
Cardiometabolic Phenotype in HFpEF: Insights from Murine Models

 , , ,
, , ,
Abstract
1. Introduction
- Clinical proof of HFpEF’ inflammatory nature
- HFpEF is a heterogeneous disease, mainly with cardiometabolic phenotype
- Therapies targeting inflammation in HF
2. HFpEF Animal Models
- (1)
- Obesity-related HFpEF (leptin-deficient ob/ob and leptin receptor-deficient db/db mutant strains; induced by high-fat diet (HFD));
- (2)
- HFpEF induced by arterial hypertension (chronic subjection to deoxycorticosterone acetate (DOCA), DOCA + angiotensin II infusion, transverse aortic constriction (TAC), chronic subjection to angiotensin II, DOCA infusion + TAC);
- (3)
- Ageing HFpEF (senescence-accelerated mouse strains).
- (1)
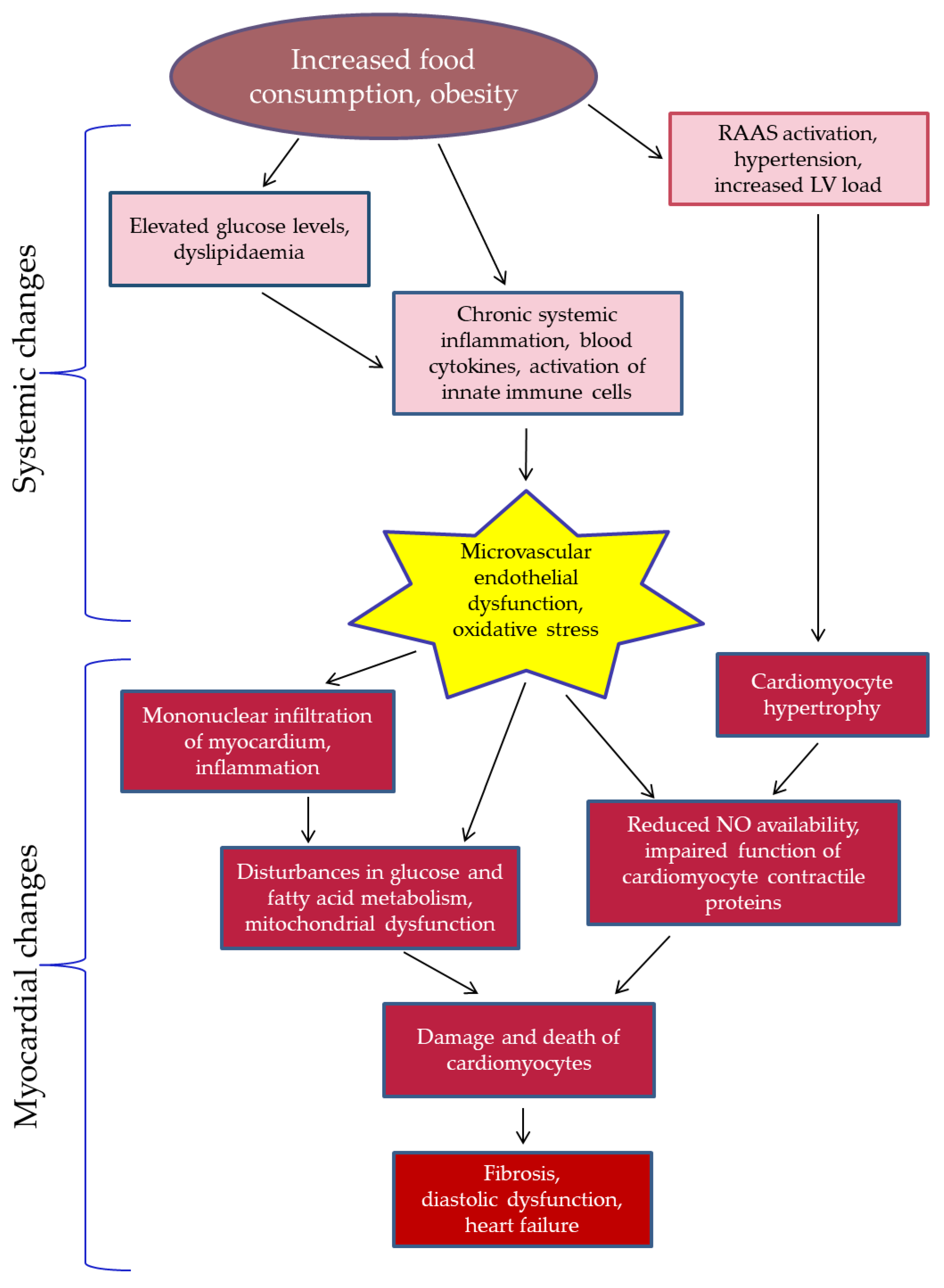
- Inflammation (production of pro-inflammatory cytokines and activation of inflammatory cells);
- (2)
- Oxidative stress (dysfunction of the antioxidant systems and overproduction of ROS, mitochondrial dysfunction);
- (3)
- Dysfunction of the NO/cyclic guanosine monophosphate (cGMP)/protein kinase G (PKG) pathway (decreased PKG and eNOS activity, activation of inducible NOS (iNOS), decreased cGMP and NO production);
- (4)
- Changes in the level of phosphorylation in cardiomyocyte proteins involved in contraction and regulation of intracellular calcium waves (titin, myosin heavy chain (MHC), sarco-endoplasmic reticulum Ca2+-ATPase (SERCA), phospholamban);
- (5)
- Changes in myocardial metabolism (suppression of glycolysis, increased fatty acid uptake and oxidation, impaired processes of oxidative phosphorylation and ATP synthesis).
2.1. Obesity-Related Models of HFpEF
2.1.1. Diabetic db/db and Obese ob/ob Mice
2.1.2. HFD
2.2. Hypertension-Induced HFpEF Models
2.2.1. DOCA + High Salt Diet + Unilateral Nephrectomy [DOCA]
2.2.2. Angiotensin II (Angiotensin II Infusion) [Ang II]
2.2.3. DOCA + High Salt Diet + Angiotensin II + Unilateral Nephrectomy [DOCA + Ang II]
2.2.4. Transverse Aortic Constriction (TAC)
2.2.5. TAC + DOCA
2.3. HFpEF as a Result of Accelerated Ageing (The Senescence-Accelerated Mice)
3. Limitations of HFpEF Murine Models
4. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADMA | Asymmetric dimethylarginine |
| ANP | Atrial natriuretic peptide |
| Bcl-2 | anti-apoptotic B-cell lymphoma 2 |
| BNP | Brain natriuretic peptide |
| cGMP | Cyclic guanosine monophosphate |
| cMYBP-C | Myosin-binding protein C |
| DOCA | Deoxycorticosterone acetate |
| eIF2α | Eukaryotic initiation factor 2 |
| eNOS | Endothelial nitric oxide synthase |
| GSH | Glutathione |
| GSSG | Glutathione disulfide |
| HF | Heart failure |
| HFD | High-fat diet |
| HFpEF | Heart failure with preserved ejection fraction |
| HFrEF | Heart failure with reduced ejection fraction |
| IL | Interleukin |
| IRE-1 | Inositol-requiring protein-1α |
| iNOS | Inducible nitric oxide synthase |
| IRS-1 | Insulin receptor substrate-1 |
| IVRT | Isovolumic relaxation time |
| LV | Left ventricular |
| MCP-1 | Monocyte chemotactic protein-1 |
| MHC | Myosin heavy chain |
| NAD+ | Nicotinamide adenine dinucleotide phosphate |
| NADH | Nicotinamide adenine dinucleotide phosphate |
| nNOS | Neuronal nitric oxide synthase |
| NO | Nitric oxide |
| NOX | Nicotinamide adenine dinucleotide phosphate oxidase |
| NT-proBNP | N-terminal fragment of brain natriuretic hormone precursor |
| PERK | Protein kinase RNA-like endoplasmic reticulum kinase |
| PKG | Protein kinase G |
| ROS | Reactive oxygen species |
| SAMP | Senescence-accelerated prone strain |
| SERCA | Sarco-endoplasmic reticulum Ca2+-ATPase |
| SIRT | Silent mating type information regulation 2 homolog 1 |
| T2DM | Type 2 diabetes mellitus |
| TAC | Transverse aortic constriction |
| TGF | Transforming growth factor |
| TNF | Tumour necrosis factor |
| XOR | Xanthine oxidoreductase |
References
- Vasan, R.S.; Xanthakis, V.; Lyass, A.; Andersson, C.; Tsao, C.; Cheng, S.; Aragam, J.; Benjamin, E.J.; Larson, M.G. Epidemiology of Left Ventricular Systolic Dysfunction and Heart Failure in the Framingham Study: An Echocardiographic Study over 3 Decades. JACC Cardiovasc. Imaging 2018, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Borlaug, B.A.; Jensen, M.D.; Kitzman, D.W.; Lam, C.; Obokata, M.; Rider, O.J. Obesity and heart failure with preserved ejection fraction: New insights and pathophysiological targets. Cardiovasc. Res. 2023, 118, 3434–3450. [Google Scholar] [CrossRef]
- Shah, K.S.; Xu, H.; Matsouaka, R.A.; Bhatt, D.L.; Heidenreich, P.A.; Hernandez, A.F.; Devore, A.D.; Yancy, C.W.; Fonarow, G.C. Heart Failure with Preserved, Borderline, and Reduced Ejection Fraction: 5-Year Outcomes. J. Am. Coll. Cardiol. 2017, 70, 2476–2486. [Google Scholar] [CrossRef]
- Heidenreich, P.A.; Bozkurt, B.; Aguilar, D.; Allen, L.A.; Byun, J.J.; Colvin, M.M.; Deswal, A.; Drazner, M.H.; Dunlay, S.M.; Evers, L.R.; et al. 2022 AHA/ACC/HFSA Guideline for the Management of Heart Failure: Executive Summary: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 2022, 145, e876–e894. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. ESC Scientific Document Group. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef] [PubMed]
- Bozkurt, B.; Coats, A.; Tsutsui, H.; Abdelhamid, C.M.; Adamopoulos, S.; Albert, N.; Anker, S.D.; Atherton, J.; Böhm, M.; Butler, J.; et al. Universal definition and classification of heart failure: A report of the Heart Failure Society of America, Heart Failure Association of the European Society of Cardiology, Japanese Heart Failure Society and Writing Committee of the Universal Definition of Heart Failure: Endorsed by the Canadian Heart Failure Society, Heart Failure Association of India, Cardiac Society of Australia and New Zealand, and Chinese Heart Failure Association. Eur. J. Heart Fail. 2021, 23, 352–380. [Google Scholar] [CrossRef]
- Shah, S.J.; Kitzman, D.W.; Borlaug, B.A.; van Heerebeek, L.; Zile, M.R.; Kass, D.A.; Paulus, W.J. Phenotype-Specific Treatment of Heart Failure with Preserved Ejection Fraction: A Multiorgan Roadmap. Circulation 2016, 134, 73–90. [Google Scholar] [CrossRef]
- Anker, S.D.; Butler, J.; Filippatos, G.; Ferreira, J.P.; Bocchi, E.; Böhm, M.; Brunner-La Rocca, H.P.; Choi, D.J.; Chopra, V.; Chuquiure-Valenzuela, E.; et al. Empagliflozin in Heart Failure with a Preserved Ejection Fraction. N. Engl. J. Med. 2021, 385, 1451–1461. [Google Scholar] [CrossRef]
- Solomon, S.D.; McMurray, J.; Claggett, B.; de Boer, R.A.; DeMets, D.; Hernandez, A.F.; Inzucchi, S.E.; Kosiborod, M.N.; Lam, C.; Martinez, F.; et al. Dapagliflozin in Heart Failure with Mildly Reduced or Preserved Ejection Fraction. N. Engl. J. Med. 2022, 387, 1089–1098. [Google Scholar] [CrossRef]
- Solomon, S.D.; McMurray, J.; Vaduganathan, M.; Claggett, B.; Jhund, P.S.; Desai, A.S.; Henderson, A.D.; Lam, C.; Pitt, B.; Senni, M.; et al. Finerenone in Heart Failure with Mildly Reduced or Preserved Ejection Fraction. N. Engl. J. Med. 2024, 391, 1475–1485. [Google Scholar] [CrossRef]
- Redfield, M.M.; Borlaug, B.A. Heart Failure with Preserved Ejection Fraction: A Review. JAMA 2023, 329, 827–838. [Google Scholar] [CrossRef] [PubMed]
- Ovchinnikov, A.G.; Arefieva, T.I.; Potekhina, A.V.; Filatova, A.Y.; Ageev, F.T.; Boytsov, S.A. The Molecular and Cellular Mechanisms Associated with a Microvascular Inflammation in the Pathogenesis of Heart Failure with Preserved Ejection Fraction. Acta Naturae 2020, 12, 40–51. [Google Scholar] [CrossRef]
- Schiattarella, G.G.; Rodolico, D.; Hill, J.A. Metabolic inflammation in heart failure with preserved ejection fraction. Cardiovasc. Res. 2021, 117, 423–434. [Google Scholar] [CrossRef]
- Schiattarella, G.G.; Alcaide, P.; Condorelli, G.; Gilette, T.G.; Heymans, S.; Jones, A.; Kallikourdis, M.; Lichtman, A.; Marelli-Berg, F.; Shah, S.; et al. Immunometabolic mechanisms of heart failure with preserved ejection fraction. Nat. Cardiovasc. Res. 2022, 1, 211–222. [Google Scholar] [CrossRef]
- Higashi, Y. Roles of oxidative stress and inflammation in vascular endothelial dysfunction-related disease. Antioxidants 2022, 11, 1958. [Google Scholar] [CrossRef] [PubMed]
- Kovács, Á.; Alogna, A.; Post, H.; Hamdani, N. Is enhancing cGMP-PKG signalling a promising therapeutic target for heart failure with preserved ejection fraction? Neth. Heart J. 2016, 24, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Putko, B.N.; Wang, Z.; Lo, J.; Anderson, T.; Becher, H.; Dyck, J.; Kassiri, Z.; Oudit, G.Y.; Alberta HEART Investigators. Circulating levels of tumor necrosis factor-alpha receptor 2 are increased in heart failure with preserved ejection fraction relative to heart failure with reduced ejection fraction: Evidence for a divergence in pathophysiology. PLoS ONE 2014, 9, e99495. [Google Scholar] [CrossRef]
- Tromp, J.; Khan, M.; Klip, I.T.; Meyer, S.; de Boer, R.A.; Jaarsma, T.; Hillege, H.; van Veldhuisen, D.J.; van der Meer, P.; Voors, A.A. Biomarker Profiles in Heart Failure Patients with Preserved and Reduced Ejection Fraction. J. Am. Heart Assoc. 2017, 6, e003989. [Google Scholar] [CrossRef]
- Santhanakrishnan, R.; Chong, J.P.; Ng, T.P.; Ling, L.H.; Sim, D.; Leong, K.T.; Yeo, P.S.; Ong, H.Y.; Jaufeerally, F.; Wong, R.; et al. Growth differentiation factor 15, ST2, high-sensitivity troponin T, and N-terminal pro brain natriuretic peptide in heart failure with preserved vs. reduced ejection fraction. Eur. J. Heart Fail. 2012, 14, 1338–1347. [Google Scholar] [CrossRef]
- Sanders-van Wijk, S.; van Empel, V.; Davarzani, N.; Maeder, M.T.; Handschin, R.; Pfisterer, M.E.; Brunner-La Rocca, H.P.; TIME-CHF Investigators. Circulating biomarkers of distinct pathophysiological pathways in heart failure with preserved vs. reduced left ventricular ejection fraction. Eur. J. Heart Fail. 2015, 17, 1006–1014. [Google Scholar] [CrossRef]
- Tromp, J.; Westenbrink, B.D.; Ouwerkerk, W.; van Veldhuisen, D.J.; Samani, N.J.; Ponikowski, P.; Metra, M.; Anker, S.D.; Cleland, J.G.; Dickstein, K.; et al. Identifying Pathophysiological Mechanisms in Heart Failure with Reduced Versus Preserved Ejection Fraction. J. Am. Coll. Cardiol. 2018, 72, 1081–1090. [Google Scholar] [CrossRef] [PubMed]
- Carris, N.W.; Mhaskar, R.; Coughlin, E.; Bracey, E.; Tipparaju, S.M.; Halade, G.V. Novel biomarkers of inflammation in heart failure with preserved ejection fraction: Analysis from a large prospective cohort study. BMC Cardiovasc. Disord. 2022, 22, 221. [Google Scholar] [CrossRef] [PubMed]
- Lakhani, I.; Wong, M.V.; Hung, J.; Gong, M.; Waleed, K.B.; Xia, Y.; Lee, S.; Roever, L.; Liu, T.; Tse, G.; et al. Diagnostic and prognostic value of serum C-reactive protein in heart failure with preserved ejection fraction: A systematic review and meta-analysis. Heart Fail. Rev. 2021, 26, 1141–1150. [Google Scholar] [CrossRef]
- Alogna, A.; Koepp, K.E.; Sabbah, M.; Espindola Netto, J.M.; Jensen, M.D.; Kirkland, J.L.; Lam, C.; Obokata, M.; Petrie, M.C.; Ridker, P.M.; et al. Interleukin-6 in Patients with Heart Failure and Preserved Ejection Fraction. JACC Heart Fail. 2023, 11, 1549–1561. [Google Scholar] [CrossRef] [PubMed]
- Abernethy, A.; Raza, S.; Sun, J.L.; Anstrom, K.J.; Tracy, R.; Steiner, J.; VanBuren, P.; LeWinter, M.M. Pro-inflammatory biomarkers in stable versus acutely decompensated heart failure with preserved ejection fraction. J. Am. Heart Assoc. 2018, 7, e007385. [Google Scholar] [CrossRef]
- Chaar, D.; Dumont, B.L.; Vulesevic, B.; Neagoe, P.-E.; Räkel, A.; White, M.; Sirois, M.G. Neutrophils and Circulating Inflammatory Biomarkers in Diabetes Mellitus and Heart Failure with Preserved Ejection Fraction. Am. J. Cardiol. 2022, 178, 80–88. [Google Scholar] [CrossRef]
- Westermann, D.; Lindner, D.; Kasner, M.; Zietsch, C.; Savvatis, K.; Escher, F.; von Schlippenbach, J.; Skurk, C.; Steendijk, P.; Riad, A.; et al. Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circ. Heart Fail. 2011, 4, 44–52. [Google Scholar] [CrossRef]
- Franssen, C.; Chen, S.; Unger, A.; Korkmaz, H.I.; De Keulenaer, G.W.; Tschöpe, C.; Leite-Moreira, A.F.; Musters, R.; Niessen, H.W.; Linke, W.A.; et al. Myocardial Microvascular Inflammatory Endothelial Activation in Heart Failure with Preserved Ejection Fraction. JACC Heart Fail. 2016, 4, 312–324. [Google Scholar] [CrossRef]
- Mohammed, S.F.; Hussain, S.F.; Mirzoev, S.A.; Edwards, W.D.; Maleszewski, J.J.; Redfield, M.M. Coronary microvascular rarefaction and myocardial fibrosis in heart failure with preserved ejection fraction. Circulation 2015, 131, 550–559. [Google Scholar] [CrossRef]
- Srivaratharajah, K.; Coutinho, T.; deKemp, R.; Liu, P.; Haddad, H.; Stadnick, E.; Davies, R.A.; Chih, S.; Dwivedi, G.; Guo, A.; et al. Reduced Myocardial Flow in Heart Failure Patients with Preserved Ejection Fraction. Circ. Heart Fail. 2016, 9, e002562. [Google Scholar] [CrossRef]
- Glezeva, N.; Voon, V.; Watson, C.; Horgan, S.; McDonald, K.; Ledwidge, M.; Baugh, J. Exaggerated inflammation and monocytosis associate with diastolic dysfunction in heart failure with preserved ejection fraction: Evidence of M2 macrophage activation in disease pathogenesis. J. Card. Fail. 2015, 21, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Ovchinnikov, A.; Filatova, A.; Potekhina, A.; Arefieva, T.; Gvozdeva, A.; Ageev, F.; Belyavskiy, E. Blood Immune Cell Alterations in Patients with Hypertensive Left Ventricular Hypertrophy and Heart Failure with Preserved Ejection Fraction. J. Cardiovasc. Dev. Dis. 2023, 10, 310. [Google Scholar] [CrossRef]
- Hahn, V.S.; Yanek, L.R.; Vaishnav, J.; Ying, W.; Vaidya, D.; Zhen Joan Lee, Y.; Riley, S.J.; Subramanya, V.; Brown, E.; Danielle Hopkins, C.; et al. Endomyocardial Biopsy Characterization of Heart Failure with Preserved Ejection Fraction and Prevalence of Cardiac Amyloidosis. JACC Heart Fail. 2020, 8, 712–724. [Google Scholar] [CrossRef] [PubMed]
- Hulsmans, M.; Sager, H.B.; Roh, J.D.; Valero-Muñoz, M.; Houstis, N.E.; Iwamoto, Y.; Sun, Y.; Wilson, R.M.; Wojtkiewicz, G.; Tricot, B.; et al. Cardiac macrophages promote diastolic dysfunction. J. Exp. Med. 2018, 215, 423–440. [Google Scholar] [CrossRef]
- Bajpai, G.; Schneider, C.; Wong, N.; Bredemeyer, A.; Hulsmans, M.; Nahrendorf, M.; Epelman, S.; Kreisel, D.; Liu, Y.; Itoh, A.; et al. The human heart contains distinct macrophage subsets with divergent origins and functions. Nat. Med. 2018, 24, 1234–1245. [Google Scholar] [CrossRef]
- Shah, S.J.; Katz, D.H.; Selvaraj, S.; Burke, M.A.; Yancy, C.W.; Cheorghiade, M.; Bonow, R.O.; Huang, C.-C.; Deo, R.C. Phenomapping for Novel Classification of Heart Failure with Preserved Ejection Fraction. Circulation 2015, 131, 269–279. [Google Scholar] [CrossRef]
- Shah, S.J.; Katz, D.H.; Deo, R.C. Phenotypic Spectrum of Heart Failure with Preserved Ejection Fraction. Heart Fail. Clin. 2014, 10, 407–418. [Google Scholar] [CrossRef]
- Teramoto, K.; Katherine Teng, T.-H.; Chandramouli, C.; Tromp, J.; Sakata, Y.; Lam, C.S. Epidemiology and Clinical Features of Heart Failure with Preserved Ejection Fraction. Card. Fail. Rev. 2022, 8, e27. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.B.; Schrauben, S.J.; Zhao, L.; Basso, M.D.; Cvijic, M.E.; Li, Z.; Yarde, M.; Wang, Z.; Bhattacharya, P.T.; Chirinos, D.A.; et al. Clinical Phenogroups in Heart Failure with Preserved Ejection Fraction: Detailed Phenotypes, Prognosis, and Response to Spironolactone. JACC Heart Fail. 2020, 8, 172–184. [Google Scholar] [CrossRef]
- Galli, E.; Bourg, C.; Kosmala, W.; Oger, E.; Donal, E. Phenomapping Heart Failure with Preserved Ejection Fraction Using Machine Learning Cluster Analysis: Prognostic and Therapeutic Implications. Heart Fail. Clin. 2021, 17, 499–518. [Google Scholar] [CrossRef]
- Bronzwaer, J.; Paulus, W.J. Diastolic and systolic heart failure: Different stages or distinct phenotypes of the heart failure syndrome? Curr. Heart Fail. Rep. 2009, 6, 281–286. [Google Scholar] [CrossRef]
- Paulus, W.J.; Tschöpe, C. A Novel Paradigm for Heart Failure with Preserved Ejection Fraction. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef]
- Kao, D.P.; Lewsey, J.D.; Anand, I.S.; Massie, B.M.; Zile, M.R.; Carson, P.E.; McKelvie, R.S.; Komajda, M.; McMurray, J.; Lindenfels, J. Characterization of subgroups of heart failure patients with preserved ejection fraction with possible implications for prognosis and treatment response. Eur. J. Heart Fail. 2015, 17, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, P. The role of adipokines in chronic inflammation. Immunotargets Ther. 2016, 5, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Lam, C.S. Diabetic cardiomyopathy: An expression of stage B heart failure with preserved ejection fraction. Diab Vasc. Dis. Res. 2015, 12, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, R.H.; Abel, E.D. Basic Mechanisms of Diabetic Heart Disease. Circ. Res. 2020, 126, 1501–1525. [Google Scholar] [CrossRef]
- Jackson, A.M.; Rørth, R.; Liu, J.; Kristensen, S.L.; Anand, I.S.; Claggett, B.L.; Cleland, J.; Chopra, V.K.; Desai, A.S.; Ge, J.; et al. Diabetes and pre-diabetes in patients with heart failure and preserved ejection fraction. Eur. J. Heart Fail. 2022, 24, 497–509. [Google Scholar] [CrossRef]
- Lehrke, M.; Marx, N. Diabetes Mellitus and Heart Failure. Am. J. Med. 2017, 130, S40–S50. [Google Scholar] [CrossRef]
- van Heerebeek, L.; Hamdani, N.; Handoko, M.L.; Falcao-Pires, I.; Musters, R.J.; Kupreishvili, K.; Ijsselmuiden, A.; Schalkwijk, C.G.; Bronzwaer, J.; Diamant, M.; et al. Diastolic stiffness of the failing diabetic heart: Importance of fibrosis, advanced glycation end products, and myocyte resting tension. Circulation 2008, 117, 43–51. [Google Scholar] [CrossRef]
- Plante, T.B.; Juraschek, S.P.; Howard, G.; Howard, V.J.; Tracy, R.P.; Olson, N.C.; Judd, S.E.; Mukaz, D.K.; Zakai, N.A.; Long, D.L.; et al. Cytokines, C-Reactive Protein, and Risk of Incident Hypertension in the REGARDS Study. Hypertension 2024, 81, 1244–1253. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, J.; Yang, P.; Song, X.; Li, Y. Elevated Th17 cell proportion, related cytokines and mRNA expression level in patients with hypertension-mediated organ damage: A case control study. BMC Cardiovasc. Disord. 2022, 22, 257. [Google Scholar] [CrossRef] [PubMed]
- Cantero-Navarro, E.; Fernández-Fernández, B.; Ramos, A.M.; Rayego-Mateos, S.; Rodrigues-Diez, R.R.; Sánchez-Niño, M.D.; Sanz, A.B.; Ruiz-Ortega, M.; Ortiz, A. Renin-angiotensin system and inflammation update. Mol. Cell Endocrinol. 2021, 529, 111254. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Harrison, D.G. Inflammation in Hypertension. Can. J. Cardiol. 2020, 36, 635–647. [Google Scholar] [CrossRef] [PubMed]
- Lam, C.; Donal, E.; Kraigher-Krainer, E.; Vasan, R.S. Epidemiology and clinical course of heart failure with preserved ejection fraction. Eur. J. Heart Fail. 2011, 13, 18–28. [Google Scholar] [CrossRef]
- Packer, M.; Lam, C.; Lund, L.H.; Maurer, M.S.; Borlaug, B.A. Characterization of the inflammatory-metabolic phenotype of heart failure with a preserved ejection fraction: A hypothesis to explain influence of sex on the evolution and potential treatment of the disease. Eur. J. Heart Fail. 2020, 22, 1551–1567. [Google Scholar] [CrossRef]
- Segar, M.W.; Patel, K.V.; Ayers, C.; Basit, M.; Tang, W.; Willett, D.; Berry, J.; Grodin, J.L.; Pandey, A. Phenomapping of patients with heart failure with preserved ejection fraction using machine learning-based unsupervised cluster analysis. Eur. J. Heart Fail. 2020, 22, 148–158. [Google Scholar] [CrossRef]
- Woolley, R.J.; Ceelen, D.; Ouwerkerk, W.; Tromp, J.; Figarska, S.M.; Anker, S.D.; Dickstein, K.; Filippatos, G.; Zannad, F.; Metra, M.; et al. Machine learning based on biomarker profiles identifies distinct subgroups of heart failure with preserved ejection fraction. Eur. J. Heart Fail. 2021, 23, 983–991. [Google Scholar] [CrossRef]
- Anker, S.D.; Butler, J.; Filippatos, G.; Shahzeb Khan, M.; Ferreira, J.P.; Bocchi, E.; Böhm, M.; Brunner-La Rocca, H.P.; Choi, D.J.; Chopra, V.; et al. Baseline characteristics of patients with heart failure with preserved ejection fraction in the EMPEROR-Preserved trial. Eur. J. Heart Fail. 2020, 22, 2383–2392. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory therapy with Canakinumab for atherosclerotic disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Van Tassell, B.W.; Buckley, L.F.; Carbone, S.; Trankle, C.R.; Canada, J.M.; Dixon, D.L.; Abouzaki, N.; Oddi-Erdle, C.; Biondi-Zoccai, G.; Arena, R.; et al. Interleukin-1 blockade in heart failure with preserved ejection fraction: Rationale and design of the Diastolic Heart Failure Anakinra Response Trial 2 (D-HART2). Clin. Cardiol. 2017, 40, 626–632. [Google Scholar] [CrossRef]
- Lam, C.; Lund, L.; Shah, S.; Voors, A.A.; Erlinge, D.; Saraste, A.; Pirazzi, C.; Grove, E.L.; Barasa, A.; Schou, M.; et al. Myeloperoxidase inhibition in heart failure with preserved or mildly reduced ejection fraction: SATELLITE trial results. J. Card. Fail. 2024, 30, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Riehle, C.; Bauersachs, J. Small animal models of heart failure. Cardiovasc. Res. 2019, 115, 1838–1849. [Google Scholar] [CrossRef]
- Miyagi, C.; Miyamoto, T.; Kuroda, T.; Karimov, J.H.; Starling, R.C.; Fukamachi, K. Large animal models of heart failure with preserved ejection fraction. Heart Fail. Rev. 2022, 27, 595–608. [Google Scholar] [CrossRef]
- Gao, S.; Liu, X.P.; Li, T.T.; Chen, L.; Feng, Y.P.; Wang, Y.K.; Yin, Y.J.; Little, P.J.; Wu, X.Q.; Xu, S.W.; et al. Animal models of heart failure with preserved ejection fraction (HFpEF): From metabolic pathobiology to drug discovery. Acta Pharmacol. Sin. 2024, 45, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Lourenço, A.P.; Leite-Moreira, A.F.; Balligand, J.L.; Bauersachs, J.; Dawson, D.; de Boer, R.A.; de Windt, L.J.; Falcão-Pires, I.; Fontes-Carvalho, R.; Franz, S.; et al. An integrative translational approach to study heart failure with preserved ejection fraction: A position paper from the Working Group on Myocardial Function of the European Society of Cardiology. Eur. J. Heart Fail. 2018, 20, 216–227. [Google Scholar] [CrossRef]
- Schauer, A.; Draskowski, R.; Jannasch, A.; Kirchhoff, V.; Goto, K.; Männel, A.; Barthel, P.; Augstein, A.; Winzer, E.; Tugtekin, M.; et al. ZSF1 rat as animal model for HFpEF: Development of reduced diastolic function and skeletal muscle dysfunction. ESC Heart Fail. 2020, 7, 2123–2134. [Google Scholar] [CrossRef] [PubMed]
- Murase, T.; Hattori, T.; Ohtake, M.; Abe, M.; Amakusa, Y.; Takatsu, M.; Murohara, T.; Nagata, K. Cardiac remodeling and diastolic dysfunction in DahlS.Z-Lepr(fa)/Lepr(fa) rats: A new animal model of metabolic syndrome. Hypertens. Res. 2012, 35, 186–193. [Google Scholar] [CrossRef]
- Schwarzl, M.; Hamdani, N.; Seiler, S.; Alogna, A.; Manninger, M.; Reilly, S.; Zirngast, B.; Kirsch, A.; Steendijk, P.; Verderber, J.; et al. A porcine model of hypertensive cardiomyopathy: Implications for heart failure with preserved ejection fraction. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1407–H1418. [Google Scholar] [CrossRef]
- Sharp, T.E., 3rd; Scarborough, A.L.; Li, Z.; Polhemus, D.J.; Hidalgo, H.A.; Schumacher, J.D.; Matsuura, T.R.; Jenkins, J.S.; Kelly, D.P.; Goodchild, T.T.; et al. Novel Göttingen Miniswine Model of Heart Failure with Preserved Ejection Fraction Integrating Multiple Comorbidities. JACC Basic. Transl. Sci. 2021, 6, 154–170. [Google Scholar] [CrossRef]
- Wang, L.; Halliday, G.; Huot, J.R.; Satoh, T.; Baust, J.J.; Fisher, A.; Cook, T.; Hu, J.; Avolio, T.; Goncharov, D.A.; et al. Treatment with Treprostinil and Metformin Normalizes Hyperglycemia and Improves Cardiac Function in Pulmonary Hypertension Associated with Heart Failure with Preserved Ejection Fraction. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1543–1558. [Google Scholar] [CrossRef]
- Deng, Y.; Xie, M.; Li, Q.; Xu, X.; Ou, W.; Zhang, Y.; Xiao, H.; Yu, H.; Zheng, Y.; Liang, Y.; et al. Targeting Mitochondria-Inflammation Circuit by β-Hydroxybutyrate Mitigates HFpEF. Circ. Res. 2021, 128, 232–245. [Google Scholar] [CrossRef] [PubMed]
- Withaar, C.; Meems, L.; Markousis-Mavrogenis, G.; Boogerd, C.J.; Silljé, H.; Schouten, E.M.; Dokter, M.M.; Voors, A.A.; Westenbrink, B.D.; Lam, C.; et al. The effects of liraglutide and dapagliflozin on cardiac function and structure in a multi-hit mouse model of heart failure with preserved ejection fraction. Cardiovasc. Res. 2021, 117, 2108–2124. [Google Scholar] [CrossRef]
- Gevaert, A.B.; Shakeri, H.; Leloup, A.J.; Van Hove, C.E.; De Meyer, G.; Vrints, C.J.; Lemmens, K.; Van Craenenbroeck, E.M. Endothelial Senescence Contributes to Heart Failure with Preserved Ejection Fraction in an Aging Mouse Model. Circ. Heart Fail. 2017, 10, e003806. [Google Scholar] [CrossRef]
- Li, Y.; Kubo, H.; Yu, D.; Yang, Y.; Johnson, J.P.; Eaton, D.M.; Berretta, R.M.; Foster, M.; McKinsey, T.A.; Yu, J.; et al. Combining three independent pathological stressors induces a heart failure with preserved ejection fraction phenotype. Am. J. Physiol. Heart Circ. Physiol. 2023, 324, H443–H460. [Google Scholar] [CrossRef] [PubMed]
- Kelley, R.C.; Betancourt, L.; Noriega, A.M.; Brinson, S.C.; Curbelo-Bermudez, N.; Hahn, D.; Kumar, R.A.; Balazic, E.; Muscato, D.R.; Ryan, T.E.; et al. Skeletal myopathy in a rat model of postmenopausal heart failure with preserved ejection fraction. J. Appl. Physiol. 2022, 132, 106–125. [Google Scholar] [CrossRef]
- Vilariño-García, T.; Polonio-González, M.L.; Pérez-Pérez, A.; Ribalta, J.; Arrieta, F.; Aguilar, M.; Obaya, J.C.; Gimeno-Orna, J.A.; Iglesias, P.; Navarro, J.; et al. Role of Leptin in Obesity, Cardiovascular Disease, and Type 2 Diabetes. Int. J. Mol. Sci. 2024, 25, 2338. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Charlat, O.; Tartaglia, L.A.; Woolf, E.A.; Weng, X.; Ellis, S.J.; Lakey, N.D.; Culpepper, J.; Moore, K.J.; Breitbart, R.E.; et al. Evidence That the Diabetes Gene Encodes the Leptin Receptor: Identification of a Mutation in the Leptin Receptor Gene in db/db Mice. Cell 1996, 84, 491–495. [Google Scholar] [CrossRef]
- Mori, J.; Patel, V.B.; Alrob, O.A.; Basu, R.; Altamimi, T.; Desaulniers, J.; Wagg, C.S.; Zamaneh, K.; Lopaschuk, G.D.; Oudit, G.Y. Angiotensin 1-7 ameliorates diabetic cardiomyopathy and diastolic dysfunction in db/db mice by reducing lipotoxicity and inflammation. Circ. Heart Fail. 2014, 7, 327–339. [Google Scholar] [CrossRef]
- Vecoli, C.; Cao, J.; Neglia, D.; Inoue, K.; Sodhi, K.; Vanells, L.; Gabrielson, K.K.; Bedja, D.; Paolocci, N.; L’abbate, A.; et al. Apolipoprotein A-I mimetic peptide L-4F prevents myocardial and coronary dysfunction in diabetic mice. J. Cell Biochem. 2011, 112, 2616–2626. [Google Scholar] [CrossRef]
- Buchanan, J.; Mazumder, P.K.; Hu, P.; Chakrabarti, G.; Roberts, M.W.; Jeong Yun, U.; Cooksey, R.C.; Litwin, S.E.; Dale Abel, E. Reduced Cardiac Efficiency and Altered Substrate Metabolism Precedes the Onset of Hyperglycemia and Contractile Dysfunction in Two Mouse Models of Insulin Resistance and Obesity. Endocrinology 2005, 146, 5341–5349. [Google Scholar] [CrossRef]
- Ge, F.; Hu, C.; Hyodo, E.; Arai, K.; Zhou, S.; Lobdell, H., 4th; Walewski, J.L.; Homma, S.; Berk, P.D. Cardiomyocyte Triglyceride Accumulation and Reduced Ventricular Function in Mice with Obesity Reflect Increased Long Chain Fatty Acid Uptake and De Novo Fatty Acid Synthesis. J. Obes. 2012, 2012, 205648. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Forte, T.M.; Taniguchi, S.; Ishida, B.Y.; Oka, K.; Chan, L. The db/db mouse, a model for diabetic dyslipidemia: Molecular characterization and effects of western diet feeding. Metabolism 2000, 49, 22–31. [Google Scholar] [CrossRef]
- Senador, D.; Kanakamedala, K.; Irigoyen, M.C.; Morris, M.; Elased, K.M. Cardiovascular and autonomic phenotype of db/db diabetic mice. Exp. Physiol. 2009, 94, 648–658. [Google Scholar] [CrossRef] [PubMed]
- Bowden, M.A.; Tesch, G.H.; Julius, T.; Rosli, S.; Love, J.E.; Ritchie, R.H. Earlier onset of diabesity-Induced adverse cardiac remodeling in female compared to male mice. Obesity (Silver Spring) 2015, 23, 1166–1177. [Google Scholar] [CrossRef] [PubMed]
- Dong, F.; Ren, J. Adiponectin improves cardiomyocyte contractile function in db/db diabetic obese mice. Obesity (Silver Spring) 2009, 17, 262–268. [Google Scholar] [CrossRef]
- Semeniuk, L.M.; Kryski, A.J.; Severson, D.L. Echocardiographic assessment of cardiac function in diabetic db/db and transgenic db/db-hGLUT4 mice. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H976–H982. [Google Scholar] [CrossRef]
- Daniels, A.; van Bilsen, M.; Janssen, B.; Brouns, A.E.; Cleutjens, J.; Roemen, T.; Schaart, G.; van der Velden, J.; van der Vusse, G.J.; van Nieuwenhoven, F.A. Impaired cardiac functional reserve in type 2 diabetic db/db mice is associated with metabolic, but not structural, remodelling. Acta Physiologica. 2010, 200, 11–22. [Google Scholar] [CrossRef]
- Alex, L.; Russo, I.; Holoborodko, V.; Frangogiannis, N.G. Characterization of a mouse model of obesity-related fibrotic cardiomyopathy that recapitulates features of human heart failure with preserved ejection fraction. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H934–H949. [Google Scholar] [CrossRef]
- Hamdani, N.; Hervent, A.-S.; Vandekerckhove, L.; Matheeussen, V.; Demolder, M.; Baerts, L.; De Meester, I.; Linke, W.A.; Paulus, W.J.; Keulenaer, G. Left ventricular diastolic dysfunction and myocardial stiffness in diabetic mice is attenuated by inhibition of dipeptidyl peptidase 4. Cardiovasc. Res. 2014, 104, 423–431. [Google Scholar] [CrossRef]
- Zhao, R.; Xie, X.; Le, K.; Li, W.; Moghadasian, M.H.; Beta, T.; Shen, G.X. Endoplasmic reticulum stress in diabetic mouse or glycated LDL-treated endothelial cells: Protective effect of Saskatoon berry powder and cyanidin glycans. J. Nutr. Biochem. 2015, 26, 1248–1253. [Google Scholar] [CrossRef]
- Koka, S.; Aluri, H.S.; Xi, L.; Lesnefsky, E.; Kukreja, R.C. Chronic inhibition of phosphodiesterase 5 with tadalafil attenuates mitochondrial dysfunction in type 2 diabetic hearts: Potential role of NO/SIRT1/PGC-1α signaling. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H1558–H1568. [Google Scholar] [CrossRef] [PubMed]
- Monma, Y.; Shindo, T.; Eguchi, K.; Kurosawa, R.; Kagaya, Y.; Ikumi, Y.; Ichijo, S.; Nakata, T.; Miyata, S.; Matsumoto, A.; et al. Low-intensity pulsed ultrasound ameliorates cardiac diastolic dysfunction in mice: A possible novel therapy for heart failure with preserved left ventricular ejection fraction. Cardiovasc. Res. 2021, 117, 1325–1338. [Google Scholar] [CrossRef] [PubMed]
- Reil, J.-C.; Honl, M.; Reil, G.-H.; Granzier, H.L.; Kratz, M.T.; Kazakov, A.; Fries, P.; Müller, A.; Lenski, M.; Custodis, F.; et al. Heart rate reduction by If-inhibition improves vascular stiffness and left ventricular systolic and diastolic function in a mouse model of heart failure with preserved ejection fraction. Eur. Heart J. 2013, 34, 2839–2849. [Google Scholar] [CrossRef] [PubMed]
- Ingalls, A.M.; Dickie, M.M.; Snell, G.D. Obese, a new mutation in the house mouse. J. Hered. 1950, 41, 317–318. [Google Scholar] [CrossRef]
- Barouch, L.A.; Berkowitz, D.E.; Harrison, R.W.; O’Donell, C.P.; Hare, J.M. Disruption of leptin signaling contributes to cardiac hypertrophy independently of body weight in mice. Circulation 2003, 108, 754–759. [Google Scholar] [CrossRef]
- Hammoudi, N.; Jeong, D.; Singh, R.; Farhat, A.; Komajda, M.; Mayoux, E.; Hajjar, R.; Lebeche, D. Empagliflozin Improves Left Ventricular Diastolic Dysfunction in a Genetic Model of Type 2 Diabetes. Cardiovasc. Drugs Ther. 2017, 31, 233–246. [Google Scholar] [CrossRef]
- Guo, W.; Jiang, T.; Lian, C.; Wang, H.; Zheng, Q.; Ma, H. QKI deficiency promotes FoxO1 mediated nitrosative stress and endoplasmic reticulum stress contributing to increased vulnerability to ischemic injury in diabetic heart. J. Mol. Cell Cardiol. 2014, 75, 131–140. [Google Scholar] [CrossRef]
- Minhas, K.M.; Khan, S.A.; Raju, S.; Phan, A.C.; Gonzalez, D.R.; Skaf, M.W.; Lee, K.; Tejani, A.D.; Saliaris, A.P.; Barouch, L.A.; et al. Leptin repletion restores depressed {beta}-adrenergic contractility in ob/ob mice independently of cardiac hypertrophy. J. Physiol. 2005, 565, 463–474. [Google Scholar] [CrossRef]
- Li, S.-Y.; Yang, X.; Ceylan-Isik, A.F.; Du, M.; Sreejayan, N.; Ren, J. Cardiac contractile dysfunction in Lep/Lep obesity is accompanied by NADPH oxidase activation, oxidative modification of sarco(endo)plasmic reticulum Ca2+-ATPase and myosin heavy chain isozyme switch. Diabetologia 2006, 49, 1434–1446. [Google Scholar] [CrossRef]
- Adingupu, D.D.; Göpel, S.O.; Grönros, J.; Behrendt, M.; Sotak, M.; Miliotis, T.; Dahlqvist, U.; Gan, L.-M.; Jönsson-Rylander, A.-C. SGLT2 inhibition with empagliflozin improves coronary microvascular function and cardiac contractility in prediabetic ob/ob-/- mice. Cardiovasc. Diabetol. 2019, 18, 16. [Google Scholar] [CrossRef]
- Christoffersen, C.; Bollano, E.; Lindegaard, M.; Bartels, E.D.; Goetze, J.P.; Andersen, C.B.; Nielsen, L.B. Cardiac lipid accumulation associated with diastolic dysfunction in obese mice. Endocrinology 2003, 144, 3483–3490. [Google Scholar] [CrossRef]
- Ye, Y.; Bajaj, M.; Yang, H.-C.; Perez-Polo, J.R.; Birnbaum, Y. SGLT-2 Inhibition with Dapagliflozin Reduces the Activation of the Nlrp3/ASC Inflammasome and Attenuates the Development of Diabetic Cardiomyopathy in Mice with Type 2 Diabetes. Further Augmentation of the Effects with Saxagliptin, a DPP4 Inhibitor. Cardiovasc. Drugs Ther. 2017, 31, 119–132. [Google Scholar] [CrossRef]
- Mariappan, N.; Elks, C.M.; Sriramula, S.; Guggilam, A.; Liu, Z.; Borkhsenious, O.; Francis, J. NF-kappaB-induced oxidative stress contributes to mitochondrial and cardiac dysfunction in type II diabetes. Cardiovasc. Res. 2010, 85, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Methawasin, M.; Strom, J.; Borkowski, T.; Hourani, Z.; Runyan, R.; Smith, J.E., 3rd; Granzier, H. Phosphodiesterase 9a Inhibition in Mouse Models of Diastolic Dysfunction. Circ. Heart Fail. 2020, 13, e006609. [Google Scholar] [CrossRef] [PubMed]
- Broderick, T.L.; Parrott, C.R.; Wang, D.; Jankowski, M.; Gutkowska, J. Expression of cardiac GATA4 and downstream genes after exercise training in the db/db mouse. Pathophysiology 2012, 19, 193–203. [Google Scholar] [CrossRef]
- Belke, D.D.; Swanson, E.A.; Dillmann, W.H. Decreased sarcoplasmic reticulum activity and contractility in diabetic db/db mouse heart. Diabetes 2004, 53, 3201–3208. [Google Scholar] [CrossRef]
- Lugnier, C.; Meyer, A.; Charloux, A.; Andrès, E.; Gény, B.; Talha, S. The Endocrine Function of the Heart: Physiology and Involvements of Natriuretic Peptides and Cyclic Nucleotide Phosphodiesterases in Heart Failure. J. Clin. Med. 2019, 8, 1746. [Google Scholar] [CrossRef]
- Juguilon, C.; Wang, Z.; Wang, Y.; Enrick, M.; Jamaiyar, A.; Xu, Y.; Gadd, J.; Chen, C.-L.W.; Pu, A.; Kolz, C.; et al. Mechanism of the switch from NO to H2O2 in endothelium-dependent vasodilation in diabetes. Basic. Res. Cardiol. 2022, 117, 2. [Google Scholar] [CrossRef] [PubMed]
- Saraiva, R.M.; Minhas, K.M.; Zheng, M.; Pitz, E.; Treuer, A.; Gonzalez, D.; Schuleri, K.H.; Vandegaer, K.M.; Barouch, L.A.; Hare, J.M. Reduced neuronal nitric oxide synthase expression contributes to cardiac oxidative stress and nitroso-redox imbalance in ob/ob mice. Nitric Oxide 2007, 16, 331–338. [Google Scholar] [CrossRef]
- Schiattarella, G.G.; Altamirano, F.; Tong, D.; French, K.M.; Villalobos, E.; Kim, S.Y.; Luo, X.; Jiang, N.; May, H.I.; Wang, Z.V.; et al. Nitrosative stress drives heart failure with preserved ejection fraction. Nature 2019, 568, 351–356. [Google Scholar] [CrossRef]
- van Heerebeek, L.; Borbély, A.; Niessen, H.; Bronzwaer, J.; van der Velden, J.; Stienen, G.; Linke, W.A.; Laarman, G.J.; Paulus, W.J. Myocardial Structure and Function Differ in Systolic and Diastolic Heart Failure. Circulation 2006, 113, 1966–1973. [Google Scholar] [CrossRef]
- Linke, W.A.; Hamdani, N. Gigantic Business: Titin properties and function through thick and thin. Circ. Res. 2014, 114, 1052–1068. [Google Scholar] [CrossRef] [PubMed]
- Greene, S.J.; Gheorghiade, M.; Borlaug, B.A.; Pieske, B.; Vaduganathan, M.; Burnett, J.C., Jr.; Roessig, L.; Stasch, J.-P.; Solomon, S.D.; Paulus, W.J.; et al. The cGMP Signaling Pathway as a Therapeutic Target in Heart Failure with Preserved Ejection Fraction. J. Am. Heart Assoc. 2013, 2, e000536. [Google Scholar] [CrossRef]
- Münzel, T.; Gori, T.; Keaney, J.F., Jr.; Maack, C.; Daiber, A. Pathophysiological role of oxidative stress in systolic and diastolic heart failure and its therapeutic implications. Eur. Heart J. 2015, 36, 2555–2564. [Google Scholar] [CrossRef] [PubMed]
- van Heerebeek, L.; Hamdani, N.; Falcão-Pires, I.; Leite-Moreira, A.F.; Begieneman, M.; Bronzwaer, J.; van der Velden, J.; Stienen, G.; Laarman, G.J.; Somsen, A.; et al. Low myocardial protein kinase G activity in heart failure with preserved ejection fraction. Circulation 2012, 126, 830–839. [Google Scholar] [CrossRef]
- Williams, T.D.; Chambers, J.B.; Roberts, L.M.; Henderson, R.P.; Overton, J.M. Diet-induced obesity and cardiovascular regulation in C57BL/6J mice. Clin. Exp. Pharmacol. Physiol. 2003, 30, 769–778. [Google Scholar] [CrossRef]
- Basheer, S.; Malik, I.R.; Awan, F.R.; Sughra, K.; Roshan, S.; Khalil, A.; Iqbal, M.J.; Parveen, Z. Histological and Microscopic Analysis of Fats in Heart, Liver Tissue, and Blood Parameters in Experimental Mice. Genes 2023, 14, 515. [Google Scholar] [CrossRef]
- Benetti, E.; Mastrocola, R.; Vitarelli, G.; Cutrin, J.C.; Nigro, D.; Chiazza, F.; Mayoux, E.; Collino, M.; Fantozzi, R. Empagliflozin Protects against Diet-Induced NLRP-3 Inflammasome Activation and Lipid Accumulation. J. Pharmacol. Exp. Ther. 2016, 359, 45–53. [Google Scholar] [CrossRef]
- Zhou, X.; Li, Z.; Qi, M.; Zhao, P.; Duan, Y.; Yang, G.; Yuan, L. Brown adipose tissue-derived exosomes mitigate the metabolic syndrome in high fat diet mice. Theranostics 2020, 10, 8197–8210. [Google Scholar] [CrossRef]
- Wang, H.-T.; Liu, C.-F.; Tsai, T.-H.; Chen, Y.-L.; Chang, H.-W.; Tsai, C.-Y.; Leu, S.; Zhen, Y.-Y.; Chai, H.-T.; Chung, S.-Y.; et al. Effect of obesity reduction on preservation of heart function and attenuation of left ventricular remodeling, oxidative stress and inflammation in obese mice. J. Transl. Med. 2012, 10, 145. [Google Scholar] [CrossRef]
- Abdurrachim, D.; Ciapaite, J.; Wessels, B.; Nabben, M.; Luiken, J.; Nicolay, K.; Prompers, J.J. Cardiac diastolic dysfunction in high-fat diet fed mice is associated with lipotoxicity without impairment of cardiac energetics in vivo. Biochim. Biophys. Acta 2014, 1842, 1525–1537. [Google Scholar] [CrossRef] [PubMed]
- Steven, S.; Dib, M.; Hausding, M.; Kashani, F.; Oelze, M.; Kröller-Schön, S.; Hanf, A.; Daub, S.; Roohani, S.; Gramlich, Y.; et al. CD40L controls obesity-associated vascular inflammation, oxidative stress, and endothelial dysfunction in high fat diet-treated and db/db mice. Cardiovasc. Res. 2018, 114, 312–323. [Google Scholar] [CrossRef]
- Li, W.; Tang, R.; Ouyang, S.; Ma, F.; Liu, Z.; Wu, J. Folic acid prevents cardiac dysfunction and reduces myocardial fibrosis in a mouse model of high-fat diet-induced obesity. Nutr. Metab. 2017, 14, 68. [Google Scholar] [CrossRef] [PubMed]
- Jeong, E.; Chung, J.; Liu, H.; Go, Y.; Gladstein, S.; Farzaneh-Far, A.; Lewandowski, E.D.; Dudley, S.C., Jr. Role of Mitochondrial Oxidative Stress in Glucose Tolerance, Insulin Resistance, and Cardiac Diastolic Dysfunction. J. Am. Heart Assoc. 2016, 5, e003046. [Google Scholar] [CrossRef]
- Carbone, S.; Mauro, A.G.; Mezzaroma, E.; Kraskauskas, D.; Marchetti, C.; Buzzetti, R.; Van Tassell, B.W.; Abbate, A.; Toldo, S. A high-sugar and high-fat diet impairs cardiac systolic and diastolic function in mice. Int. J. Cardiol. 2015, 198, 66–69. [Google Scholar] [CrossRef]
- Roche, C.; Besnier, M.; Cassel, R.; Harouki, N.; Coquerel, D.; Guerrot, D.; Nicol, L.; Loizon, E.; Remy-Jouet, I.; Morisseau, C.; et al. Soluble epoxide hydrolase inhibition improves coronary endothelial function and prevents the development of cardiac alterations in obese insulin-resistant mice. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H1020–H1029. [Google Scholar] [CrossRef]
- Heinzel, F.R.; Shah, S.J. The future of heart failure with preserved ejection fraction: Deep phenotyping for targeted therapeutics. Herz 2022, 47, 308–323. [Google Scholar] [CrossRef]
- Xia, N.; Weisenburger, S.; Koch, E.; Burkart, M.; Reifenberg, G.; Förstermann, U.; Li, H. Restoration of perivascular adipose tissue function in diet-induced obese mice without changing bodyweight. Br. J. Pharmacol. 2017, 174, 3443–3453. [Google Scholar] [CrossRef]
- Smart, C.D.; Fehrenbach, D.J.; Wassenaar, J.W.; Agrawal, V.; Fortune, N.L.; Dixon, D.D.; Cottam, M.A.; Hasty, A.H.; Hemnes, A.R.; Doran, A.C.; et al. Immune profiling of murine cardiac leukocytes identifies triggering receptor expressed on myeloid cells 2 as a novel mediator of hyper-tensive heart failure. Cardiovasc. Res. 2023, 119, 2312–2328. [Google Scholar] [CrossRef]
- Yan, W.; Bi, H.-L.; Liu, L.-X.; Li, N.-N.; Liu, Y.; Du, J.; Wang, H.-Z.; Li, H.-H. Knockout of immunoproteasome subunit β2i ameliorates cardiac fibrosis and inflammation in DOCA/Salt hypertensive mice. Biochem. Biophys. Res. Commun. 2017, 490, 84–90. [Google Scholar] [CrossRef]
- Cai, R.; Hao, Y.; Liu, Y.-Y.; Huang, L.; Yao, Y.; Zhou, M.-S. Tumor Necrosis Factor Alpha Deficiency Improves Endothelial Function and Cardiovascular Injury in Deoxycorticosterone Acetate/Salt-Hypertensive Mice. Biomed. Res. Int. 2020, 2020, 3921074. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Carretero, O.A.; Xu, J.; Rhaleb, N.-E.; Wang, F.; Lin, C.; Yang, J.J.; Pagano, P.J.; Yang, X.-P. Lack of inducible NO synthase reduces oxidative stress and enhances cardiac response to isoproterenol in mice with deoxycorticosterone acetate-salt hypertension. Hypertension 2005, 46, 1355–1361. [Google Scholar] [CrossRef]
- Silberman, G.A.; Fan, T.-H.M.; Liu, H.; Jiao, Z.; Xiao, H.D.; Lovelock, J.D.; Boulden, B.M.; Widder, J.; Fredd, S.; Bernstein, K.E.; et al. Uncoupled cardiac nitric oxide synthase mediates diastolic dysfunction. Circulation 2010, 121, 519–528. [Google Scholar] [CrossRef]
- Karatas, A.; Hegner, B.; Windt, L.J.; Luft, F.C.; Schubert, C.; Gross, V.; Akashi, Y.J.; Gürgen, D.; Kintscher, U.; da Costa Goncalves, A.C.; et al. Deoxycorticosterone acetate-salt mice exhibit blood pressure-independent sexual dimorphism. Hypertension 2008, 51, 1177–1183. [Google Scholar] [CrossRef]
- Hartner, A.; Cordasic, N.; Klanke, B.; Veelken, R.; Hilgers, K.F. Strain differences in the development of hypertension and glomerular lesions induced by deoxycorticosterone acetate salt in mice. Nephrol. Dial. Transpl. 2003, 18, 1999–2004. [Google Scholar] [CrossRef]
- Lin, Y.; Fu, S.; Yao, Y.; Li, Y.; Zhao, Y.; Luo, L. Heart failure with preserved ejection fraction based on aging and comorbidities. J. Transl. Med. 2021, 19, 291. [Google Scholar] [CrossRef]
- Schnelle, M.; Catibog, N.; Zhang, M.; Nabeebaccus, A.A.; Anderson, G.; Richards, D.A.; Sawyer, G.; Zhang, X.; Toischer, K.; Hasenfuss, G.; et al. Echocardiographic evaluation of diastolic function in mouse models of heart disease. J. Mol. Cell Cardiol. 2018, 114, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, S.F.; Ohtani, T.; Korinek, J.; Lam, C.; Larsen, K.; Simari, R.D.; Valenchik, M.L.; Burnett, J.C., Jr.; Redfiled, M.M. Mineralocorticoid accelerates transition to heart failure with preserved ejection fraction via “nongenomic effects”. Circulation 2010, 122, 370–378. [Google Scholar] [CrossRef]
- Kirchhoff, F.; Krebs, C.; Abdulhag, U.N.; Meyer-Schwesinger, C.; Maas, R.; Helmchen, U.; Hilgers, K.F.; Wolf, G.; Stahl, R.; Wenzel, U. Rapid development of severe end-organ damage in C57BL/6 mice by combining DOCA salt and angiotensin II. Kidney Int. 2008, 73, 643–650. [Google Scholar] [CrossRef]
- Wang, Q.; Euy-Myoung, J.; Liu, H.; Gu, L.; Dudley, S.C.; Yu, J. Astragaloside IV improves left ventricular diastolic dysfunction in hypertensive mice by increasing the phosphorylation of endothelial nitric oxide synthase. J. Hypertens. 2016, 34, e48–e49. [Google Scholar] [CrossRef]
- Landmesser, U.; Dikalov, S.; Price, S.R.; McCann, L.; Fukai, T.; Holland, S.M.; Mitch, W.E.; Harrison, D.G. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J. Clin. Investig. 2003, 111, 1201–1209. [Google Scholar] [CrossRef] [PubMed]
- Guzik, T.J.; Hoch, N.E.; Brown, K.A.; McCann, L.A.; Rahman, A.; Dikalov, S.; Goronzy, J.; Weyand, C.; Harrison, D.G. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J. Exp. Med. 2007, 204, 2449–2460. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Luo, J.; Xu, Q.; Liu, Y.; Cai, R.; Zhou, M.-S. Macrophage depletion protects against endothelial dysfunction and cardiac remodeling in angiotensin II hypertensive mice. Clin. Exp. Hypertens. 2021, 43, 699–706. [Google Scholar] [CrossRef]
- Regan, J.A.; Mauro, A.G.; Carbone, S.; Marchetti, C.; Gill, R.; Mezzaroma, E.; Raleigh, J.V.; Salloum, F.N.; Van Tassel, B.W.; Abbate, A.; et al. A mouse model of heart failure with preserved ejection fraction due to chronic infusion of a low subpressor dose of angiotensin II. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H771–H778. [Google Scholar] [CrossRef]
- Wang, B.; Xu, M.; Li, W.; Li, X.; Zheng, Q.; Niu, X. Aerobic exercise protects against pressure overload-induced cardiac dysfunction and hypertrophy via β3-AR-nNOS-NO activation. PLoS ONE 2017, 12, e0179648. [Google Scholar] [CrossRef]
- Chen, Y.; Guo, H.; Xu, D.; Xu, X.; Wang, H.; Hu, X.; Lu, Z.; Kwak, D.; Xu, Y.; Gunther, R.; et al. Left ventricular failure produces profound lung remodeling and pulmonary hypertension in mice: Heart failure causes severe lung disease. Hypertension 2012, 59, 1170–1178. [Google Scholar] [CrossRef]
- An, D.; Zeng, Q.; Zhang, P.; Ma, Z.; Zhang, H.; Liu, Z.; Li, J.; Ren, H.; Xu, D. Alpha-ketoglutarate ameliorates pressure overload-induced chronic cardiac dysfunction in mice. Redox Biol. 2021, 46, 102088. [Google Scholar] [CrossRef] [PubMed]
- Tsujita, Y.; Kato, T.; Sussman, M.A. Evaluation of left ventricular function in cardiomyopathic mice by tissue Doppler and color M-mode Doppler echocardiography. Echocardiography 2005, 22, 245–253. [Google Scholar] [CrossRef]
- Lu, Z.; Xu, X.; Hu, X.; Zhu, G.; Zhang, P.; van Deel, E.D.; French, J.P.; Fassett, J.T.; Oury, T.D.; Bache, R.J.; et al. Extracellular superoxide dismutase deficiency exacerbates pressure overload-induced left ventricular hypertrophy and dysfunction. Hypertension 2008, 51, 19–25. [Google Scholar] [CrossRef]
- Zi, M.; Stafford, N.; Prehar, S.; Baudoin, F.; Oceandy, D.; Wang, X.; Bui, T.; Shaheen, M.; Neyses, L.; Cartwright, E.J. Cardiac hypertrophy or failure?—A systematic evaluation of the transverse aortic constriction model in C57BL/6NTac and C57BL/6J substrains. Curr. Res. Physiol. 2019, 1, 1–10. [Google Scholar] [CrossRef]
- Baier, M.J.; Klatt, S.; Hammer, K.P.; Maier, L.S.; Rokita, A.G. Ca2+/calmodulin-dependent protein kinase II is essential in hyperacute pressure overload. J. Mol. Cell Cardiol. 2020, 138, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Slater, R.E.; Strom, J.G.; Methawasin, M.; Liss, M.; Gotthardt, M.; Sweitzer, N.; Granzier, H.L. Metformin improves diastolic function in an HFpEF-like mouse model by increasing titin compliance. J. Gen. Physiol. 2019, 151, 42–52. [Google Scholar] [CrossRef]
- Qiu, Z.; Fan, Y.; Wang, Z.; Huang, F.; Li, Z.; Sun, Z.; Hua, S.; Jin, W.; Chen, Y. Catestatin Protects Against Diastolic Dysfunction by Attenuating Mitochondrial Reactive Oxygen Species Generation. J. Am. Heart Assoc. 2023, 12, e029470. [Google Scholar] [CrossRef]
- Methawasin, M.; Granzier, H. Experimentally Increasing the Compliance of Titin Through RNA Binding Motif-20 (RBM20) Inhibition Improves Diastolic Function in a Mouse Model of Heart Failure with Preserved Ejection Fraction. Circulation 2016, 134, 1085–1099. [Google Scholar] [CrossRef]
- Dimitriadis, K.; Theofilis, P.; Koutsopoulos, G.; Pyrpyris, N.; Beneki, E.; Tatakis, F.; Tsioufis, P.; Chrysohoou, C.; Fragkoulis, C.; Tsioufis, K. The role of coronary microcirculation in heart failure with preserved ejection fraction: An unceasing odyssey. Heart Fail. Rev. 2025, 30, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Marie, A.; Larroze-Chicot, P.; Cosnier-Pucheu, S.; Gonzalez-Gonzalez, S. Senescence-accelerated mouse prone 8 (SAMP8) as a model of age-related hearing loss. Neurosci. Lett. 2017, 656, 138–143. [Google Scholar] [CrossRef]
- Reed, A.L.; Tanaka, A.; Sorescu, D.; Liu, H.; Jeong, E.-M.; Strudy, M.; Walp, E.R.; Dudley, S.C., Jr.; Sutliff, R.L. Diastolic dysfunction is associated with cardiac fibrosis in the senescence-accelerated mouse. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H824–H831. [Google Scholar] [CrossRef]
- Rodriguez-Calvo, R.; Serrano, L.; Barroso, E.; Coll, T.; Palomer, X.; Camins, A.; Sánchez, R.M.; Alegret, M.; Merlos, M.; Pallàs, M.; et al. Peroxisome Proliferator-Activated Receptor Down-Regulation Is Associated with Enhanced Ceramide Levels in Age-Associated Cardiac Hypertrophy. J. Gerontol. A Biol. Sci. Med. Sci. 2007, 62, 1326–1336. [Google Scholar] [CrossRef] [PubMed]
- Giridharan, V.V.; Karupppagounder, V.; Arumugam, S.; Nakamura, Y.; Guha, A.; Barichello, T.; Quevedo, J.; Watanabe, K.; Konishi, T.; Thandavarayan, R.A. 3,4-Dihydroxybenzalacetone (DBL) Prevents Aging-Induced Myocardial Changes in Senescence-Accelerated Mouse-Prone 8 (SAMP8) Mice. Cells 2020, 9, 597. [Google Scholar] [CrossRef]
- Forman, K.; Vara, E.; García, C.; Kireev, R.; Cuesta, S.; Escames, G.; Tresquerres, J. Effect of a Combined Treatment with Growth Hormone and Melatonin in the Cardiological Aging on Male SAMP8 Mice. J. Gerontol. A Biol. Sci. Med. Sci. 2011, 66A, 823–834. [Google Scholar] [CrossRef]
- Takeda, T.; Matsushita, T.; Kurozumi, M.; Takemura, K.; Higuchi, K.; Hosokawa, M. Pathobiology of the senescence-accelerated mouse (SAM). Exp. Gerontol. 1997, 32, 117–127. [Google Scholar] [CrossRef]
- Zhou, H.; Peng, W.; Li, F.; Wang, Y.; Wang, B.; Ding, Y.; Lin, Q.; Zhao, Y.; Pan, G.; Wang, X. Effect of Sodium-Glucose Cotransporter 2 Inhibitors for Heart Failure with Preserved Ejection Fraction: A Systematic Review and Meta-Analysis of Randomized Clinical Trials. Front. Cardiovasc. Med. 2022, 4, 875327. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Cai, Z.; Yuan, J.; Liu, H.; Pang, X.; Chen, Q.; Tang, X.; Geng, Q.; Dong, S. Effect of pharmacological treatment on outcomes of heart failure with preserved ejection fraction: An updated systematic review and network meta-analysis of randomized controlled trials. Cardiovasc. Diabetol. 2022, 21, 237. [Google Scholar] [CrossRef]
- Croteau, D.; Baka, T.; Young, S.; He, H.; Chambers, J.M.; Qin, F.; Panagia, M.; Pimentel, D.R.; Balschi, J.A.; Colucci, W.S.; et al. SGLT2 inhibitor ertugliflozin decreases elevated intracellular sodium, and improves energetics and contractile function in diabetic cardiomyopathy. Biomed. Pharmacother. 2023, 160, 114310. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Ye, T.; Tian, H.; Song, H.; Kan, C.; Han, F.; Hou, N.; Sun, X.; Zhang, J. Empagliflozin alleviates obesity-related cardiac dysfunction via the activation of SIRT3-mediated autophagosome formation. Lipids Health Dis. 2024, 23, 308. [Google Scholar] [CrossRef] [PubMed]
- Moellmann, J.; Klinkhammer, B.M.; Droste, P.; Kappel, B.; Haj-Yehia, E.; Maxeiner, S.; Artati, A.; Adamski, J.; Boor, P.; Schütt, K.; et al. Empagliflozin improves left ventricular diastolic function of db/db mice. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165807. [Google Scholar] [CrossRef]
- Wang, J.; Huang, X.; Liu, H.; Chen, Y.; Li, P.; Liu, L.; Li, J.; Ren, Y.; Huang, J.; Xiong, E.; et al. Empagliflozin Ameliorates Diabetic Cardiomyopathy via Attenuating Oxidative Stress and Improving Mitochondrial Function. Oxid. Med. Cell Longev. 2022, 2022, 1122494. [Google Scholar] [CrossRef]
- Byrne, N.J.; Parajuli, N.; Levasseur, J.L.; Boisvenue, J.; Beker, D.L.; Masson, G.; Fedak, P.W.M.; Verma, S.; Dyck, J.R.B. Empagliflozin Prevents Worsening of Cardiac Function in an Experimental Model of Pressure Overload-Induced Heart Failure. JACC Basic. Transl. Sci. 2017, 2, 347–354. [Google Scholar] [CrossRef]
- Karakasis, P.; Fragakis, N.; Patoulias, D.; Theofilis, P.; Sagris, M.; Koufakis, T.; Vliachakis, P.K.; Rangraze, I.R.; Tanani, M.; Tsioufis, K.; et al. The emerging role of glucagon-like peptide-1 receptor agonists in the management of obesity-related heart failure with preserved ejection fraction: Benefits beyond what scales can measure? Biomedicines 2024, 12, 2112. [Google Scholar] [CrossRef]
- Ni, X.-Y.; Feng, X.-J.; Wang, Z.-H.; Zhang, Y.; Little, P.J.; Cao, Y.; Xu, S.-W.; Tang, L.-Q.; Weng, J.-P. Empagliflozin and liraglutide ameliorate HFpEF in mice via augmenting the Erbb4 signaling pathway. Acta Pharmacol. Sin. 2024, 45, 1604–1617. [Google Scholar] [CrossRef]
- Koike, M.; Saito, H.; Kohno, G.; Takubo, M.; Watanabe, K.; Ishihara, H. Effects of GLP-1RA and SGLT2i, alone or in combination, on mouse models of type 2 diabetes representing different disease stages. Int. J. Mol. Sci. 2021, 22, 11463. [Google Scholar] [CrossRef] [PubMed]
- Withaar, C.; Meems, L.; Nollet, E.E.; Schouten, E.M.; Schroeder, M.A.; Knudsen, L.B.; Niss, K.; Madsen, C.T.; Hoegl, A.; Mazzoni, G.; et al. The cardioprotective effects of semaglutide exceed those of dietary weight loss in mice with HFpEF. JACC Basic. Transl. Sci. 2023, 8, 1298–1314. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| db/db | ob/ob | HFD | DOCA | Ang II | DOCA + Ang II | TAC | TAC + DOCA | SAMP | |
|---|---|---|---|---|---|---|---|---|---|
| Systemic inflammation signs | + | no data | |||||||
| Myocardial inflammation signs | + | +/− (limited data) | no data | + | |||||
| Myocardial hypertrophy | Described for all models | ||||||||
| Myocardial oxidative stress | + | no data | +/− (limited data) | ||||||
| Myocardial NO-cGMP-PKG signalling pathway dysfunction | + | +/− (limited data) | no data | +/− (limited data) | no data | +/− (limited data) | |||
| Structural and functional alterations in cardiomyocyte contractile proteins | + | +/− (limited data) | no data | ||||||
| Myocardial fibrosis signs | + | +/− (limited data) | + | ||||||
| Diastolic dysfunction signs (according to echocardiographic data) | + | +/− (limited data) | no data | +/− (limited data) | |||||
| Elevated ANP/BNP in myocardial tissue and/or blood | + | no data | + | +/− (limited data) | + | no data | +/− (limited data) | ||
| Gender differences | + (females show more pronounced changes) | +/− (limited data, females show more pronounced changes) | no data | +/− (limited data, males show more pronounced changes) | no data | ||||
| Model Limitations in Data Translation to Humans | Challenges of Practical Application | |
|---|---|---|
| db/db, ob/ob | Rather rapid development of obesity and insulin resistance, which is unlike typical human conditions | Homozygote infertility increases breeding costs |
| HFD | Possible development of systolic dysfunction | Duration of follow-up |
| DOCA, Ang II, DOCA + Ang II | Elevated blood pressure is a significant factor in the onset of human damage, including the progression to heart failure | Duration of follow-up (the C57BL/6 strain is fairly resistant to arterial hypertension development and hypertensive damage) |
| TAC | Primarily hypertensive organ damage, with indication of systolic dysfunction | The need for surgical intervention |
| TAC + DOCA | Primarily hypertensive organ damage | |
| SAMP | Early manifestations of inflammatory damage in various organs, neurodegeneration, age-associated lymphoproliferative conditions preceding the development of heart failure | Difficulties in breeding and housing animals due to short fertility period and high mortality rate |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ogurtsova, E.; Arefieva, T.; Filatova, A.; Radyukhina, N.; Ovchinnikov, A. Cardiometabolic Phenotype in HFpEF: Insights from Murine Models. Biomedicines 2025, 13, 744. https://doi.org/10.3390/biomedicines13030744
Ogurtsova E, Arefieva T, Filatova A, Radyukhina N, Ovchinnikov A. Cardiometabolic Phenotype in HFpEF: Insights from Murine Models. Biomedicines. 2025; 13(3):744. https://doi.org/10.3390/biomedicines13030744
Chicago/Turabian StyleOgurtsova, Ekaterina, Tatiana Arefieva, Anastasiia Filatova, Natalya Radyukhina, and Artem Ovchinnikov. 2025. "Cardiometabolic Phenotype in HFpEF: Insights from Murine Models" Biomedicines 13, no. 3: 744. https://doi.org/10.3390/biomedicines13030744
APA StyleOgurtsova, E., Arefieva, T., Filatova, A., Radyukhina, N., & Ovchinnikov, A. (2025). Cardiometabolic Phenotype in HFpEF: Insights from Murine Models. Biomedicines, 13(3), 744. https://doi.org/10.3390/biomedicines13030744