
Dyslipidemia, Diabetes and Atherosclerosis: Role of Inflammation and ROS-Redox-Sensitive Factors



{kind=link}
Abstract
:1. Introduction
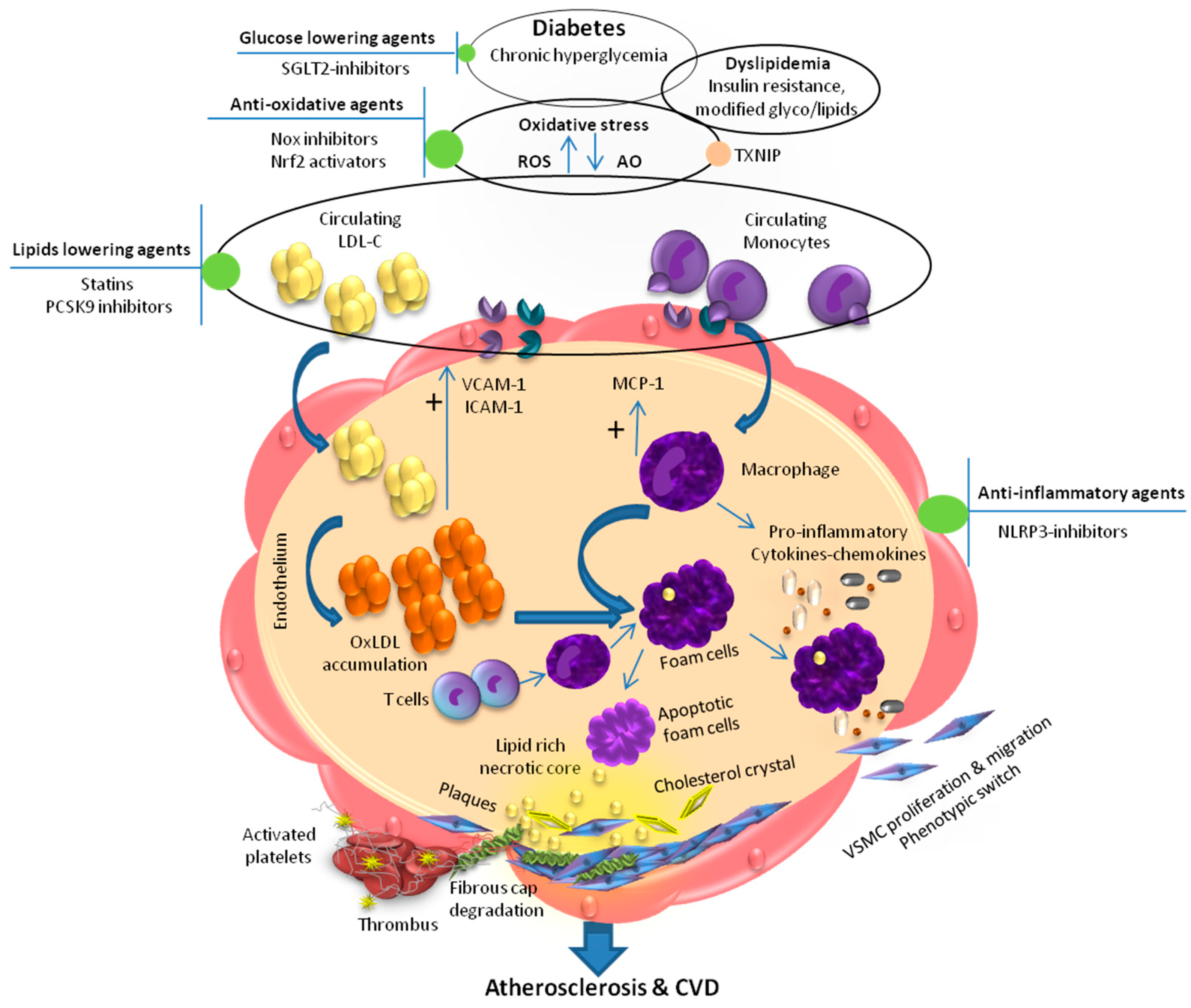
2. Dyslipidemia and Inflammation in Diabetic Atherosclerosis
3. Dyslipidemia and ROS-Redox-Sensitive Factors in Diabetic Atherosclerosis
4. Therapeutic Approaches to Combat Atherosclerosis in Diabetes
4.1. Glucose-Lowering Agents
4.2. Lipid-Lowering Agents
4.2.1. Statins
4.2.2. Fibrates
4.2.3. PCSK9 Inhibitors
4.3. Anti-Inflammatory Agents
4.4. Antioxidants and Nrf2 Activators
4.5. Agent Targeting the Source of ROS (NOX Inhibitors)
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Charlton, A.; Garzarella, J.; Jandeleit-Dahm, K.A.; Jha, J.C. Oxidative stress and inflammation in renal and cardiovascular complications of diabetes. Biology 2021, 10, 18. [Google Scholar]
- Harding, J.L.; Pavkov, M.E.; Magliano, D.J.; Shaw, J.E.; Gregg, E.W. Global trends in diabetes complications: A review of current evidence. Diabetologia 2019, 62, 3–16. [Google Scholar] [CrossRef] [Green Version]
- Teodoro, J.S.; Nunes, S.; Rolo, A.P.; Reis, F.; Palmeira, C.M. Therapeutic options targeting oxidative stress, mitochondrial dysfunction and inflammation to hinder the progression of vascular complications of diabetes. Front. Physiol. 2019, 9, 1857. [Google Scholar] [CrossRef] [PubMed]
- Mostofizadeh, N.; Hashemipour, M.; Roostazadeh, M.; Hashemi-Dehkordi, E.; Shahsanai, A.; Reisi, M. The impact of poor glycemic control on lipid profile variables in children with type 1 diabetes mellitus. J. Educ. Health Promot. 2019, 8, 6. [Google Scholar] [PubMed]
- Begum, A.; Irfan, S.R. Lipid profile abnormalities in Bangladeshi type 2 diabetic patients attending a tertiary care hospital: A cross-sectional study. Bangladesh Crit. Care J. 2019, 7, 44–47. [Google Scholar] [CrossRef]
- Wen, J.; Huang, Y.; Lu, Y.; Yuan, H. Associations of non-high-density lipoprotein cholesterol, triglycerides and the total cholesterol/HDL-c ratio with arterial stiffness independent of low-density lipoprotein cholesterol in a Chinese population. Hypertens. Res. 2019, 42, 1223–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, J. Diabetes: Managing dyslipidaemia. BMJ Clin. Evid. 2008, 2008, 0610. [Google Scholar] [PubMed]
- Athyros, V.G.; Doumas, M.; Imprialos, K.P.; Stavropoulos, K.; Georgianou, E.; Katsimardou, A.; Karagiannis, A. Diabetes and lipid metabolism. Hormones 2018, 17, 61–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jialal, I.; Bajaj, M. Therapy and clinical trials: Management of diabetic dyslipidemia. Curr. Opin. Lipidol. 2009, 20, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulut, T.; Demirel, F.; Metin, A. The prevalence of dyslipidemia and associated factors in children and adolescents with type 1 diabetes. J. Pediatr. Endocrinol. Metab. 2017, 30, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Homma, T.K.; Endo, C.M.; Saruhashi, T.; Mori, A.P.I.; De Noronha, R.M.; Monte, O.; Calliari, L.E.P. Dyslipidemia in young patients with type 1 diabetes mellitus. Arch. Endocrinol. Metab. 2015, 59, 215–219. [Google Scholar] [CrossRef] [Green Version]
- Shah, V.N.; Wu, M.; Polsky, S.; Snell-Bergeon, J.K.; Sherr, J.L.; Cengiz, E.; DiMeglio, L.A.; Pop-Busui, R.; Mizokami-Stout, K.; Foster, N.C. Gender differences in diabetes self-care in adults with type 1 diabetes: Findings from the T1D Exchange clinic registry. J. Diabetes Complicat. 2018, 32, 961–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poznyak, A.; Grechko, A.V.; Poggio, P.; Myasoedova, V.A.; Alfieri, V.; Orekhov, A.N. The diabetes mellitus–atherosclerosis connection: The role of lipid and glucose metabolism and chronic inflammation. Int. J. Mol. Sci. 2020, 21, 1835. [Google Scholar] [CrossRef] [Green Version]
- Zernecke, A.; Winkels, H.; Cochain, C.; Williams, J.W.; Wolf, D.; Soehnlein, O.; Robbins, C.S.; Monaco, C.; Park, I.; McNamara, C.A. Meta-analysis of leukocyte diversity in atherosclerotic mouse aortas. Circ. Res. 2020, 127, 402–426. [Google Scholar] [CrossRef]
- Lin, P.; Ji, H.-H.; Li, Y.-J.; Guo, S.-D. Macrophage plasticity and atherosclerosis therapy. Front. Mol. Biosci. 2021, 8, 324. [Google Scholar] [CrossRef] [PubMed]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- Groenen, A.G.; Halmos, B.; Tall, A.R.; Westerterp, M. Cholesterol efflux pathways, inflammation, and atherosclerosis. Crit. Rev. Biochem. Mol. Biol. 2021, 56, 426–439. [Google Scholar] [CrossRef] [PubMed]
- Rocha, N.A.; East, C.; Zhang, J.; McCullough, P.A. ApoCIII as a cardiovascular risk factor and modulation by the novel lipid-lowering agent volanesorsen. Curr. Atheroscler. Rep. 2017, 19, 62. [Google Scholar] [CrossRef] [PubMed]
- Størling, J.; Juntti-Berggren, L.; Olivecrona, G.; Prause, M.C.; Berggren, P.-O.; Mandrup-Poulsen, T. Apolipoprotein CIII reduces proinflammatory cytokine-induced apoptosis in rat pancreatic islets via the Akt prosurvival pathway. Endocrinology 2011, 152, 3040–3048. [Google Scholar] [CrossRef] [Green Version]
- Katakami, N. Mechanism of development of atherosclerosis and cardiovascular disease in diabetes mellitus. J. Atheroscler. Thromb. 2017, 25, RV17014. [Google Scholar] [CrossRef] [Green Version]
- Sorokin, V.; Vickneson, K.; Kofidis, T.; Woo, C.C.; Lin, X.Y.; Foo, R.; Shanahan, C.M. Role of vascular smooth muscle cell plasticity and interactions in vessel wall inflammation. Front. Immunol. 2020, 11, 3053. [Google Scholar] [CrossRef] [PubMed]
- Rattik, S.; Engelbertsen, D.; Wigren, M.; Ljungcrantz, I.; Östling, G.; Persson, M.; Nordin Fredrikson, G.; Bengtsson, E.; Nilsson, J.; Björkbacka, H. Elevated circulating effector memory T cells but similar levels of regulatory T cells in patients with type 2 diabetes mellitus and cardiovascular disease. Diabetes Vasc. Dis. Res. 2019, 16, 270–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meshkani, R.; Vakili, S. Tissue resident macrophages: Key players in the pathogenesis of type 2 diabetes and its complications. Clin. Chim. Acta 2016, 462, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Hoseini, Z.; Sepahvand, F.; Rashidi, B.; Sahebkar, A.; Masoudifar, A.; Mirzaei, H. NLRP3 inflammasome: Its regulation and involvement in atherosclerosis. J. Cell. Physiol. 2018, 223, 2116–2132. [Google Scholar] [CrossRef] [PubMed]
- Calling, S.; Johansson, S.-E.; Wolff, M.; Sundquist, J.; Sundquist, K. Total cholesterol/HDL-C ratio versus non-HDL-C as predictors for ischemic heart disease: A 17-year follow-up study of women in southern Sweden. BMC Cardiovasc. Disord. 2021, 21, 163. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Park, J.T.; Kim, H.W.; Chang, T.I.; Kang, E.W.; Ahn, C.; Oh, K.H.; Lee, J.; Chung, W.; Kim, Y.S. Inflammation alters relationship between high-density lipoprotein cholesterol and cardiovascular risk in patients with chronic kidney disease: Results from KNOW-CKD. J. Am. Heart Assoc. 2021, 10, e021731. [Google Scholar] [CrossRef] [PubMed]
- Van Capelleveen, J.C.; Bernelot Moens, S.J.; Yang, X.; Kastelein, J.J.; Wareham, N.J.; Zwinderman, A.H.; Stroes, E.S.; Witztum, J.L.; Hovingh, G.K.; Khaw, K.-T. Apolipoprotein C-III levels and incident coronary artery disease risk: The EPIC-Norfolk prospective population study. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1206–1212. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Shim, D.; Lee, J.S.; Zaitsev, K.; Williams, J.W.; Kim, K.-W.; Jang, M.-Y.; Seok Jang, H.; Yun, T.J.; Lee, S.H. Transcriptome analysis reveals nonfoamy rather than foamy plaque macrophages are proinflammatory in atherosclerotic murine models. Circ. Res. 2018, 123, 1127–1142. [Google Scholar] [CrossRef]
- Bojanin, D.; Vekic, J.; Milenkovic, T.; Vukovic, R.; Zeljkovic, A.; Stefanovic, A.; Janac, J.; Ivanisevic, J.; Mitrovic, K.; Miljkovic, M. Association between proprotein convertase subtilisin/kexin 9 (PCSK9) and lipoprotein subclasses in children with type 1 diabetes mellitus: Effects of glycemic control. Atherosclerosis 2019, 280, 14–20. [Google Scholar] [CrossRef] [Green Version]
- Levenson, A.E.; Shah, A.S.; Khoury, P.R.; Kimball, T.R.; Urbina, E.M.; de Ferranti, S.D.; Maahs, D.M.; Dolan, L.M.; Wadwa, R.P.; Biddinger, S.B. Obesity and type 2 diabetes are associated with elevated PCSK9 levels in young women. Pediatr. Diabetes 2017, 18, 755–760. [Google Scholar] [CrossRef]
- Guo, W.; Gong, Y.; Li, J.; Qin, P.; Lu, J.; Li, X.; Zhu, W.; Xu, N.; Zhou, H.; Zhang, Q. Association of serum proprotein convertase subtilisin/kexin type 9 with early atherosclerosis in newly diagnosed type 2 diabetes mellitus. Nutr. Metab. Cardiovasc. Dis. 2019, 29, 815–821. [Google Scholar] [CrossRef]
- Artha, I.M.J.R.; Bhargah, A.; Dharmawan, N.K.; Pande, U.W.; Triyana, K.A.; Mahariski, P.A.; Yuwono, J.; Bhargah, V.; Prabawa, I.P.Y.; Manuaba, I.B.A.P. High level of individual lipid profile and lipid ratio as a predictive marker of poor glycemic control in type-2 diabetes mellitus. Vasc. Health Risk Manag. 2019, 15, 149. [Google Scholar] [CrossRef] [Green Version]
- Kitasato, L.; Tojo, T.; Hatakeyama, Y.; Kameda, R.; Hashikata, T.; Yamaoka-Tojo, M. Postprandial hyperglycemia and endothelial function in type 2 diabetes: Focus on mitiglinide. Cardiovasc. Diabetol. 2012, 11, 79. [Google Scholar] [CrossRef] [Green Version]
- Klisic, A.; Kavaric, N.; Jovanovic, M.; Zvrko, E.; Skerovic, V.; Scepanovic, A.; Medin, D.; Ninic, A. Association between unfavorable lipid profile and glycemic control in patients with type 2 diabetes mellitus. J. Res. Med. Sci. 2017, 22, 122. [Google Scholar] [CrossRef]
- Poznyak, A.V.; Grechko, A.V.; Orekhova, V.A.; Khotina, V.; Ivanova, E.A.; Orekhov, A.N. NADPH oxidases and their role in atherosclerosis. Biomedicines 2020, 8, 206. [Google Scholar] [CrossRef] [PubMed]
- Bubb, K.J.; Drummond, G.R.; Figtree, G.A. New opportunities for targeting redox dysregulation in cardiovascular disease. Cardiovasc. Res. 2020, 116, 532–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Costa, R.M.; Rodrigues, D.; Pereira, C.A.; Silva, J.F.; Alves, J.V.; Lobato, N.S.; Tostes, R.C. Nrf2 as a potential mediator of cardiovascular risk in metabolic diseases. Front. Pharmacol. 2019, 10, 382. [Google Scholar] [CrossRef] [Green Version]
- Yuan, T.; Yang, T.; Chen, H.; Fu, D.; Hu, Y.; Wang, J.; Yuan, Q.; Yu, H.; Xu, W.; Xie, X. New insights into oxidative stress and inflammation during diabetes mellitus-accelerated atherosclerosis. Redox Biol. 2019, 20, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-Q.; Yan, J.-C.; Sun, Z.; Bao, Z.-Y.; Shao, C.; Pang, Q.-W.; Geng, Y.; Zhang, L.-L.; Li, L.-H. Role of AGEs in the progression and regression of atherosclerotic plaques. Glycoconj. J. 2018, 35, 443–450. [Google Scholar] [CrossRef]
- Pennathur, S.; Heinecke, J.W. Mechanisms for oxidative stress in diabetic cardiovascular disease. Antioxid. Redox Signal. 2007, 9, 955–969. [Google Scholar] [CrossRef]
- Yamagishi, S.-I.; Matsui, T. Role of hyperglycemia-induced Advanced Glycation End product (AGE) accumulation in atherosclerosis. Ann. Vasc. Dis. 2018, 11, 253–258. [Google Scholar] [CrossRef] [Green Version]
- Daiber, A.; Steven, S.; Vujacic-Mirski, K.; Kalinovic, S.; Oelze, M.; Di Lisa, F.; Münzel, T. Regulation of vascular function and inflammation via cross talk of reactive oxygen and nitrogen species from mitochondria or NADPH oxidase—Implications for diabetes progression. Int. J. Mol. Sci. 2020, 21, 3405. [Google Scholar] [CrossRef]
- Kaur, K.K.; Allahbadia, G.; Singh, M. The role of Diabetes Mellitus (both T1D and T2D) in the atherosclerosis development—A systematic review with part of inflammation along with altered glucose and lipid metabolism for forming therapeutic aproaches. EC Diabetes Metab. Res. 2020, 4, 27–43. [Google Scholar]
- Ling, P.; Shan, W.; Zhai, G.; Qiu, C.; Liu, Y.; Xu, Y.; Yang, X. Association between glutathione peroxidase-3 activity and carotid atherosclerosis in patients with type 2 diabetes mellitus. Brain Behav. 2020, 10, e01773. [Google Scholar] [CrossRef] [PubMed]
- Barteková, M.; Adameová, A.; Görbe, A.; Ferenczyová, K.; Pecháňová, O.; Lazou, A.; Dhalla, N.S.; Ferdinandy, P.; Giricz, Z. Natural and synthetic antioxidants targeting cardiac oxidative stress and redox signaling in cardiometabolic diseases. Free Radic. Biol. Med. 2021, 169, 446–477. [Google Scholar] [CrossRef] [PubMed]
- Akoumianakis, I.; Antoniades, C. Impaired vascular redox signaling in the vascular complications of obesity and diabetes mellitus. Antioxid. Redox Signal. 2019, 30, 333–353. [Google Scholar] [CrossRef]
- Gray, S.P.; Di Marco, E.; Okabe, J.; Szyndralewiez, C.; Heitz, F.; Montezano, A.C.; de Haan, J.B.; Koulis, C.; El-Osta, A.; Andrews, K.L. NADPH oxidase 1 plays a key role in diabetes mellitus–accelerated atherosclerosis. Circulation 2013, 127, 1888–1902. [Google Scholar] [CrossRef] [Green Version]
- Gray, S.P.; Jha, J.C.; Kennedy, K.; van Bommel, E.; Chew, P.; Szyndralewiez, C.; Touyz, R.M.; Schmidt, H.; Cooper, M.E.; Jandeleit-Dahm, K.A.M. Combined NOX1/4 inhibition with GKT137831 in mice provides dose-dependent reno- and atheroprotection even in established micro- and macrovascular disease. Diabetologia 2017, 60, 927–937. [Google Scholar] [CrossRef] [Green Version]
- Oppi, S.; Lüscher, T.F.; Stein, S. Mouse models for atherosclerosis research—Which is my line? Front. Cardiovasc. Med. 2019, 6, 46. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, A.M. Diabetes mellitus and cardiovascular disease: Emerging therapeutic approaches. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 558–568. [Google Scholar] [CrossRef] [Green Version]
- Maqbool, A.; Watt, N.T.; Haywood, N.; Viswambharan, H.; Skromna, A.; Makava, N.; Visnagri, A.; Shawer, H.M.; Bridge, K.; Muminov, S.K. Divergent effects of genetic and pharmacological inhibition of Nox2 NADPH oxidase on insulin resistance-related vascular damage. Am. J. Physiol. Cell Physiol. 2020, 319, C64–C74. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Li, S.; Wu, H.; Hu, P.; Chen, L.; Zeng, C.; Tong, X. Endothelial Nox4 dysfunction aggravates atherosclerosis by inducing endoplasmic reticulum stress and soluble epoxide hydrolase. Free Radic. Biol. Med. 2021, 164, 44–57. [Google Scholar] [CrossRef]
- Gajos-Draus, A.; Duda, M.; Beręsewicz, A. Cardiac and renal upregulation of Nox2 and NF-κB and repression of Nox4 and Nrf2 in season-and diabetes-mediated models of vascular oxidative stress in guinea-pig and rat. Physiol. Rep. 2017, 5, e13474. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, J.F.; Alves, J.V.; Silva-Neto, J.A.; Costa, R.M.; Neves, K.B.; Alves-Lopes, R.; Carmargo, L.L.; Rios, F.J.; Montezano, A.C.; Touyz, R.M. Lysophosphatidylcholine induces oxidative stress in human endothelial cells via NOX5 activation–implications in atherosclerosis. Clin. Sci. 2021, 135, 1845–1858. [Google Scholar] [CrossRef] [PubMed]
- Poznyak, A.V.; Grechko, A.V.; Orekhova, V.A.; Chegodaev, Y.S.; Wu, W.-K.; Orekhov, A.N. Oxidative stress and antioxidants in atherosclerosis development and treatment. Biology 2020, 9, 60. [Google Scholar] [CrossRef] [Green Version]
- Touyz, R.M.; Anagnostopoulou, A.; Camargo, L.L.; Rios, F.J.; Montezano, A.C. Vascular biology of superoxide-generating NADPH oxidase 5—Implications in hypertension and cardiovascular disease. Antioxid. Redox Signal. 2019, 30, 1027–1040. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.-J.; Zhao, C.-L.; Ouyang, S.; Deng, K.-Q.; Zhu, L.; Montezano, A.C.; Zhang, C.; Hu, F.; Zhu, X.-Y.; Tian, S. Ca2+-dependent NOX5 (NADPH Oxidase 5) exaggerates cardiac hypertrophy through reactive oxygen species production. Hypertension 2020, 76, 827–838. [Google Scholar] [CrossRef]
- Harja, E.; Chang, J.S.; Lu, Y.; Leitges, M.; Zou, Y.S.; Schmidt, A.M.; Yan, S.-F. Mice deficient in PKC β and apolipoprotein E display decreased atherosclerosis. FASEB J. 2009, 23, 1081–1091. [Google Scholar] [CrossRef] [Green Version]
- Durpès, M.-C.; Morin, C.; Paquin-Veillet, J.; Beland, R.; Paré, M.; Guimond, M.-O.; Rekhter, M.; King, G.L.; Geraldes, P. PKC-β activation inhibits IL-18-binding protein causing endothelial dysfunction and diabetic atherosclerosis. Cardiovasc. Res. 2015, 106, 303–313. [Google Scholar] [CrossRef]
- Byon, C.H.; Han, T.; Wu, J.; Hui, S.T. Txnip ablation reduces vascular smooth muscle cell inflammation and ameliorates atherosclerosis in apolipoprotein E knockout mice. Atherosclerosis 2015, 241, 313–321. [Google Scholar] [CrossRef] [Green Version]
- Bajic, V.P.; Van Neste, C.; Obradovic, M.; Zafirovic, S.; Radak, D.; Bajic, V.B.; Essack, M.; Isenovic, E.R. Glutathione “redox homeostasis” and its relation to cardiovascular disease. Oxid. Med. Cell. Longev. 2019, 2019, 5028181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chawla, R.; Loomba, R.; Guru, D.; Loomba, V. Ischemia Modified Albumin (IMA)-a marker of glycaemic control and vascular complications in type 2 diabetes mellitus. JCDR 2016, 10, BC13. [Google Scholar] [CrossRef] [PubMed]
- Femlak, M.; Gluba-Brzózka, A.; Ciałkowska-Rysz, A.; Rysz, J. The role and function of HDL in patients with diabetes mellitus and the related cardiovascular risk. Lipids Health Dis. 2017, 16, 207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Ji, X.; Zhang, Z.; Xue, F. Relationship between lipid profiles and glycemic control among patients with type 2 diabetes in Qingdao, China. Int. J. Environ. Res. Public Health 2020, 17, 5317. [Google Scholar] [CrossRef]
- Schofield, J.D.; Liu, Y.; Rao-Balakrishna, P.; Malik, R.A.; Soran, H. Diabetes dyslipidemia. Diabetes Ther. 2016, 7, 203–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goderis, G.; Vaes, B.; Mamouris, P.; van Craeyveld, E.; Mathieu, C. Prevalence of atherosclerotic cardiovascular disease, heart failure, and chronic kidney disease in patients with type 2 Diabetes Mellitus: A primary care research network-based study. Exp. Clin. Endocrinol. Diabetes 2021. [Google Scholar] [CrossRef]
- Ghosh-Swaby, O.R.; Goodman, S.G.; Leiter, L.A.; Cheng, A.; Connelly, K.A.; Fitchett, D.; Juni, P.; Farkouh, M.E.; Udell, J.A. Glucose-lowering drugs or strategies, atherosclerotic cardiovascular events, and heart failure in people with or at risk of type 2 diabetes: An updated systematic review and meta-analysis of randomised cardiovascular outcome trials. Lancet Diabetes Endocrinol. 2020, 8, 418–435. [Google Scholar] [CrossRef]
- Lee, S. Update on SGLT2 inhibitors-new data released at the American Diabetes Association. Crit. Pathw. Cardiol. 2017, 16, 93–95. [Google Scholar] [CrossRef] [PubMed]
- Association, A.D. Diabetes care in the hospital: Standards of medical care in diabetes—2019. Diabetes Care 2019, 42, S173–S181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stancu, C.; Sima, A. Statins: Mechanism of action and effects. J. Cell. Mol. Med. 2001, 5, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Palasubramaniam, J.; Wang, X.; Peter, K. Myocardial infarction—From atherosclerosis to thrombosis: Uncovering new diagnostic and therapeutic approaches. Arter. Thromb. Vasc. Biol. 2019, 39, e176–e185. [Google Scholar] [CrossRef] [PubMed]
- Arrigoni, E.; Del Re, M.; Fidilio, L.; Fogli, S.; Danesi, R.; Di Paolo, A. Pharmacogenetic foundations of therapeutic efficacy and adverse events of statins. Int. J. Mol. Sci. 2017, 18, 104. [Google Scholar] [CrossRef] [PubMed]
- Devaraj, S.; Rogers, J.; Jialal, I. Statins and biomarkers of inflammation. Curr. Atheroscler. Rep. 2007, 9, 33–41. [Google Scholar] [CrossRef]
- Neil, H.; DeMicco, D.; Luo, D.; Betteridge, D.; Colhoun, H.; Durrington, P.; Livingstone, S.; Fuller, J.; Hitman, G. CARDS study investigators: Analysis of efficacy and safety in patients aged 65–75 years at randomization: Collaborative Atorvastatin Diabetes Study (CARDS). Diabetes Care 2006, 29, 2378–2384. [Google Scholar] [CrossRef] [Green Version]
- Mancini, G.J.; Hegele, R.A.; Leiter, L.A. Dyslipidemia. Can. J. Diabetes 2018, 42, S178–S185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stegman, B.; Puri, R.; Cho, L.; Shao, M.; Ballantyne, C.M.; Barter, P.J.; Chapman, M.J.; Erbel, R.; Libby, P.; Raichlen, J.S. High-intensity statin therapy alters the natural history of diabetic coronary atherosclerosis: Insights from SATURN. Diabetes Care 2014, 37, 3114–3120. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; Danielson, E.; Fonseca, F.A.; Genest, J.; Gotto Jr, A.M.; Kastelein, J.J.; Koenig, W.; Libby, P.; Lorenzatti, A.J.; MacFadyen, J.G. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N. Engl. J. Med. 2008, 359, 2195–2207. [Google Scholar] [CrossRef] [Green Version]
- Thompson, P.D.; Panza, G.; Zaleski, A.; Taylor, B. Statin-associated side effects. J. Am. Coll. Cardiol. 2016, 67, 2395–2410. [Google Scholar] [CrossRef] [PubMed]
- Grundy, S.M.; Stone, N.J.; Bailey, A.L.; Beam, C.; Birtcher, K.K.; Blumenthal, R.S.; Braun, L.T.; De Ferranti, S.; Faiella-Tommasino, J.; Forman, D.E. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA guideline on the management of blood cholesterol: Executive summary: A report of the American College of Cardiology/American Heart Association Task Force on clinical practice guidelines. J. Am. Coll. Cardiol. 2019, 73, 3168–3209. [Google Scholar] [CrossRef] [PubMed]
- Staels, B.; Auwerx, J. Regulation of apo AI gene expression by fibrates. Atherosclerosis 1998, 137, S19–S23. [Google Scholar] [CrossRef]
- Superko, H.R.; Berneis, K.K.; Williams, P.T.; Rizzo, M.; Wood, P.D. Gemfibrozil reduces small low-density lipoprotein more in normolipemic subjects classified as low-density lipoprotein pattern B compared with pattern A. Am. J. Cardiol. 2005, 96, 1266–1272. [Google Scholar] [CrossRef] [PubMed]
- Barter, P. Anti-atherogenic effects of fibrates in type 2 diabetes. Trials 2001, 2, 218. [Google Scholar] [CrossRef]
- Keech, A. FIELD study investigators: Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus: Randomised controlled trial. Lancet 2005, 366, 1849–1861. [Google Scholar] [PubMed]
- Orringer, C.E.; Jacobson, T.A.; Saseen, J.J.; Brown, A.S.; Gotto, A.M.; Ross, J.L. Underberg, J.A. Update on the use of PCSK9 inhibitors in adults: Recommendations from an expert panel of the National Lipid Association. J. Clin. Lipidol. 2017, 11, 880–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholls, S.; Puri, R.; Anderson, T.; Ballantyne, C.; Cho, L.; Kastelein, J. Effect of evolocumab on progression of coronary disease. The GLAGOV randomized clinical trial in statin-treated patients. JAMA 2016, 316, 2373–2384. [Google Scholar] [CrossRef] [PubMed]
- Sabatine, M.S.; Giugliano, R.; Keech, A.C.; Honarpour, N.; Wiviott, S.D.; Murphy, S.A.; Kuder, J.F.; Wang, H.; Liu, T.; Wasserman, S.M.; et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N. Engl. J. Med. 2017, 376, 1713–1722. [Google Scholar] [CrossRef] [PubMed]
- Hlatky, M.; Kazi, D. PCSK9 inhibitors: Economics and policy. J. Am. Coll. Cardiol. 2017, 70, 2677–2687. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Choi, J.S.; Stefanovic, N.; Al-Sharea, A.; Simpson, D.S.; Mukhamedova, N.; Jandeleit-Dahm, K.; Murphy, A.J.; Sviridov, D.; Vince, J.E. Specific NLRP3 inhibition protects against diabetes-associated atherosclerosis. Diabetes 2021, 70, 772–787. [Google Scholar] [CrossRef] [PubMed]
- Tsalamandris, S.; Antonopoulos, A.S.; Oikonomou, E.; Papamikroulis, G.-A.; Vogiatzi, G.; Papaioannou, S.; Deftereos, S.; Tousoulis, D. The role of inflammation in diabetes: Current concepts and future perspectives. Eur. Cardiol. 2019, 14, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsui, T.; Nakamura, N.; Ojima, A.; Nishino, Y.; Yamagishi, S.-I. Sulforaphane reduces advanced glycation end products (AGEs)-induced inflammation in endothelial cells and rat aorta. Nutr. Metab. Cardiovasc. Dis. 2016, 26, 797–807. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Shao, W.; Chiang, Y.; Foltz, W.; Zhang, Z.; Ling, W.; Fantus, I.; Jin, T. Oltipraz upregulates the nuclear factor (erythroid-derived 2)-like 2 [corrected](NRF2) antioxidant system and prevents insulin resistance and obesity induced by a high-fat diet in C57BL/6J mice. Diabetologia 2010, 54, 922–934. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Zhang, Z.-Z.; Wu, Y.; Wang, R.-Q.; Chen, J.-W.; Chen, J.; Zhang, Y.; Chen, Y.-J.; Geng, M.; Xu, Z.-D. (–)-Epigallocatechin-3-Gallate ameliorates atherosclerosis and modulates hepatic lipid metabolic gene expression in apolipoprotein E knockout mice: Involvement of TTC39B. Front. Pharmacol. 2018, 9, 195. [Google Scholar] [CrossRef] [Green Version]
- Vendrov, A.E.; Madamanchi, N.R.; Niu, X.-L.; Molnar, K.C.; Runge, M.; Szyndralewiez, C.; Page, P.; Runge, M.S. NADPH oxidases regulate CD44 and hyaluronic acid expression in thrombin-treated vascular smooth muscle cells and in atherosclerosis. J. Biol. Chem. 2010, 285, 26545–26557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cifuentes-Pagano, M.E.; Meijles, D.N.; Pagano, P.J. Nox inhibitors & therapies: Rational design of peptidic and small molecule inhibitors. Curr. Pharm. Des. 2015, 21, 6032–6035. [Google Scholar]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hasheminasabgorji, E.; Jha, J.C. Dyslipidemia, Diabetes and Atherosclerosis: Role of Inflammation and ROS-Redox-Sensitive Factors. Biomedicines 2021, 9, 1602. https://doi.org/10.3390/biomedicines9111602
Hasheminasabgorji E, Jha JC. Dyslipidemia, Diabetes and Atherosclerosis: Role of Inflammation and ROS-Redox-Sensitive Factors. Biomedicines. 2021; 9(11):1602. https://doi.org/10.3390/biomedicines9111602
Chicago/Turabian StyleHasheminasabgorji, Elham, and Jay C. Jha. 2021. "Dyslipidemia, Diabetes and Atherosclerosis: Role of Inflammation and ROS-Redox-Sensitive Factors" Biomedicines 9, no. 11: 1602. https://doi.org/10.3390/biomedicines9111602
APA StyleHasheminasabgorji, E., & Jha, J. C. (2021). Dyslipidemia, Diabetes and Atherosclerosis: Role of Inflammation and ROS-Redox-Sensitive Factors. Biomedicines, 9(11), 1602. https://doi.org/10.3390/biomedicines9111602