.; Kinscherf, R.; et al. Osteosarcoma microenvironment: Whole-slide imaging and optimized antigen detection overcome major limitations in immunohistochemical quantification. PLoS ONE 2014, 9, e90727. [Google Scholar] [CrossRef]
- Li, X.; Chen, Y.; Liu, X.; Zhang, J.; He, X.; Teng, G.; Yu, D. Tim3/Gal9 interactions between T cells and monocytes result in an immunosuppressive feedback loop that inhibits Th1 responses in osteosarcoma patients. Int. Immunopharmacol. 2017, 44, 153–159. [Google Scholar] [CrossRef]
- Meraviglia, S.; Di Carlo, P.; Pampinella, D.; Guadagnino, G.; Presti, E.L.; Orlando, V.; Marchetti, G.; Dieli, F.; Sergi, C. T-Cell Subsets (TCM, TEM, TEMRA) and Poly-Functional Immune Response in Patients with Human Immunodeficiency Virus (HIV) Infection and Different T-CD4 Cell Response. Ann. Clin. Lab. Sci. 2019, 49, 519–528. Available online: https://www.ncbi.nlm.nih.gov/pubmed/31471343 (accessed on 30 November 2024).
- Solomon, I.; Amann, M.; Goubier, A.; Arce Vargas, F.; Zervas, D.; Qing, C.; Henry, J.Y.; Ghorani, E.; Akarca, A.U.; Marafioti, T.; et al. CD25-T(reg)-depleting antibodies preserving IL-2 signaling on effector T cells enhance effector activation and antitumor immunity. Nat. Cancer 2020, 1, 1153–1166. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Xu, Z.; Li, T.; Thakur, A.; Wen, Y.; Zhang, K.; Liu, Y.; Liang, Q.; Liu, W.; Qin, J.J.; et al. Nanomaterial-encapsulated STING agonists for immune modulation in cancer therapy. Biomark. Res. 2024, 12, 2. [Google Scholar] [CrossRef]
- Chang, X.; Ma, Z.; Zhu, G.; Lu, Y.; Yang, J. New perspective into mesenchymal stem cells: Molecular mechanisms regulating osteosarcoma. J. Bone Oncol. 2021, 29, 100372. [Google Scholar] [CrossRef]
- Sarhadi, V.K.; Daddali, R.; Seppanen-Kaijansinkko, R. Mesenchymal Stem Cells and Extracellular Vesicles in Osteosarcoma Pathogenesis and Therapy. Int. J. Mol. Sci. 2021, 22, 11035. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Scholtemeijer, M.; Shah, K. Mesenchymal Stem Cell Immunomodulation: Mechanisms and Therapeutic Potential. Trends Pharmacol. Sci. 2020, 41, 653–664. [Google Scholar] [CrossRef] [PubMed]
- Davis, K.L.; Fox, E.; Merchant, M.S.; Reid, J.M.; Kudgus, R.A.; Liu, X.; Minard, C.G.; Voss, S.; Berg, S.L.; Weigel, B.J.; et al. Nivolumab in children and young adults with relapsed or refractory solid tumours or lymphoma (ADVL1412): A multicentre, open-label, single-arm, phase 1-2 trial. Lancet Oncol. 2020, 21, 541–550. [Google Scholar] [CrossRef]
- Que, Y.; Hu, Y.; Hong, D.; Zhang, Y. Trends in clinical development of pediatric cancer for PD-1 and PD-L1 inhibitors: An analysis of ClinicalTrials.gov. J. Immunother. Cancer 2021, 9, e002920. [Google Scholar] [CrossRef]
- Xin Yu, J.; Hodge, J.P.; Oliva, C.; Neftelinov, S.T.; Hubbard-Lucey, V.M.; Tang, J. Trends in clinical development for PD-1/PD-L1 inhibitors. Nat. Rev. Drug Discov. 2020, 19, 163–164. [Google Scholar] [CrossRef]
- Kraehenbuehl, L.; Weng, C.H.; Eghbali, S.; Wolchok, J.D.; Merghoub, T. Enhancing immunotherapy in cancer by targeting emerging immunomodulatory pathways. Nat. Rev. Clin. Oncol. 2022, 19, 37–50. [Google Scholar] [CrossRef]
- Jia, D.; Li, S.; Li, D.; Xue, H.; Yang, D.; Liu, Y. Mining TCGA database for genes of prognostic value in glioblastoma microenvironment. Aging 2018, 10, 592–605. [Google Scholar] [CrossRef]
- Wang, B.; Cheng, D.; Ma, D.; Chen, R.; Li, D.; Zhao, W.; Fang, C.; Ji, M. Mutual regulation of PD-L1 immunosuppression between tumor-associated macrophages and tumor cells: A critical role for exosomes. Cell Commun. Signal 2024, 22, 21. [Google Scholar] [CrossRef] [PubMed]
- Mahadevia, H.; Uson Junior, P.L.S.; Wang, J.; Borad, M.; Babiker, H. An overview of up-and-coming immune checkpoint inhibitors for pancreatic cancer. Expert Opin. Pharmacother. 2024, 25, 79–90. [Google Scholar] [CrossRef]
- Meng, L.; Wu, H.; Wu, J.; Ding, P.; He, J.; Sang, M.; Liu, L. Mechanisms of immune checkpoint inhibitors: Insights into the regulation of circular RNAS involved in cancer hallmarks. Cell Death Dis. 2024, 15, 3. [Google Scholar] [CrossRef] [PubMed]
- Galle, P.; Finn, R.S.; Mitchell, C.R.; Ndirangu, K.; Ramji, Z.; Redhead, G.S.; Pinato, D.J. Treatment-emergent antidrug antibodies related to PD-1, PD-L1, or CTLA-4 inhibitors across tumor types: A systematic review. J. Immunother. Cancer 2024, 12, e008266. [Google Scholar] [CrossRef]
- Koirala, P.; Roth, M.E.; Gill, J.; Piperdi, S.; Chinai, J.M.; Geller, D.S.; Hoang, B.H.; Park, A.; Fremed, M.A.; Zang, X.; et al. Immune infiltration and PD-L1 expression in the tumor microenvironment are prognostic in osteosarcoma. Sci. Rep. 2016, 6, 30093. [Google Scholar] [CrossRef] [PubMed]
- Vaddepally, R.K.; Kharel, P.; Pandey, R.; Garje, R.; Chandra, A.B. Review of Indications of FDA-Approved Immune Checkpoint Inhibitors per NCCN Guidelines with the Level of Evidence. Cancers 2020, 12, 738. [Google Scholar] [CrossRef]
- Boye, K.; Longhi, A.; Guren, T.; Lorenz, S.; Naess, S.; Pierini, M.; Taksdal, I.; Lobmaier, I.; Cesari, M.; Paioli, A.; et al. Pembrolizumab in advanced osteosarcoma: Results of a single-arm, open-label, phase 2 trial. Cancer Immunol. Immunother. 2021, 70, 2617–2624. [Google Scholar] [CrossRef]
- Geoerger, B.; Zwaan, C.M.; Marshall, L.V.; Michon, J.; Bourdeaut, F.; Casanova, M.; Corradini, N.; Rossato, G.; Farid-Kapadia, M.; Shemesh, C.S.; et al. Atezolizumab for children and young adults with previously treated solid tumours, non-Hodgkin lymphoma, and Hodgkin lymphoma (iMATRIX): A multicentre phase 1–2 study. Lancet Oncol. 2020, 21, 134–144. [Google Scholar] [CrossRef]
- Xie, L.; Xu, J.; Sun, X.; Guo, W.; Gu, J.; Liu, K.; Zheng, B.; Ren, T.; Huang, Y.; Tang, X.; et al. Apatinib plus camrelizumab (anti-PD1 therapy, SHR-1210) for advanced osteosarcoma (APFAO) progressing after chemotherapy: A single-arm, open-label, phase 2 trial. J. Immunother. Cancer. 2020, 8, e000798. [Google Scholar] [CrossRef]
- Ding, X.; Zhang, W.J.; You, R.; Zou, X.; Wang, Z.Q.; Ouyang, Y.F.; Peng, L.; Liu, Y.P.; Duan, C.Y.; Yang, Q.; et al. Camrelizumab Plus Apatinib in Patients with Recurrent or Metastatic Nasopharyngeal Carcinoma: An Open-Label, Single-Arm, Phase II Study. J. Clin. Oncol. 2023, 10, 2571–2582. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Richards, A.L.; Conley, A.P.; Woo, H.J.; Dickson, M.A.; Gounder, M.; Kelly, C.; Keohan, M.L.; Movva, S.; Thornton, K.; et al. Pilot study of bempegaldesleukin in combination with nivolumab in patients with metastatic sarcoma. Nat. Commun. 2022, 13, 3477. [Google Scholar] [CrossRef] [PubMed]
- Lussier, D.M.; O’Neill, L.; Nieves, L.M.; McAfee, M.S.; Holechek, S.A.; Collins, A.W.; Dickman, P.; Jacobsen, J.; Hingorani, P.; Blattman, J.N. Enhanced T-cell immunity to osteosarcoma through antibody blockade of PD-1/PD-L1 interactions. J. Immunother. 2015, 38, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Lussier, D.M.; Johnson, J.L.; Hingorani, P.; Blattman, J.N. Combination immunotherapy with α-CTLA-4 and α-PD-L1 antibody blockade prevents immune escape and leads to complete control of metastatic osteosarcoma. J. Immunother. Cancer 2015, 3, 21. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Mahoney, M.R.; Van Tine, B.A.; Atkins, J.; Milhem, M.M.; Jahagirdar, B.N.; Antonescu, C.R.; Horvath, E.; Tap, W.D.; Schwartz, G.K.; et al. Nivolumab with or without ipilimumab treatment for metastatic sarcoma (Alliance A091401): Two open-label, non-comparative, randomised, phase 2 trials. Lancet Oncol. 2018, 19, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Sterz, U.; Grube, M.; Herr, W.; Menhart, K.; Wendl, C.; Vogelhuber, M. Case Report: Dual Checkpoint Inhibition in Advanced Metastatic Osteosarcoma Results in Remission of All Tumor Manifestations-A Report of a Stunning Success in a 37-Year-Old Patient. Front. Oncol. 2021, 11, 684733. [Google Scholar] [CrossRef]
- Nuytemans, L.; Sys, G.; Creytens, D.; Lapeire, L. NGS-analysis to the rescue: Dual checkpoint inhibition in metastatic osteosarcoma—A case report and review of the literature. Acta Clin. Belg. 2021, 76, 162–167. [Google Scholar] [CrossRef]
- Balta, E.; Wabnitz, G.H.; Samstag, Y. Hijacked Immune Cells in the Tumor Microenvironment: Molecular Mechanisms of Immunosuppression and Cues to Improve T Cell-Based Immunotherapy of Solid Tumors. Int. J. Mol. Sci. 2021, 22, 5736. [Google Scholar] [CrossRef]
- Campagnari, A.; Belver, L. NOTCH1-Induced T-Cell Acute Lymphoblastic Leukemia In Vivo Models. Methods Mol. Biol. 2024, 2773, 9–24. [Google Scholar] [CrossRef]
- Kandasamy, G.; Maity, D. Inorganic nanocarriers for siRNA delivery for cancer treatments. Biomed. Mater. 2024, 19, 022001. [Google Scholar] [CrossRef]
- Geyer, M.B.; Brentjens, R.J. Review: Current clinical applications of chimeric antigen receptor (CAR) modified T cells. Cytotherapy 2016, 18, 1393–1409. [Google Scholar] [CrossRef]
- Hong, M.; Clubb, J.D.; Chen, Y.Y. Engineering CAR-T Cells for Next-Generation Cancer Therapy. Cancer Cell 2020, 38, 473–488. [Google Scholar] [CrossRef] [PubMed]
- Lei, L.; Huang, D.; Gao, H.; He, B.; Cao, J.; Peppas, N.A. Hydrogel-guided strategies to stimulate an effective immune response for vaccine-based cancer immunotherapy. Sci. Adv. 2022, 8, eadc8738. [Google Scholar] [CrossRef]
- Zhao, Q.; Jiang, Y.; Xiang, S.; Kaboli, P.J.; Shen, J.; Zhao, Y.; Wu, X.; Du, F.; Li, M.; Cho, C.H.; et al. Engineered TCR-T Cell Immunotherapy in Anticancer Precision Medicine: Pros and Cons. Front. Immunol. 2021, 12, 658753. [Google Scholar] [CrossRef]
- Brameshuber, M.; Kellner, F.; Rossboth, B.K.; Ta, H.; Alge, K.; Sevcsik, E.; Göhring, J.; Axmann, M.; Baumgart, F.; Gascoigne, N.R.J.; et al. Monomeric TCRs drive T cell antigen recognition. Nat. Immunol. 2018, 19, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.; Adams, K.J.; Bossi, G.; Wright, D.E.; Stacey, A.R.; Bedke, N.; Martinez-Hague, R.; Blat, D.; Humbert, L.; Buchanan, H.; et al. An approved in vitro approach to preclinical safety and efficacy evaluation of engineered T cell receptor anti-CD3 bispecific (ImmTAC) molecules. PLoS ONE 2018, 13, e0205491. [Google Scholar] [CrossRef] [PubMed]
- Iura, K.; Kohashi, K.; Ishii, T.; Maekawa, A.; Bekki, H.; Otsuka, H.; Yamada, Y.; Yamamoto, H.; Matsumoto, Y.; Iwamoto, Y.; et al. MAGEA4 expression in bone and soft tissue tumors: Its utility as a target for immunotherapy and diagnostic marker combined with NY-ESO-1. Virchows Arch. Int. J. Pathol. 2017, 471, 383–392. [Google Scholar] [CrossRef]
- Robbins, P.F.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; Dudley, M.E.; Wunderlich, J.R.; Nahvi, A.V.; Helman, L.J.; Mackall, C.L.; et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J. Clin. Oncol. 2011, 29, 917–924. [Google Scholar] [CrossRef]
- Lu, Y.C.; Parker, L.L.; Lu, T.; Zheng, Z.; Toomey, M.A.; White, D.E.; Yao, X.; Li, Y.F.; Robbins, P.F.; Feldman, S.A.; et al. Treatment of Patients with Metastatic Cancer Using a Major Histocompatibility Complex Class II-Restricted T-Cell Receptor Targeting the Cancer Germline Antigen MAGE-A3. J. Clin. Oncol. 2017, 35, 3322–3329. [Google Scholar] [CrossRef]
- Tsukahara, T.; Kawaguchi, S.; Torigoe, T.; Kimura, S.; Murase, M.; Ichimiya, S.; Wada, T.; Kaya, M.; Nagoya, S.; Ishii, T.; et al. Prognostic impact and immunogenicity of a novel osteosarcoma antigen, papillomavirus binding factor, in patients with osteosarcoma. Cancer Sci. 2008, 99, 368–375. [Google Scholar] [CrossRef]
- Watanabe, K.; Tsukahara, T.; Toji, S.; Saitoh, S.; Hirohashi, Y.; Nakatsugawa, M.; Kubo, T.; Kanaseki, T.; Kameshima, H.; Terui, T.; et al. Development of a T-cell receptor multimer with high avidity for detecting a naturally presented tumor-associated antigen on osteosarcoma cells. Cancer Sci. 2019, 110, 40–51. [Google Scholar] [CrossRef]
- Wang, Z.; Li, B.; Ren, Y.; Ye, Z. T-Cell-Based Immunotherapy for Osteosarcoma: Challenges and Opportunities. Front. Immunol. 2016, 7, 353. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, Z.; Li, B.; Wang, S.; Chen, T.; Ye, Z. Innate Immune Cells: A Potential and Promising Cell Population for Treating Osteosarcoma. Front. Immunol. 2019, 10, 1114. [Google Scholar] [CrossRef]
- Suryawanshi, A.; Manicassamy, S. Tumors induce immune tolerance through activation of β-catenin/TCF4 signaling in dendritic cells: A novel therapeutic target for cancer immunotherapy. Oncoimmunology 2015, 4, e1052932. [Google Scholar] [CrossRef] [PubMed]
- Cornwall, S.M.; Wikstrom, M.; Musk, A.W.; Alvarez, J.; Nowak, A.K.; Nelson, D.J. Human mesothelioma induces defects in dendritic cell numbers and antigen-processing function which predict survival outcomes. Oncoimmunology 2016, 5, e1082028. [Google Scholar] [CrossRef] [PubMed]
- Rizell, M.; Sternby Eilard, M.; Andersson, M.; Andersson, B.; Karlsson-Parra, A.; Suenaert, P. Phase 1 Trial with the Cell-Based Immune Primer Ilixadencel, Alone, and Combined with Sorafenib, in Advanced Hepatocellular Carcinoma. Front. Oncol. 2019, 9, 19. [Google Scholar] [CrossRef]
- Tsukahara, T.; Emori, M.; Murata, K.; Mizushima, E.; Shibayama, Y.; Kubo, T.; Kanaseki, T.; Hirohashi, Y.; Yamashita, T.; Sato, N.; et al. The future of immunotherapy for sarcoma. Expert. Opin. Biol. Ther. 2016, 16, 1049–1057. [Google Scholar] [CrossRef]
- Krishnadas, D.K.; Shusterman, S.; Bai, F.; Diller, L.; Sullivan, J.E.; Cheerva, A.C.; George, R.E.; Lucas, K.G. A phase I trial combining decitabine/dendritic cell vaccine targeting MAGE-A1, MAGE-A3 and NY-ESO-1 for children with relapsed or therapy-refractory neuroblastoma and sarcoma. Cancer Immunol. Immunother. 2015, 64, 1251–1260. [Google Scholar] [CrossRef]
- Esmaily, M.; Masjedi, A.; Hallaj, S.; Nabi Afjadi, M.; Malakotikhah, F.; Ghani, S.; Ahmadi, A.; Sojoodi, M.; Hassannia, H.; Atyabi, F.; et al. Blockade of CTLA-4 increases anti-tumor response inducing potential of dendritic cell vaccine. J. Control. Release 2020, 326, 63–74. [Google Scholar] [CrossRef]
- Altvater, B.; Pscherer, S.; Landmeier, S.; Kailayangiri, S.; Savoldo, B.; Juergens, H.; Rossig, C. Activated human γδ T cells induce peptide-specific CD8+ T-cell responses to tumor-associated self-antigens. Cancer Immunol. Immunother. 2012, 61, 385–396. [Google Scholar] [CrossRef]
- Dyson, K.A.; Stover, B.D.; Grippin, A.; Mendez-Gomez, H.R.; Lagmay, J.; Mitchell, D.A.; Sayour, E.J. Emerging trends in immunotherapy for pediatric sarcomas. J. Hematol. Oncol. 2019, 12, 78. [Google Scholar] [CrossRef]
- Senzer, N.; Barve, M.; Kuhn, J.; Melnyk, A.; Beitsch, P.; Lazar, M.; Lifshitz, S.; Magee, M.; Oh, J.; Mill, S.W.; et al. Phase I trial of “bi-shRNAi(furin)/GMCSF DNA/autologous tumor cell” vaccine (FANG) in advanced cancer. Mol. Ther. 2012, 20, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Flesner, B.K.; Wood, G.W.; Gayheart-Walsten, P.; Sonderegger, F.L.; Henry, C.J.; Tate, D.J.; Bechtel, S.M.; Donnelly, L.L.; Johnson, G.C.; Kim, D.Y.; et al. Autologous cancer cell vaccination, adoptive T-cell transfer, and interleukin-2 administration results in long-term survival for companion dogs with osteosarcoma. J. Vet. Intern. Med. 2020, 34, 2056–2067. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Toji, S.; Watanabe, K.; Torigoe, T.; Tsukahara, T. Identification of novel human leukocyte antigen-A*11:01-restricted cytotoxic T-lymphocyte epitopes derived from osteosarcoma antigen papillomavirus binding factor. Cancer Sci. 2019, 110, 1156–1168. [Google Scholar] [CrossRef]
- Schlegel, P. T cells for advanced synovial sarcoma or myxoid round cell liposarcoma. Lancet 2024, 403, 1421–1423. [Google Scholar] [CrossRef]
- Hashimoto, K.; Nishimura, S.; Ito, T.; Akagi, M. Clinicopathological Assessment of Cancer/Testis Antigens NY-ESO-1 and MAGE-A4 in Highly Aggressive Soft Tissue Sarcomas. Diagnostics 2022, 12, 733. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Class | Target | Drug | Indications |
|---|---|---|---|
| CTLA-4-I | CTLA-4 | Ipilimumab | Melanoma |
| PD-1-I | PD-1 | Cemiplimab | Metastatic cutaneous SqCC |
| Nivolumab | NSCLC, SCLC, RCC, HCC, melanoma, | ||
| Hodgkin’s lymphoma (CHL), HNC (SqCC), | |||
| Metastatic CRC with high MSI or MMRD | |||
| Urothelial carcinoma | |||
| Pembrolizumab | NSCLC, LBCL (mediastinal), Hodgkin’s | ||
| Lymphoma, gastric cancer, melanoma | |||
| Cervical carcinoma, urothelial carcinoma | |||
| PD-L1-I | PD-L1 | Avelumab | Merkel cell carcinoma, urothelial carcinoma |
| Atezolizumab | NSCLC, urothelial carcinoma | ||
| Durvalumab | NSCLC, urothelial carcinoma | ||
| CTLA-4-I + PD-1-I | CTLA-4 PD-1 | Ipilimumab AND Nivolumab | CRC (some subtypes), RCC, melanoma |
| Class | Advantages | Disadvantages |
|---|---|---|
| DNA vaccine | Simple and safe to manufacture and stable for storage, both cellular and humoral immunity, and long-term expression potential | Lack of positioning effect and targeting, gene integration with subsequent oncogenic potential, low uptake, transfection, and enzyme degradation |
| RNA vaccine | Safe, simple, and flexible to manufacture, no risk of genetic integration, and short half-life | Internal instability, low transfection efficiency |
| Peptide vaccine | Better safety and specificity, bypass the APC presenting process, simple, convenient, and economical preparation process | Weak immunogenicity for fewer epitopes, immunological tolerance for short peptides, delivery instability |
| Tumor lysates vaccine | Simple to manufacture without additional synthesis, it includes most of the potential tumor antigens | Immunosuppression inducer, weak antitumor immunity response due to interfering cellular content |
| DC vaccine | Distinct antitumor ability with better clinical efficacy | High costs with regard to treatment and manufacturing, low efficiency in vitro-induced DC mortality |
| Tumor cell membrane vaccine | Antitumor immunity promoter with multiple antigen motifs, avoids the interference of cell contents | Complexity with regard to the membrane structure limiting industrial and clinical applications |
| ICD-induced antigen vaccine | Strong operability, multiple induced methods, and ability to personalize vaccines (personalized vaccine therapy) | More energy consumption and cellular content interference |
| NCT # | Name | Status | Res | Interventions | Phases | # | Type | Study Design | Start Date | End Date |
|---|---|---|---|---|---|---|---|---|---|---|
| 06669013 | A | RECR. | NO | Dinutuximab beta | 3 | 40 | INT. | Random., IM | 5/20/2021 | 2025-09 |
| 06202599 | B | COMPL. | NO | Fruquintinib | N.A. | 124 | OBS. | Observational Model | 11/25/2021 | 11/15/2023 |
| 06171282 | C | RECR. | NO | Oncolytic Recombinant HSV-1, R130 | Early 1 | 9 | INT. | SGIM | 7/12/2023 | 7/12/2026 |
| 06114225 | D | RECR. | NO | Metastasectomy and pre-op. IT (gemcitabine and penpulimab) and stereotactic body RT | 2 | 43 | INT. | IM | 6/1/2023 | 12/30/2026 |
| 05851456 | E | RECR. | NO | Oncolytic Rec.HSV-1, R130 | Early 1 | 20 | INT. | IM | 4/10/2023 | 2026-04 |
| 05726383 | F | RECR. | NO | DRUG: Iscador*P | 2 | 32 | INT. | IM | 5/14/2024 | 5/11/2027 |
| 05660408 | G | N.A. | NO | pp65 RNA | 1–2 | 36 | INT. | IM | 2025-01 | 2035-10 |
| 05241132 | H | N.A. | NO | Tislelizumab plus chemotherapy | 2 | 27 | INT. | IM | 11/12/2021 | 10/31/2024 |
| 04751383 | I | TERM. | NO | Dinutuximab, Magrolimab | 1 | 12 | INT. | IM | 8/31/2021 | 9/30/2024 |
| 04730349 | J | TERM. | YES | Nivolumab|, NKTR-214 | 1–2 | 15 | INT. | NRIM | 6/3/2021 | 6/22/2022 |
| 04616248 | K | RECR. | NO | Anti-CD40 Agonist, Pembrolizumab, Tocilizumab | 1 | 18 | INT. | NRIM | 1/9/2023 | 1/9/2027 |
| 04483778 | L | N.A. | NO | Pembrolizumab | 1 | 68 | INT. | NRIM | 7/13/2020 | 2040-12 |
| 04433221 | M | N.A. | NO | Multiple sarcoma-specific CAR-T cells and sarcoma vaccines | 1–2 | 20 | INT. | IM | 7/1/2020 | 12/31/2023 |
| 03842865 | N | N.A. | NO | Vigil | N.A. | N.A. | N.A. | |||
| 03782363 | O | N.A. | NO | Autologous CIK | 1 | 0 | INT. | NRIM | 12/18/2020 | 4/1/2023 |
| 03635632 | P | N.A. | NO | C7R-GD2.CART cells | 1 | 94 | INT. | NRIM | 4/23/2019 | 5/16/2038 |
| 03618381 | Q | RECR. | NO | 2nd Gen 4-1BBÎ, EGFR806-EGFRt (CD19-Her2tG) | 1 | 44 | INT. | IM | 6/18/2019 | 2040-06 |
| 03013127 | R | TERM. | NO | DRUG: Pembrolizumab | 2 | 12 | INT. | IM | 5/30/2017 | 1/31/2019 |
| 03006848 | S | COMPL. | YES | Avelumab | 2 | 19 | INT. | IM | 2/16/2017 | 3/18/2020 |
| 02982486 | T | N.A. | NO | Ipilimumab, Nivolumab | 2 | 60 | INT. | IM | 2017-12 | 2020-12 |
| 02173093 | U | N.A. | NO | IL-2, GD2Bi-aATC, GM-CSF | 1–2 | 40 | INT. | IM | 2014-11 | 2019-12 |
| 02107963 | V | COMPL. | NO | Anti-GD2-CAR engineered T cells, AP1903, Cyclophosphamide | 1 | 15 | INT. | NRIM | 2/28/2014 | 1/31/2017 |
| 02100891 | W | COMPL. | NO | Allogeneic HCT (Donor NK Cell Infusion) | 2 | 15 | INT. | IM | 3/20/2013 | 7/15/2020 |
| 00001564 | X | COMPL. | NO | EF-1 Peptide, EF-2 Peptide, PXFK Peptide, E7 Peptide, IL-2, IL-4, GM-CSF, CD40 Ligand | 2 | 30 | INT. | IM | 12/23/1996 | 10/25/2007 |
| 00001566 | Y | COMPL. | YES | Autologous dendritic cells (indinavir sulfate) | 2 | 42 | INT. | IM | 1996-12 | 2008-09 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sergi, C.M.; Burnett, M.; Jantuan, E.; Hakoum, M.; Beug, S.T.; Leng, R.; Shen, F. Digging Through the Complexities of Immunological Approaches in Emerging Osteosarcoma Therapeutics: A Comprehensive Narrative Review with Updated Clinical Trials. Biomedicines 2025, 13, 664. https://doi.org/10.3390/biomedicines13030664
Sergi CM, Burnett M, Jantuan E, Hakoum M, Beug ST, Leng R, Shen F. Digging Through the Complexities of Immunological Approaches in Emerging Osteosarcoma Therapeutics: A Comprehensive Narrative Review with Updated Clinical Trials. Biomedicines. 2025; 13(3):664. https://doi.org/10.3390/biomedicines13030664
Chicago/Turabian StyleSergi, Consolato M., Mervin Burnett, Eugeniu Jantuan, Mariam Hakoum, Shawn T. Beug, Roger Leng, and Fan Shen. 2025. "Digging Through the Complexities of Immunological Approaches in Emerging Osteosarcoma Therapeutics: A Comprehensive Narrative Review with Updated Clinical Trials" Biomedicines 13, no. 3: 664. https://doi.org/10.3390/biomedicines13030664
APA StyleSergi, C. M., Burnett, M., Jantuan, E., Hakoum, M., Beug, S. T., Leng, R., & Shen, F. (2025). Digging Through the Complexities of Immunological Approaches in Emerging Osteosarcoma Therapeutics: A Comprehensive Narrative Review with Updated Clinical Trials. Biomedicines, 13(3), 664. https://doi.org/10.3390/biomedicines13030664