


Molecular Model Construction and Optimization Study of Gas Coal in the Huainan Mining Area

Abstract
:1. Introduction
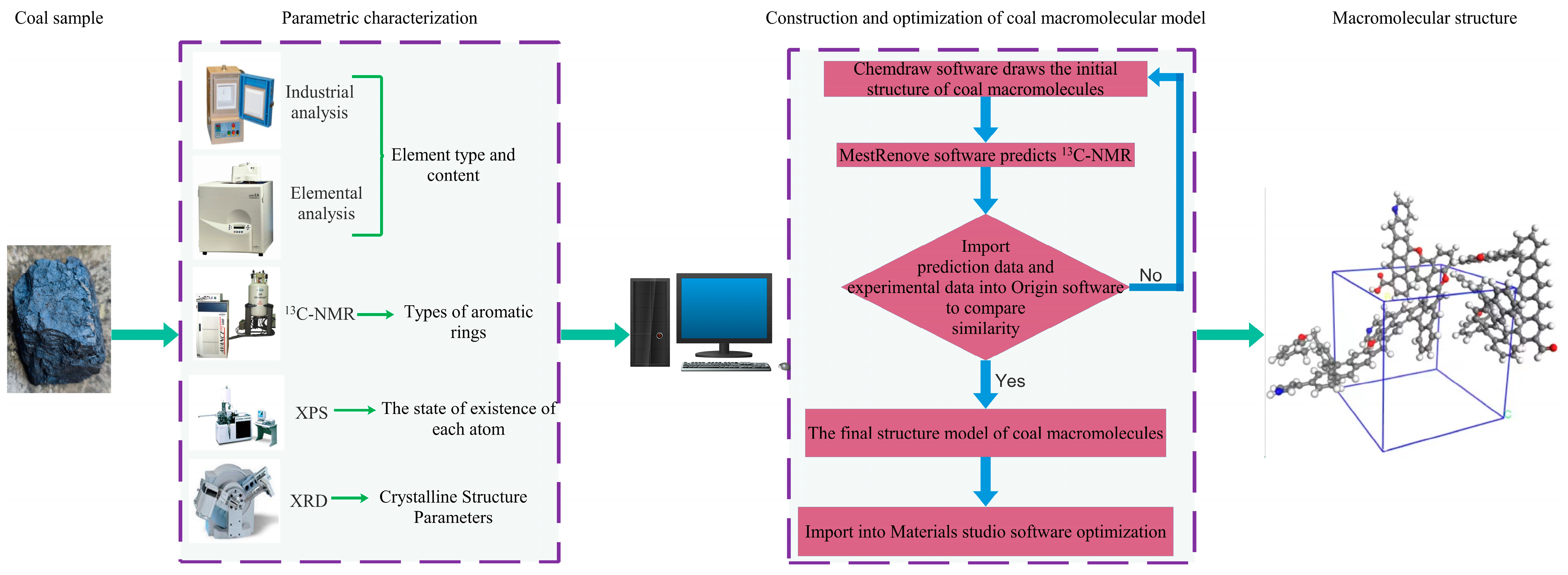
2. Experimental Section
2.1. Elemental and Industrial Analysis Tests
2.2. Experimental Protocol
- (1)
- 13C-NMR spectroscopy (13C-NMR)
- (2)
- X-ray diffraction (XRD)
- (3)
- X-ray photoelectron spectroscopy (XPS)
3. Results and Discussions
3.1. The Analysis of the Elemental Normalization and the Atomic Ratio
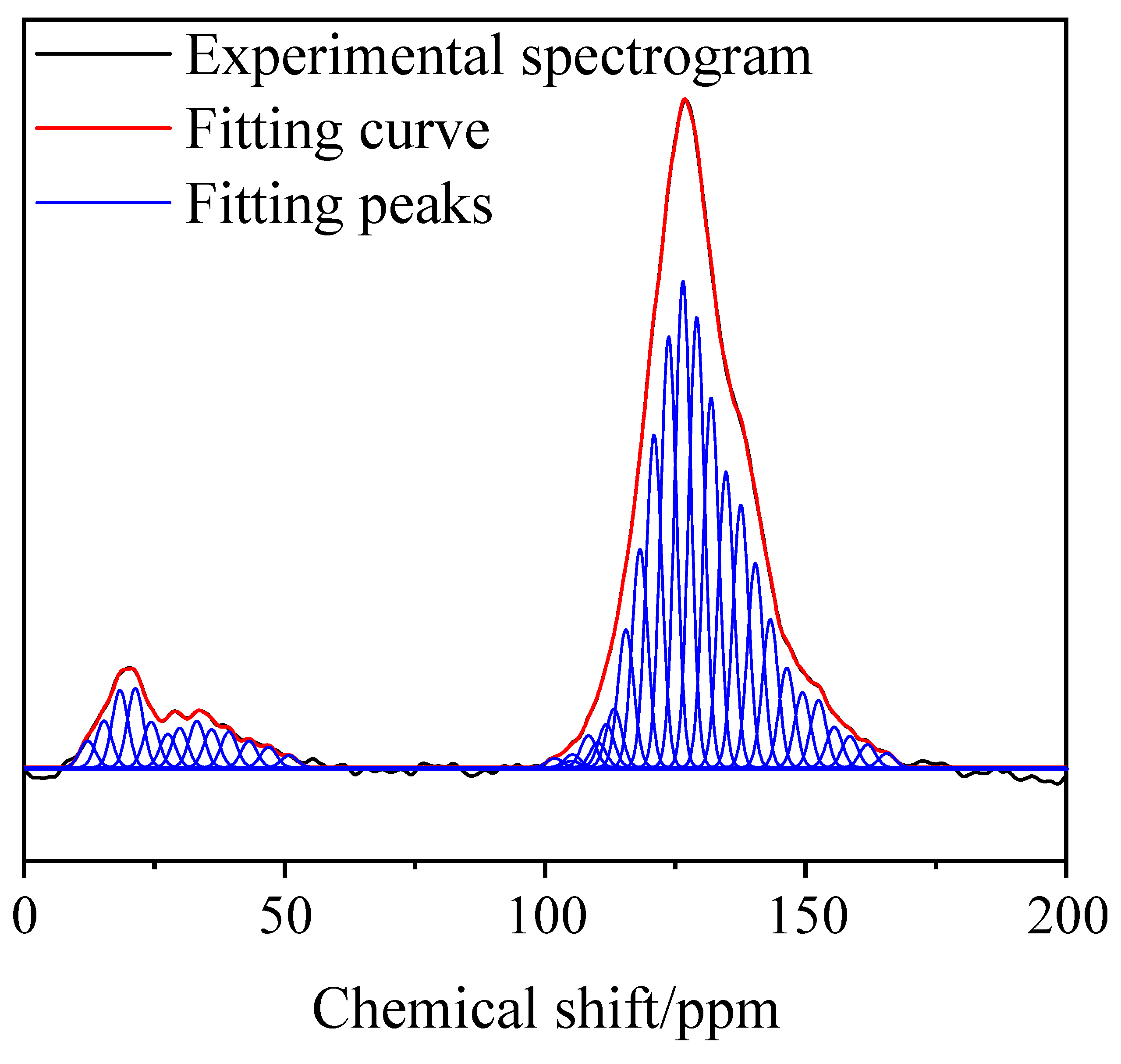
3.2. 13C-NMR Test Results and Analysis
3.3. XRD Test Results and Analyses
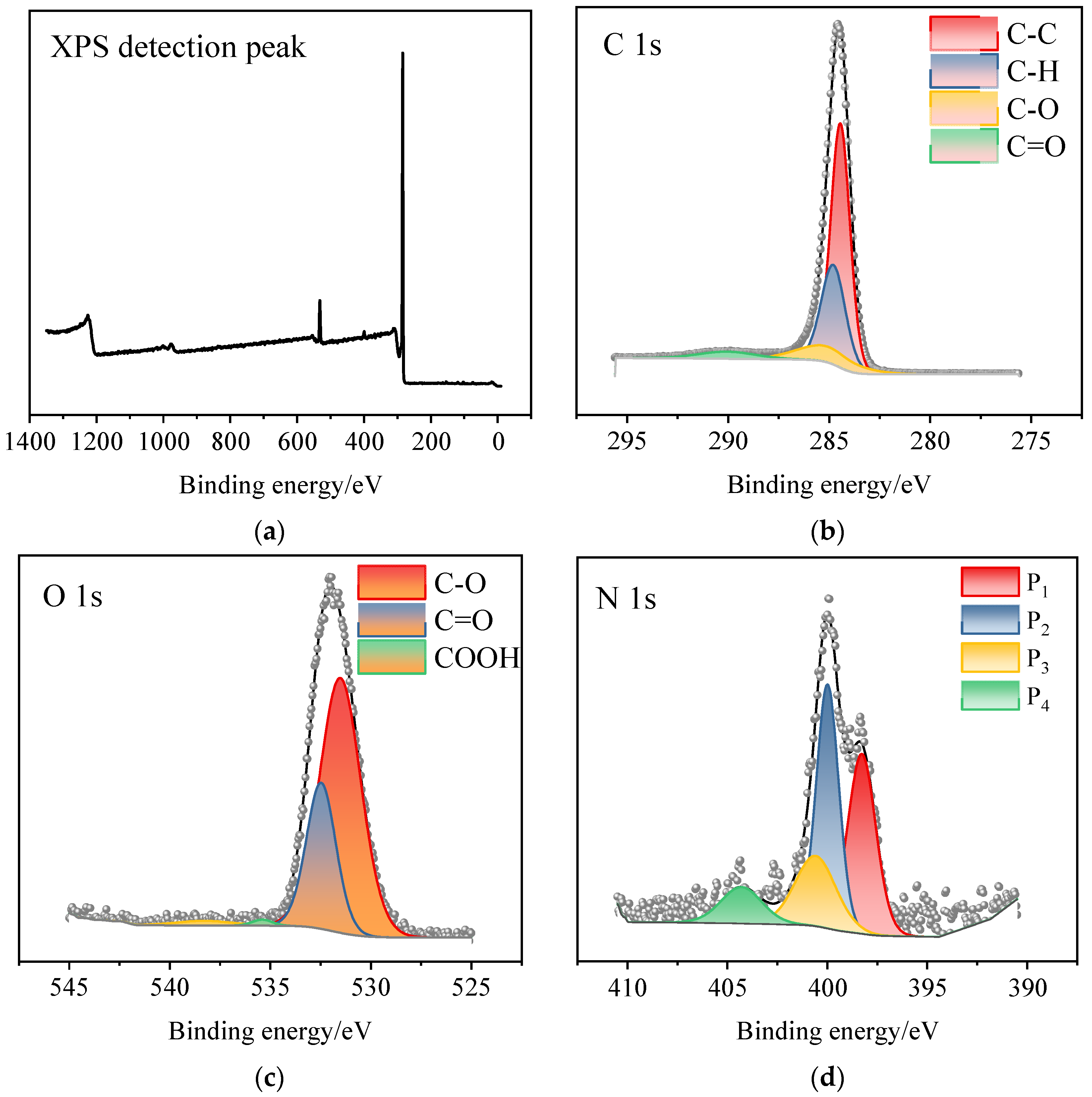
3.4. XPS Test Results and Analyses
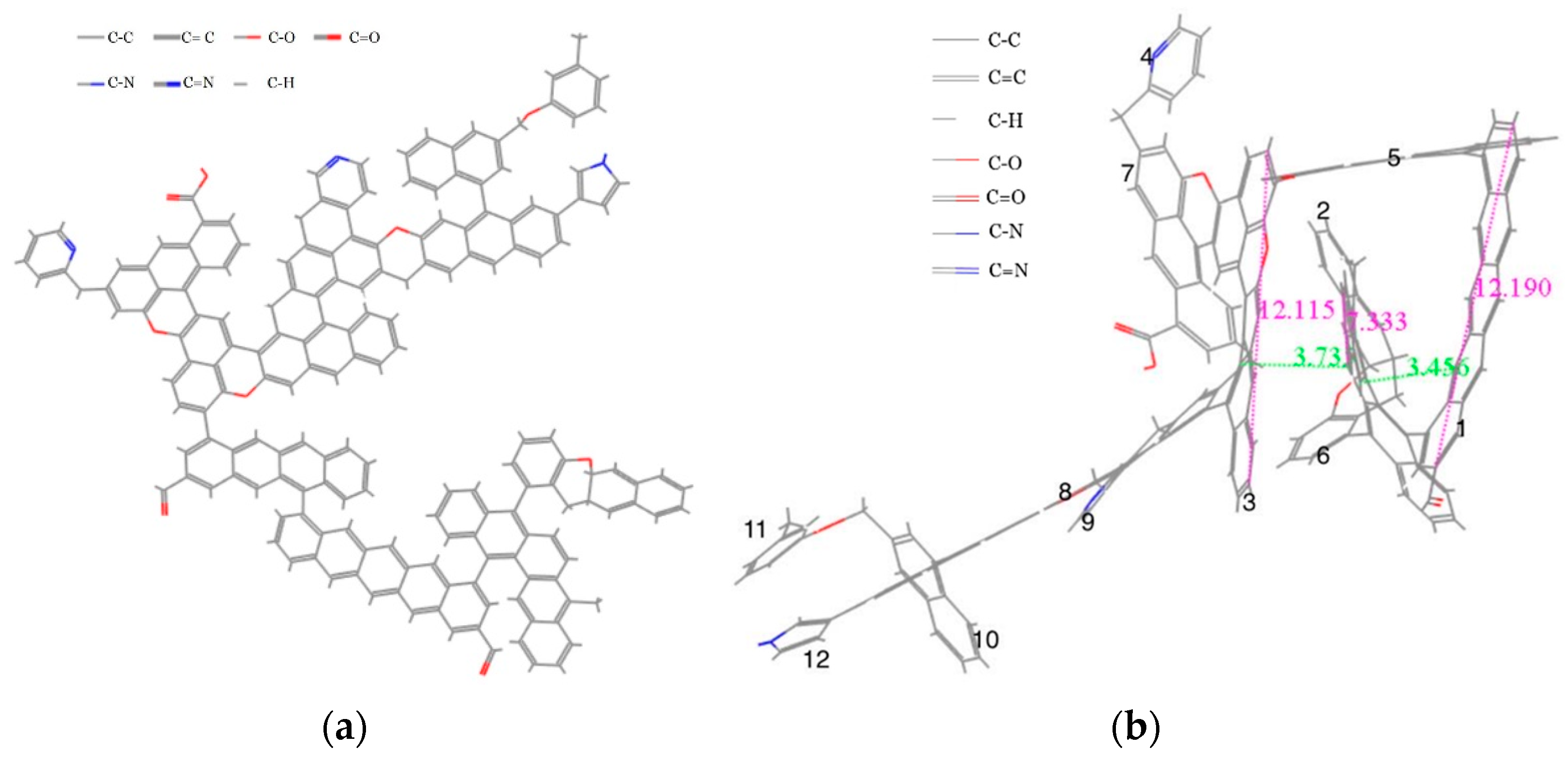
3.5. Construction of the Molecular Structure Model of Gas Coal
3.5.1. Aromatic Structures
3.5.2. Aliphatic Carbon Structures
3.5.3. Heteroatom Structures
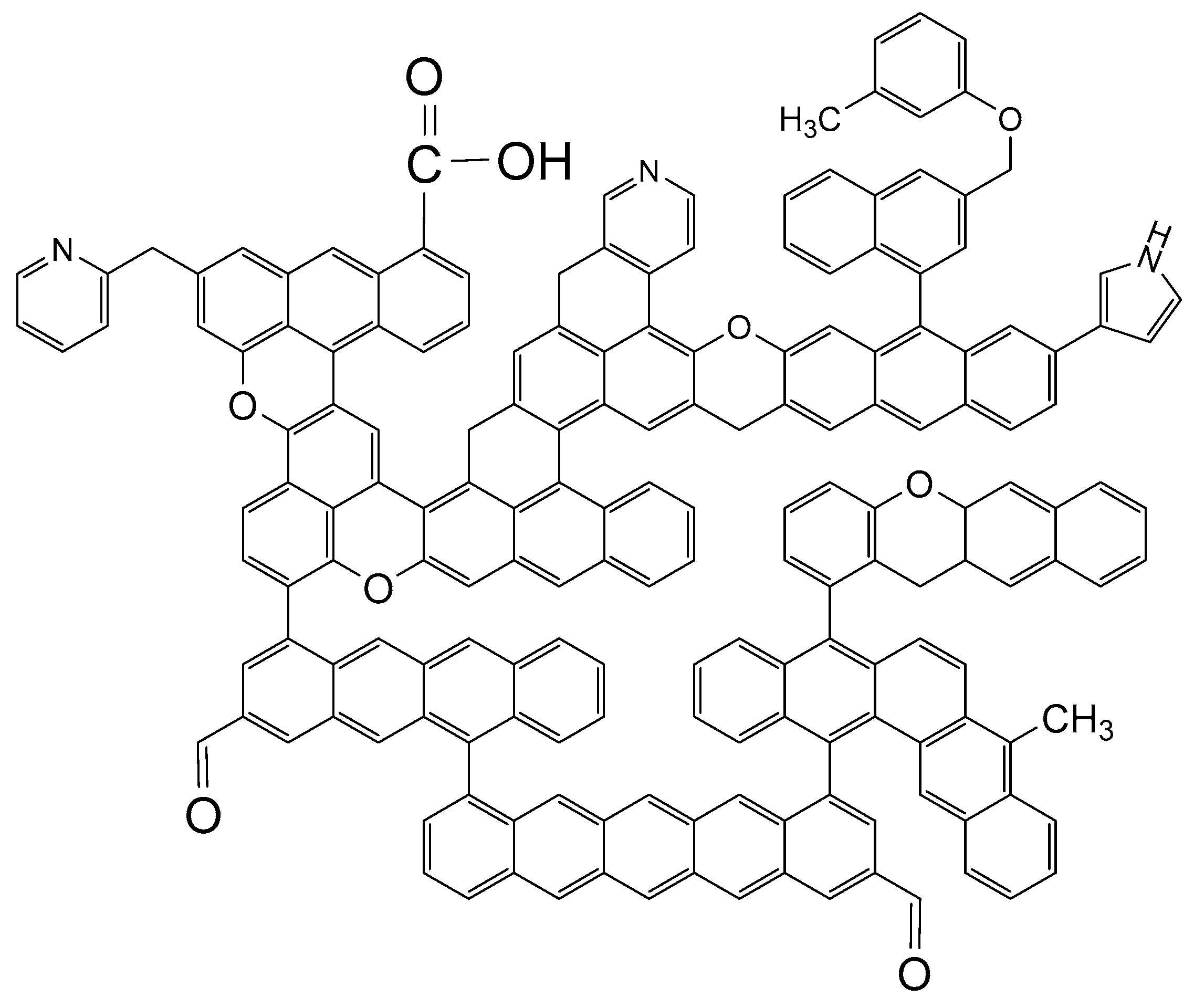
3.5.4. Construction of the Coal Molecular Structure
4. Optimization and Validation of the Coal Macromolecule Model
4.1. Optimal Structure
4.1.1. Optimized Method and Parameter Setting
4.1.2. Energy-Minimizing Geometrical Configuration and Microcrystalline Structure Parameters
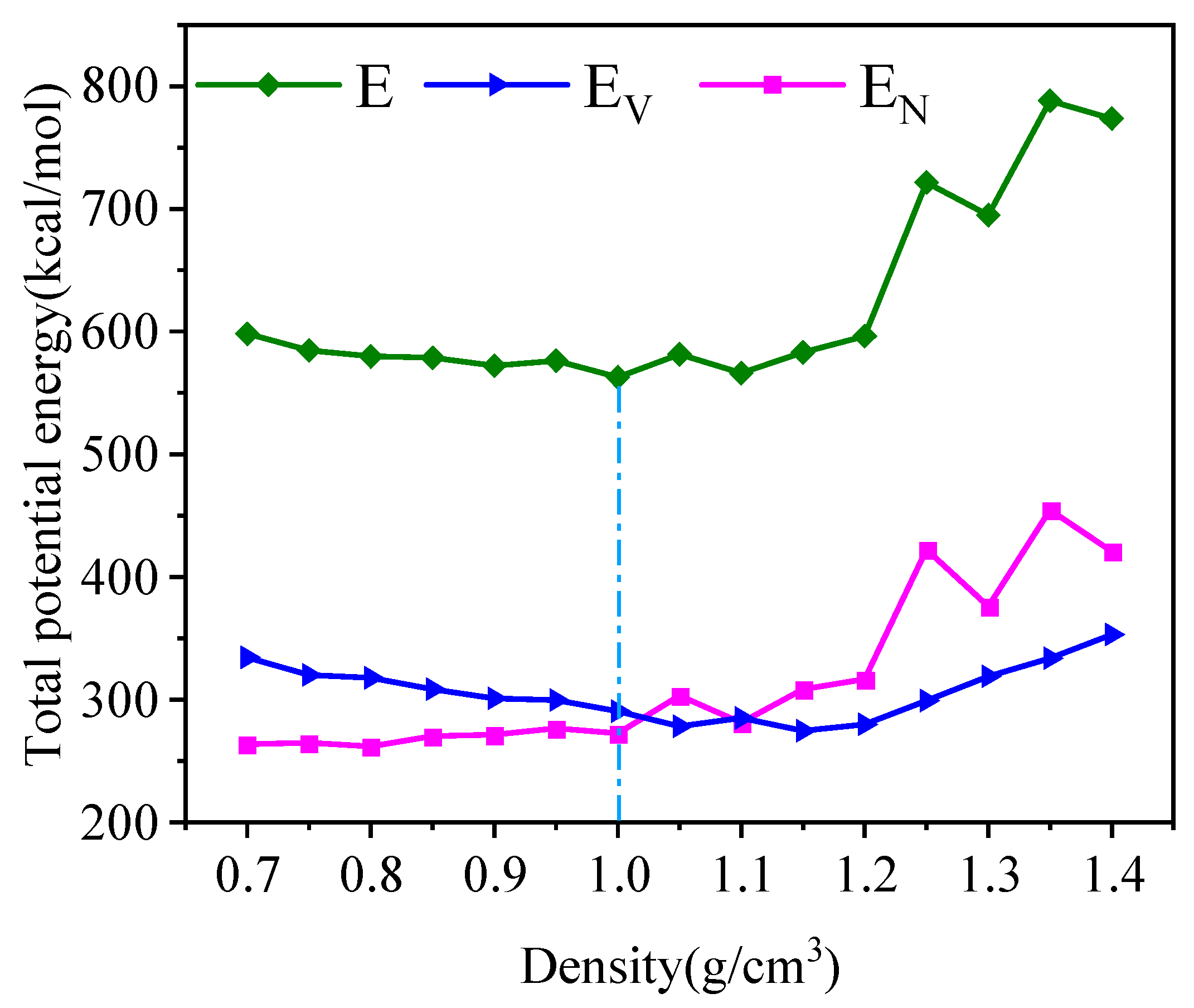
4.2. Density Simulation
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Xu, J.L. Research and progress of coal mine green mining in 20 years. Coal Sci. Technol. 2020, 48, 1–15. [Google Scholar]
- Peng, S.P.; Zhang, B.; Wang, T. China’s Coal Resources: Octothorpe Shaped Distribution Characteristics and Sustainable Development Strategies. Strateg. Study CAE 2015, 17, 45–51. [Google Scholar]
- Zhang, Y.; Yang, C.; Li, Y.; Huang, Y.; Zhang, J.; Zhang, Y.; Li, Q. Ultrasonic extraction and oxidation characteristics of functional groups during coal spontaneous combustion. Fuel 2019, 242, 287–294. [Google Scholar] [CrossRef]
- Zhang, Y.N.; Liu, C.H.; Song, J.J.; Wang, N.P. Study on transfer law of main functional groups in low temperature oxidation of long flame coal. Coal Sci. Technol. 2020, 48, 188–196. [Google Scholar]
- Ma, L.; Wang, D.; Yang, W.; Dou, G.; Xin, H. Synchronous thermal analyses and kinetic studies on a caged-wrapping and sustained-release type of composite inhibitor retarding the spontaneous combustion of low-rank coal. Fuel Process. Technol. 2017, 157, 65–75. [Google Scholar] [CrossRef]
- Zhang, J.X.; Li, B.; Liu, Y.W.; Li, P.; Fu, J.W.; Chen, L.; Ding, P.C. Dynamic multifield coupling model of gas drainage and a new remedy method for borehole leakage. Acta Geotech. 2022, 17, 4699–4715. [Google Scholar] [CrossRef]
- Zhang, J.X.; Liu, Y.W.; Ren, P.L.; Han, H.K.; Zhang, S. A fully multifield coupling model of gas extraction and air leakage for in–seam borehole. Energy Rep. 2021, 7, 1293–1305. [Google Scholar] [CrossRef]
- Li, B.; Zhang, J.X.; Liu, Y.W.; Qu, L.N.; Liu, Q.; Sun, Y.X.; Xu, G. Interfacial porosity model and modification mechanism of broken coal grouting: A theoretical and experimental study. Surf. Interfaces 2022, 33, 102286. [Google Scholar] [CrossRef]
- Deng, J.; Li, Y.Q.; Zhang, Y.T. Effects of hydroxyl on oxidation characteristics of side chain active groups in coal. J. China Coal Soc. 2020, 45, 232–240. [Google Scholar]
- Wang, S.Z. A study of the influence of the teaching time sequence of the modules “Structure and Properties of Substances” and “Fundamentals of Organic Chemistry” on the study of “Molecular Structure and Properties of Organic Substances”. Educ. Chem. 2014, 6, 16–19. [Google Scholar]
- Xie, K.C. Coal Structure and Reactivity, 3rd ed.; Science Press: Beijing, China, 2002. [Google Scholar]
- Zhang, R.; Xia, Y.C.; Tan, J.L.; Ding, S.H.; Xing, Y.W.; Gui, X.H. Analysis and research on low rank coal carbon structure. China Coal 2018, 44, 88–94. [Google Scholar]
- Bhoi, S.; Banerjee, T.; Mohanty, K. Molecular dynamic simulation of spontaneous combustion and pyrolysis of brown coal using ReaxFF. Fuel 2014, 136, 326–333. [Google Scholar] [CrossRef]
- Li, X.; Zeng, F.G.; Wang, W.; Dong, K. FTIR characterization of structural evolution in low-middle rank coals. J. China Coal Soc. 2015, 40, 2900–2908. [Google Scholar]
- Li, X.; Zeng, F.G.; Wang, W.; Dong, K. XRD characterization of structural evolution in low-middle rank coals. J. Fuel Chem. Technol. 2016, 44, 777–783. [Google Scholar]
- Zhang, Y.L.; Wang, J.F.; Xue, S.; Wu, J.M.; Chang, L.P.; Li, Z.F. Kinetic study on changes in methyl and methylene groups during low-temperature oxidation of coal via in-situ FTIR. Int. J. Coal Geol. 2016, 154, 155–164. [Google Scholar] [CrossRef]
- Zhang, Z.; Kang, Q.; Wei, S.; Yun, T.; Yan, G.; Yan, K. Large Scale Molecular Model Construction of Xishan Bituminous Coal. Energy Fuels 2017, 31, 1310–1317. [Google Scholar] [CrossRef]
- Li, Z.M.; Wang, Y.M.; Li, P.; Li, H.P.; Bai, H.C.; Guo, Q.J. Macromolecular model construction and quantum chemical calculation of Ningdong Hongshiwan coal. CIESC J. 2018, 69, 2208–2216. [Google Scholar]
- Wiser, W. Reported in division of fuel chemistry. Preprints 1975, 20, 122. [Google Scholar]
- Given, P. The distribution of hydrogen in coals and its relation to coal structure. Fuel 1960, 39, 147–153. [Google Scholar]
- Shinn, J.H. From coal to single-stage and two-stage products: A reactive model of coal structure. Fuel 1984, 63, 1187–1196. [Google Scholar] [CrossRef]
- Xiao, Y.; Ye, X.X.; Liu, K.H.; Chen, L.G. Transformation law of key functional groups in the process of coal secondary oxidation. J. China Coal Soc. 2021, 46, 989–1000. [Google Scholar]
- Jiang, Y.; Zong, P.; Tian, B.; Xu, F.; Tian, Y.; Qiao, Y.; Zhang, J. Pyrolysis behaviors and product distribution of Shenmu coal at high heating rate: A study using TG-FTIR and Py-GC/MS. Energy Convers. Manag. 2019, 179, 72–80. [Google Scholar] [CrossRef]
- Tian, Y.Y.; Xie, K.C.; Qiao, Y.Y.; Tian, B. Construction and application of coal chemistry research system based on chemical group composition. J. China Coal Soc. 2021, 46, 1137–1145. [Google Scholar]
- Xie, K.C. Structure and Reactivity of Coal; Springer: Heidelberg, Germany, 2015; Volume 10, pp. 978–983. [Google Scholar]
- Carlson, G. Computer simulation of the molecular structure of bituminous coal. Energy Fuels 1992, 6, 771–778. [Google Scholar] [CrossRef]
- Wu, S.; Jin, Z.; Deng, C. Molecular simulation of coal-fired plant flue gas competitive adsorption and diffusion on coal. Fuel 2019, 239, 87–96. [Google Scholar] [CrossRef]
- Hong, D.; Liu, L.; Wang, C.; Si, T.; Guo, X. Construction of a coal char model and its combustion and gasification characteristics: Molecular dynamic simulations based on ReaxFF. Fuel 2021, 300, 120972. [Google Scholar] [CrossRef]
- Hao, M.; Qiao, Z.; Zhang, H.; Wang, Y.; Li, Y. Thermodynamic analysis of CH4/CO2/N2 adsorption on anthracite coal: Investigated by molecular simulation. Energy Fuels 2021, 35, 4246–4257. [Google Scholar] [CrossRef]
- Meng, J.; Niu, J.; Meng, H.; Xia, J.; Zhong, R. Insight on adsorption mechanism of coal molecules at different ranks. Fuel 2020, 267, 117234. [Google Scholar] [CrossRef]
- Feng, W.; Gao, H.F.; Wang, G.; Wu, L.L.; Xu, J.Q.; Li, Z.M.; Li, P.; Bai, H.C.; Guo, Q.J. Molecular model and pyrolysis simulation of Zaoquan coal. CIESC J. 2019, 70, 1522–1531. [Google Scholar]
- Chai, S.Q.; Zeng, Q. Molecular model construction and structural characteristics analysis of Wucaiwan coal in Eastern Junggar Coalfeild based on quantum chemistry theory. J. China Coal Soc. 2022, 1–12. [Google Scholar]
- Ping, A.; Xia, W.C.; Peng, Y.L.; Xie, G.Y. Construction of bituminous coal vitrinite and inertinite molecular assisted by 13C NMR, FTIR and XPS. J. Mol. Struct. 2020, 1222, 128959. [Google Scholar] [CrossRef]
- Jia, J.B.; Zeng, F.G.; Sun, B.L. Construction and modification of macromolecular structure mode for vitrinite from Shengdong2-2 coal. J. Fuel Chem. Technol. 2011, 39, 652–657. [Google Scholar]
- Wang, Q.; Wang, Z.C.; Jia, C.X.; Gong, G.X. Study on structural features of oil sands with solid state 13C-NMR. Chem. Ind. Eng. Prog. 2014, 33, 1392–1396. [Google Scholar]
- Solum, M.S.; Pugmire, R.J.; Grant, D.M.; Kelemen, S.R.; Gorbaty, M.L.; Wind, R.A. 15N CPMAS NMR of the Argonne premium coals. Energy Fuels 1997, 11, 491–494. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, P.Z.; Zheng, M. Study on structural characterization of three Chinese coals of high organic sulphur content using XPS and solid-state NMR spectrescopy. J. Fuel Chem. Technol. 1996, 24, 539–5434. [Google Scholar]
- Budinova, T.; Peyrov, N.; Minkova, V. Computer programmers for radial distribution analyses of X-rays. Fuel 1998, 77, 577–581. [Google Scholar] [CrossRef]
- Senneca, O.; Salatino, P.; Masi, S. Combustion rates of chars from high-volatile fuels for FBC application. Fuel 1998, 77, 1483–1489. [Google Scholar] [CrossRef]
- Jia, J.B. Construction of Structural Model and Molecular Simulation of Methane Formation Mechanism during Coal Pyrolysis for Shendon Vitrinite. Doctoral Dissertation, Taiyuan University of Technology, Taiyuan, China, 2010. [Google Scholar]
- Yu, S.; Bo, J.; Jiahong, L. Retraction Note to: Simulations and experimental investigations of the competitive adsorption of CH 4 and CO 2 on low-rank coal vitrinite. J. Mol. Model. 2019, 25, 178. [Google Scholar] [CrossRef] [PubMed]
- Xiang, J.; Zeng, F.; Liang, H.; Li, B.; Song, X. Molecular simulation of the CH4/CO2/H2O adsorption onto the molecular structure of coal. Sci. China Earth Sci. 2014, 57, 1749–1759. [Google Scholar] [CrossRef]
- Nos’E, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511. [Google Scholar] [CrossRef] [Green Version]
- Quinga, E.; Larsen, J.W. Noncovalent interactions in high-rank coals. Energy Fuels 1987, 1, 300–304. [Google Scholar] [CrossRef]
- Zhu, H.Q.; He, X.; Huo, Y.F.; Xie, Y.Y.; Wang, W.; Fang, S.H. Construction and optimization of lignite molecular structure model. J. Min. Sci. Technol. 2021, 6, 429–437. [Google Scholar]
- Zhang, X.D.; Hao, Z.C.; Zhang, S.; Yang, Y.H.; Yang, H.L.; Fang, S.H. Difference of nano-scale pore changes and its control mechanism for tectonic coal under solvent reconstruction. J. China Univ. Min. Technol. 2017, 46, 7. [Google Scholar]
- Li, Z.; Ward, C.R.; Gurba, L.W. Occurrence of non-mineral inorganic elements in macerals of low-rank coals. Int. J. Coal Geol. 2010, 81, 242–250. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proximate Analysis (w%, daf) | Ultimate Analysis (wt%, daf) | |||||||
|---|---|---|---|---|---|---|---|---|
| Mad | Aad | Vdaf | FCad | Cad | Had | Oad | Nad | St,d |
| 0.79 | 8.288 | 17.3 | 70.922 | 66.12 | 4.56 | 4.37 | 1.28 | 0.18 |
| Normalization (w%, daf) | Atomic Ratio | |||||||
|---|---|---|---|---|---|---|---|---|
| C | H | O | N | S | H/C | O/C | N/C | S/C |
| 86.42 | 5.96 | 5.71 | 1.67 | 0.24 | 0.828 | 0.050 | 0.017 | 0.001 |
| Peak | Area | Center (ppm) | Relative Area | Attribution | Peak | Area | Center (ppm) | Relative Area | Attribution |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 0.5375 | 12.16 | 0.006 | A1 | 20 | 0.1183 | 113.22 | 0.013 | A6 |
| 2 | 0.9461 | 15.30 | 0.011 | A1 | 21 | 0.279 | 115.48 | 0.031 | A6 |
| 3 | 1.565 | 18.37 | 0.018 | A2 | 22 | 0.4417 | 118.19 | 0.050 | A6 |
| 4 | 1.603 | 21.32 | 0.018 | A2 | 23 | 0.6737 | 120.89 | 0.076 | A6 |
| 5 | 0.9237 | 24.36 | 0.010 | A3 | 24 | 0.8716 | 123.70 | 0.098 | A6 |
| 6 | 0.6759 | 27.55 | 0.008 | A3 | 25 | 0.9847 | 126.44 | 0.111 | A6 |
| 7 | 0.7956 | 29.85 | 0.009 | A3 | 26 | 0.9119 | 129.09 | 0.102 | A7 |
| 8 | 0.9374 | 33.14 | 0.011 | A3 | 27 | 0.7487 | 131.85 | 0.084 | A7 |
| 9 | 0.7669 | 35.97 | 0.009 | A3 | 28 | 0.5977 | 134.70 | 0.067 | A7 |
| 10 | 0.723 | 39.29 | 0.008 | A4 | 29 | 0.5317 | 137.56 | 0.060 | A8 |
| 11 | 0.5363 | 43.15 | 0.006 | A4 | 30 | 0.4133 | 140.35 | 0.046 | A8 |
| 12 | 0.4164 | 46.89 | 0.005 | A4 | 31 | 0.2996 | 143.23 | 0.034 | A8 |
| 13 | 0.243 | 50.88 | 0.003 | A5 | 32 | 0.2015 | 146.39 | 0.023 | A8 |
| 14 | 0.1836 | 101.81 | 0.002 | A6 | 33 | 0.152 | 149.39 | 0.017 | A9 |
| 15 | 0.1305 | 105.13 | 0.001 | A6 | 34 | 0.1359 | 152.47 | 0.015 | A9 |
| 16 | 0.2635 | 105.25 | 0.003 | A6 | 35 | 0.08195 | 155.50 | 0.009 | A9 |
| 17 | 0.6444 | 108.31 | 0.007 | A6 | 36 | 0.06333 | 158.49 | 0.007 | A9 |
| 18 | 0.5065 | 110.12 | 0.006 | A6 | 37 | 0.0462 | 161.88 | 0.005 | A9 |
| 19 | 0.8758 | 111.72 | 0.010 | A6 | 38 | 0.02881 | 165.59 | 0.003 | A10 |
| Type | Symbol | Value | Type | Symbol | Value |
|---|---|---|---|---|---|
| Total aromatic carbon | fa | 0.880 | Alkylated aromatic | faS | 0.179 |
| Carbonyl groups | faC | 0.003 | Aromatic bridgehead | faB | 0.253 |
| In an aromatic ring | fa′ | 0.877 | Total aliphatic carbon | fa1 | 0.120 |
| Nonprotonated and aromatic | faN | 0.470 | CH3 | fa1 * | 0.046 |
| Protonated and aromatic | faH | 0.407 | CH or CH2 | fa1H | 0.065 |
| Phenolic or phenolic ether | faP | 0.037 | Bonded to oxygen | fa10 | 0.009 |
| Type | Number | Type | Number |
|---|---|---|---|
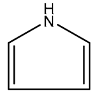 | 1 | 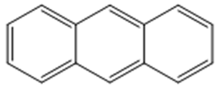 | 3 |
 | 2 | 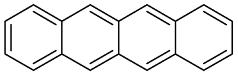 | 1 |
 | 2 | 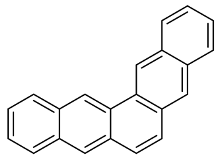 | 1 |
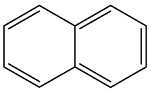 | 3 |  | 1 |
| E (kcal·mol−1) | Ev (kcal·mol−1) | EN (kcal·mol−1) | ||||||
|---|---|---|---|---|---|---|---|---|
| EB | EA | ET | EI | EH | Evan | EE | ||
| Initial | 8263.8600 | 3222.464 | 38.336 | 166.689 | 4.085 | 0 | 4850.632 | −18.35 |
| Final | 684.560 | 84.553 | 51.754 | 131.287 | 3.120 | 0 | 418.828 | −4.982 |
| Density (g/cm3) | E (kcal/mol) | EV | EN | |||||
|---|---|---|---|---|---|---|---|---|
| EB | EA [32] | ET | EI | EH | Evan | EE | ||
| 1.0 | 562.944 | 87.167 | 52.554 | 128.714 | 4.002 | 0 | 326.40 | −27.967 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qu, L.; Liu, L.; Chen, J.; Wang, Z. Molecular Model Construction and Optimization Study of Gas Coal in the Huainan Mining Area. Processes 2023, 11, 73. https://doi.org/10.3390/pr11010073
Qu L, Liu L, Chen J, Wang Z. Molecular Model Construction and Optimization Study of Gas Coal in the Huainan Mining Area. Processes. 2023; 11(1):73. https://doi.org/10.3390/pr11010073
Chicago/Turabian StyleQu, Lina, Long Liu, Jinhao Chen, and Zhenzhen Wang. 2023. "Molecular Model Construction and Optimization Study of Gas Coal in the Huainan Mining Area" Processes 11, no. 1: 73. https://doi.org/10.3390/pr11010073
APA StyleQu, L., Liu, L., Chen, J., & Wang, Z. (2023). Molecular Model Construction and Optimization Study of Gas Coal in the Huainan Mining Area. Processes, 11(1), 73. https://doi.org/10.3390/pr11010073