

The Effect of the New Imidazole Derivatives Complexation with Betacyclodextrin, on the Antifungal Activity in Oropharyngeal Infections
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
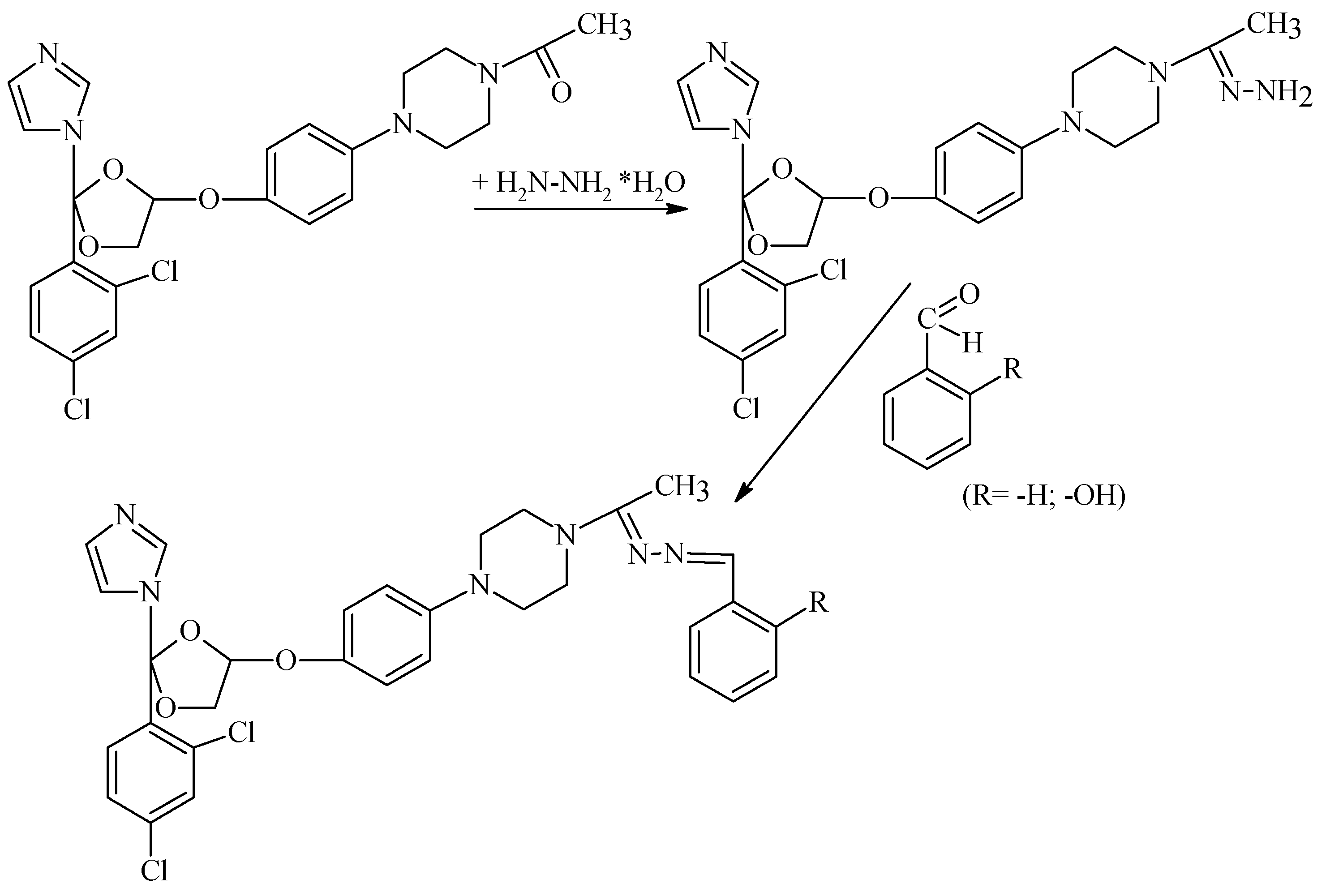
2.1. General Process for Obtaining Ketoconazole Condensation Hydrazones
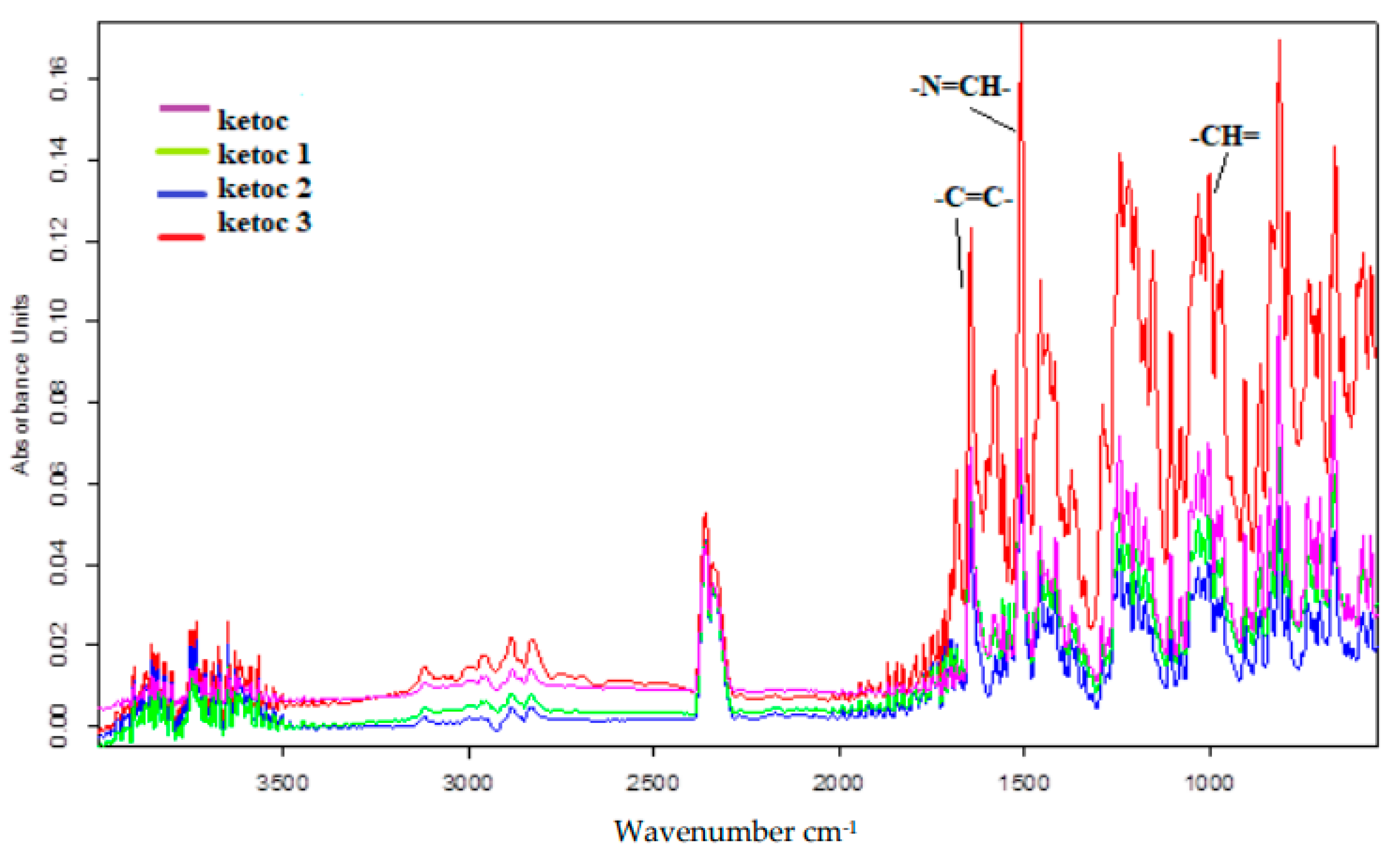
2.2. Physico-Chemical Characterization and Spectral Confirmation of Intermediate and Final Compounds
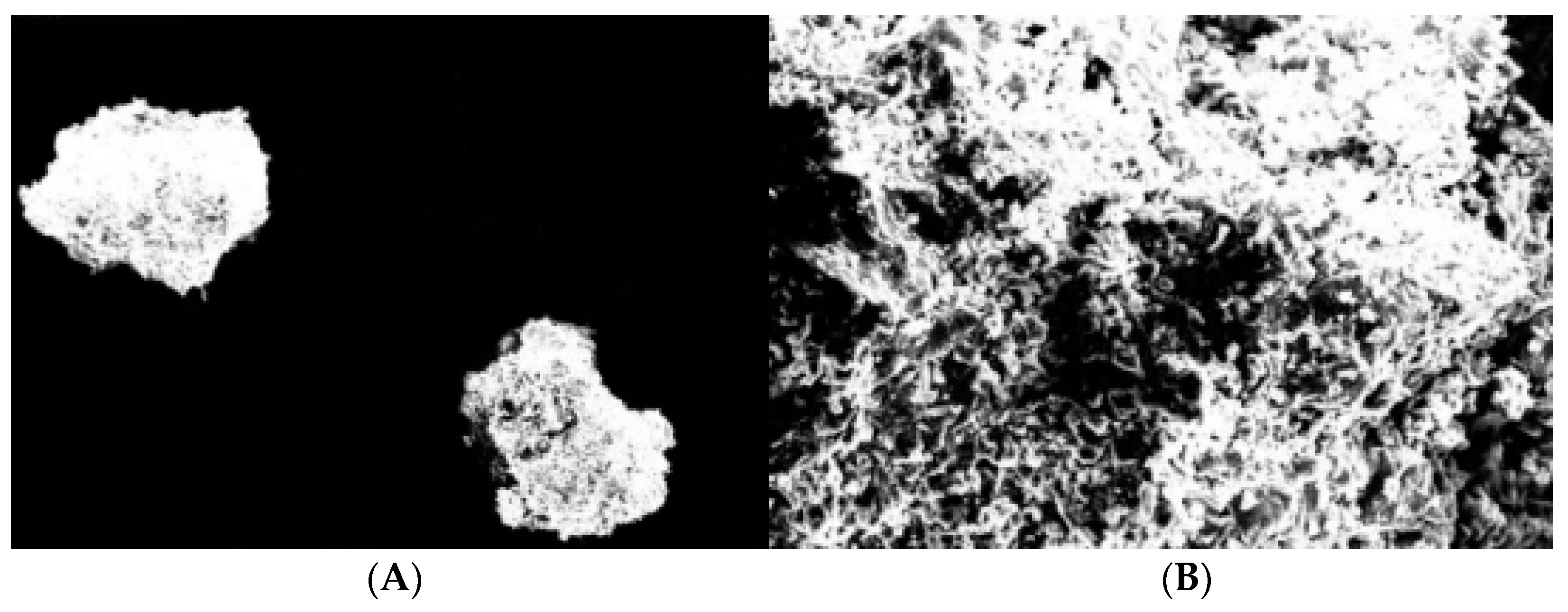
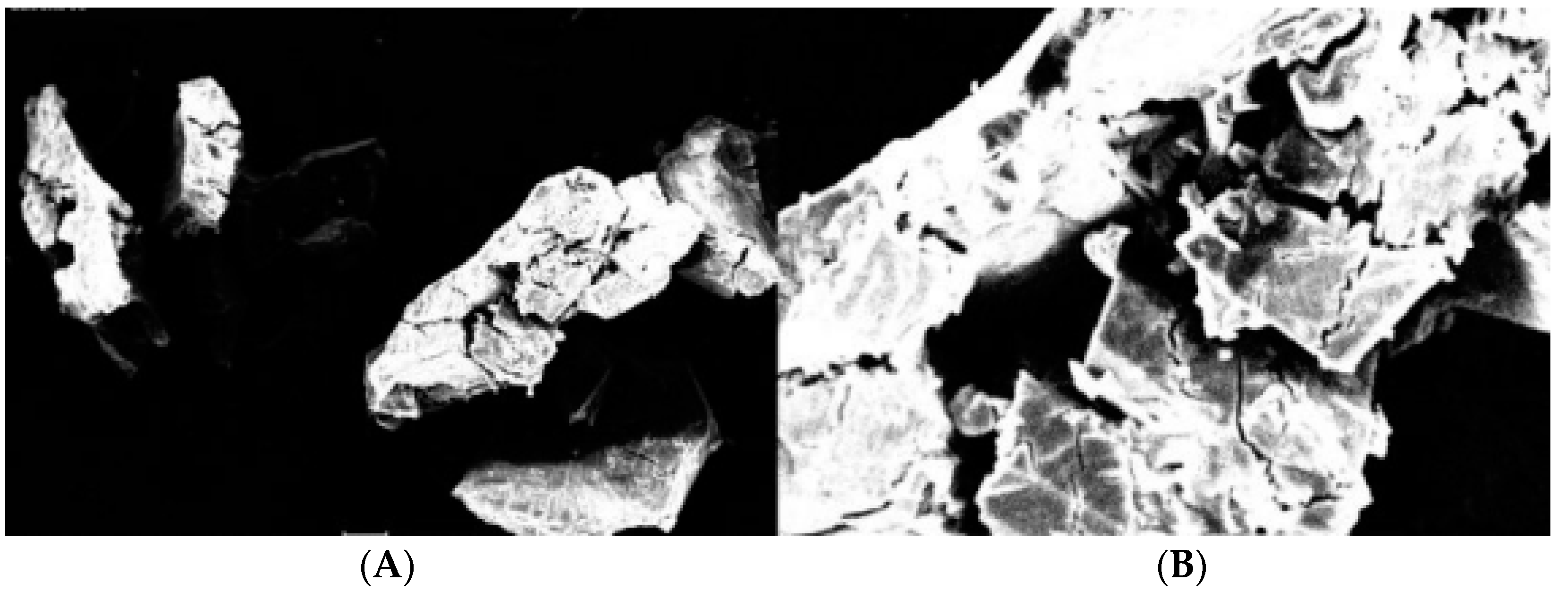
2.3. Obtaining and SEM Morphology of Inclusion Complexes with Betacyclodextrin
2.3.1. Physical Mixture Method
2.3.2. Kneading Method
2.3.3. Co-Precipitation Method
2.3.4. Solvent Evaporation Method
2.4. Evaluation of the Antifungal Activity of Ketoconazole Derivatives and their BCD Inclusion Complexes
3. Results
3.1. Physico-Chemical Characterisation and Structural Confirmation of Ketoconazole and Its New Derivatives
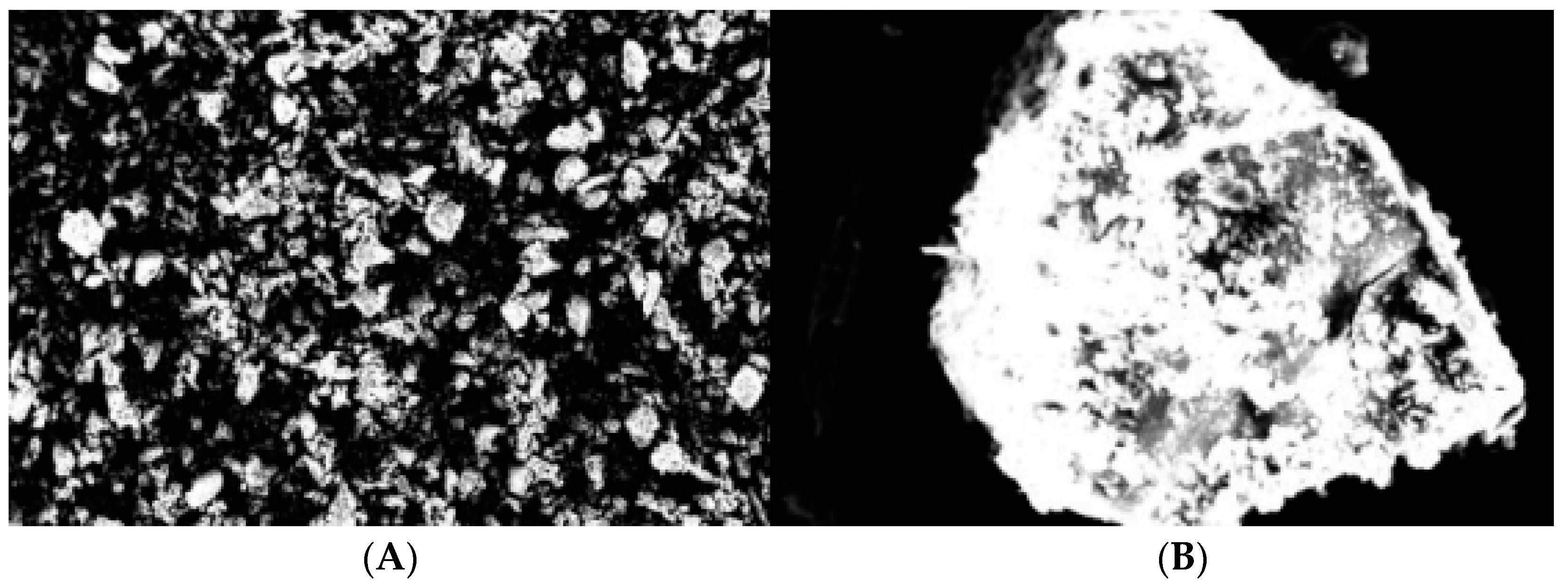
3.2. SEM Morphology of Inclusion Complexes with Betacyclodextrin
3.2.1. Physical Mixture Method
3.2.2. Kneading Method
3.2.3. Co-Precipitation Method
3.2.4. Solvent Evaporation Method
3.3. Antifungal Activity Evaluation of 1% Ketoconazole Derivatives by Determining the Diameter of the Inhibition Zone (Free and Complexed Forms)
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Alexander, B.D.; Pfaller, M.A. Contemporary Tools for the Diagnosis and Management of Invasive Mycoses, Laboratory Tools for Invasive Mycoses. Clin. Infect. Dis. 2006, 43, 15–27. [Google Scholar] [CrossRef] [Green Version]
- Sawant, B.; Khan, T. Recent advances in delivery of antifungal agents for therapeutic management of candidiasis. Biomed. Pharmacother. 2017, 96, 1478–1490. [Google Scholar] [CrossRef] [PubMed]
- Brand, S.R.; Degenhardt, T.P.; Person, K.; Sobel, J.D.; Nyirjesy, P.; Schotzinger, R.J.; Tavakkol, A. A phase 2, randomized, double-blind, placebo-controlled, dose-ranging study to evaluate the efficacy and safety of VT-1161 oral tablets in the treatment of patients with recurrent vulvovaginal candidiasis. Am. J. Obstet. Gynecol. 2014, 218, 624.e1–624.e9. [Google Scholar] [CrossRef] [PubMed]
- Garvey, E.P.; Hoekstra, W.J.; Schotzinger, R.J.; Sobel, J.D.; Lilly, E.A.; Fidel, P.L., Jr. Efficacy of the clinical agent VT-1161 against fluconazole-sensitive and -resistant Candida albicans in a murine model of vaginal candidiasis. Antimicrob. Agents Chemother. 2015, 59, 5567–5573. [Google Scholar] [CrossRef] [Green Version]
- NCT02244606. Oral SCY-078 vs. Standard-of-Care Following IV Echinocandin in the Treatment of Invasive Candidiasis. 2014. Available online: https://www.aspergillus.org.uk/new_antifungals/oral-scy-078-vs-standard-of-care-following-iv-echinocandin-in-the-treatment-of-invasive-candidiasis/ (accessed on 1 September 2022).
- Lamoth, F.; Alexander, B.D. Antifungal activities of SCY-078 (MK-3118) and standard antifungal agents against clinical non-Aspergillus mold isolates. Antimicrob. Agents Chemother. 2015, 59, 4308–4311. [Google Scholar] [CrossRef] [Green Version]
- Van Tyle, J.H. Ketoconazole; mechanism of action, spectrum of activity, pharmacokinetics, drug interactions, adverse reactions and therapeutic use. Pharmacotherapy. J. Hum. Pharmacol. Drug Ther. 1984, 4, 343–373. [Google Scholar] [CrossRef]
- Gupta, A.K.; Lyons, D.C. The rise and fall of oral ketoconazole. J. Cutan. Med. Surg. 2015, 19, 352–357. [Google Scholar] [CrossRef]
- Popa, G.; Dragostin, O.M.; Buzia, O.D.; Tartau, L.M. Studies on obtaining and characterization a pregabalin-cyclodextrin complex for taste masking purpose. Rev. Chim. 2017, 68, 337–340. [Google Scholar] [CrossRef]
- Ghosh, A.; Biswas, S.; Ghosh, T. Preparation and evaluation of silymarin β-cyclodextrin molecular inclusion complexes. J. Young Pharm. 2011, 3, 205–210. [Google Scholar] [CrossRef] [Green Version]
- Sapte, S.; Pore, Y. Inclusion complexes of cefuroximeaxetil with β-cyclodextrin: Physicochemical characterization, molecular modeling and effect of L-arginine on complexation. J. Pharm. Anal. 2016, 6, 300–306. [Google Scholar] [CrossRef]
- Nicodemus, G.D.; Bryant, S.J. Cell encapsulation in biodegradable hydrogels for tissue engineering applications. Tissue Eng. Part B 2008, 14, 149–165. [Google Scholar] [CrossRef] [PubMed]
- Dragostin, I.; Dragostin, O.M.; Samal, S.K.; Dash, S.; Tatia, R.; Dragan, M.; Confederat, L.; Ghiciuc, C.M.; Diculencu, D.; Lupușoru, C.E.; et al. New Isoniazid Derivatives with Improved Pharmaco Toxicological Profile: Obtaining, Characterization and Biological Evaluation. Eur. J. Pharm. Sci. 2019, 137, 104974. [Google Scholar] [CrossRef] [PubMed]
- Iovu, M.; Nicolescu, T.O. Organic Chemestry: Experimental Methods; Carol Davila University Publishing House: Bucharest, Romania, 2009. [Google Scholar]
- Clinical and Laboratory Standard Institute. Performance Standards for Antimicrobial Susceptibility Testing, 29th ed.; Method for Antifungal Disk Diffusion Susceptibility Testing of Yeasts; Clinical and Laboratory Standard Institute Accredited by the American National Standards Institute: Malvern, PA, USA, 2009. [Google Scholar]
- Armstrong-James, D.; Meintjes, G.; Brown, G.D. A neglected epidemic: Fungal infections in HIV/AIDS. Trends Microbiol. 2014, 22, 120–127. [Google Scholar] [CrossRef]
- Joshua, P.; Choi, B.; Spellberg, B. Nosocomial fungal infections: Epidemiology, diagnosis, and treatment. Med. Mycol. 2007, 45, 321–346. [Google Scholar] [CrossRef]
- Lass-Flörl, C.; Perkhofer, S.; Mayr, A. In vitro susceptibility testing in fungi: A global perspective on a variety of methods. Mycoses 2010, 53, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ben-Ami, R.; Kontoyiannis, D.P. Resistance to Antifungal Drugs. Infect. Dis. Clin. N. Am. 2021, 35, 279–311. [Google Scholar] [CrossRef]
- van den Bossche, H. Biochemical effects of miconazole on fungi. I. Effects on the uptake and or utilization of purines, pyrimidines, nucleosides, amino acids and glucose by Candida albicans. Biochem. Pharmacol. 2002, 23, 887–899. [Google Scholar] [CrossRef]
- Seeliger, V.H.P.R. Pilzhemmende Wirkung eines neuen Benzimidazol-Derivates. Mycoses 1958, 1, 162–171. [Google Scholar] [CrossRef]
- Shafiei, M.; Peyton, L.; Hashemzadeh, M.; Foroumadi, A. History of the development of antifungal azoles: A review on structures, SAR, and mechanism of action. Bioorg. Chem. 2020, 104, 104240. [Google Scholar] [CrossRef]
- Koparde, S.; Hosamani, K.M.; Kulkarni, V.; Joshi, S.D. Synthesis of coumarin-piperazine derivatives as potent anti-microbial and anti-inflammatory agents, and molecular docking studies. Chem. Data Collect. 2018, 15–16, 197–206. [Google Scholar] [CrossRef]
- Sanka, B.M.; Tadesse, D.M.; Bedada, E.T.; Mengesha, E.T.; Neelaiah Babu, G. Design, synthesis, biological screening and molecular docking studies of novel multifunctional 1,4-di(aryl/heteroaryl) substituted piperazine derivatives as potential antitubercular and antimicrobial agents. Bioorg. Chem. 2022, 119, 105568. [Google Scholar] [CrossRef] [PubMed]
- Heeres, J.; Backx, J.L.; Mostmans, J.; Van Cutsem, J. Antimycotic imidazoles. Part 4. Synthesis and antifungal activity of ketoconazole, a new potent orally active broad-spectrum antifungal agent. J. Med. Chem. 1979, 22, 1003–1005. [Google Scholar] [CrossRef] [PubMed]
- Režen, T.N.; Debeljak, D. Kordiš, D. Rozman. New aspects on lanosterol 14α-de-methylase and cytochrome P450 evolution: Lanosterol/cycloartenol diversifica- tion and lateral transfer. J. Mol. Evol. 2004, 59, 51–58. [Google Scholar] [CrossRef]
- Starosta, R.; Almeida, R.F.M. Luminescence properties of the antifungal agent ketoconazole and its diphenylphosphane derivatives. J. Lumin. 2020, 220, 116956. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Appearance | Molecular Formula MF | Molecular Weight MW | Reaction Yield ƞ (%) | m.p. (°C) |
|---|---|---|---|---|---|
| Ketoconazo l | White amorphous powder | C26H28Cl2 N4O4 | 531.4 | - | 148–150 |
| Ketoc 1 | White-yellow amorphous powder | C26H30Cl2 N6O3 | 543.97 | 74.07 | 150–151 |
| Ketoc 2 | White-pink crystalline powder | C33H35Cl2 N6O2 | 616.98 | 81.54 | 145–148 |
| Ketoc 3 | Orange crystalline powder | C33H35Cl2 N6O3 | 634.54 | 89.13 | 130 |
| Nr. Crt. | Compound/Complex | Reference Strain Results (mm) | Strain from Patient 1 Results (mm) | Strain from Patient 2 Results (mm) | Strain from Patient 3 Results (mm) | Strain from Patient 4 Results (mm) |
|---|---|---|---|---|---|---|
| 1. | Ketoc 1/Ketoc 1:B-CD | 24/50 | 20/22 | 21/26 | 22/24 | 24/40 |
| 2. | Ketoc 2/Ketoc 2:B-CD | 20/24 | 20/22 | 22/24 | 20/24 | 24/32 |
| 3. | Ketoc 3/Ketoc 3:B-CD | 14/24 | 18/20 | 20/24 | 24/24 | 22/24 |
| 4. | Ketoc/Ketoc: B-CD | 20/20 | 18/19 | 20/20 | 20/21 | 20/20 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lisa, E.L.; Dragostin, O.M.; Petroaie, A.D.; Gurau, G.; Cristea, A.; Pavel, A.; Bonifate, F.; Popa, P.S.; Matei, M. The Effect of the New Imidazole Derivatives Complexation with Betacyclodextrin, on the Antifungal Activity in Oropharyngeal Infections. Processes 2022, 10, 2697. https://doi.org/10.3390/pr10122697
Lisa EL, Dragostin OM, Petroaie AD, Gurau G, Cristea A, Pavel A, Bonifate F, Popa PS, Matei M. The Effect of the New Imidazole Derivatives Complexation with Betacyclodextrin, on the Antifungal Activity in Oropharyngeal Infections. Processes. 2022; 10(12):2697. https://doi.org/10.3390/pr10122697
Chicago/Turabian StyleLisa, Elena Lacramioara, Oana Maria Dragostin, Antoneta Dacia Petroaie, Gabriela Gurau, Alina Cristea, Alexandra Pavel, Florina Bonifate, Paul Serban Popa, and Madalina Matei. 2022. "The Effect of the New Imidazole Derivatives Complexation with Betacyclodextrin, on the Antifungal Activity in Oropharyngeal Infections" Processes 10, no. 12: 2697. https://doi.org/10.3390/pr10122697