Abstract
The molecular structure of cationic surfactants is closely related to their flotation performance. In this paper, three cationic surfactants with different head group structures were selected as collectors of kaolinite, and the substituent effects were studied by the DFT method. The DFT calculation results showed that increasing the number of substituents in the dodecylamine head group can significantly increase its surface and head group charge. Dodecylamine has the lowest LUMO orbital energy, so dodecylamine has the strongest electron attraction ability and the strongest interaction with kaolinite. Electron density differential showed that there was an area of electron aggregation between the collector and the surface of the kaolinite. The interaction energy of DDA on kaolinite surfaces was greater than that of the other two collectors, indicating that the adsorption of DDA on the surface of kaolinite was more stable. Flotation results showed that higher a kaolinite yield was obtained in the presence of dodecyl dihydroxyethyl methyl ammonium chloride. The calculated results of the solvent-accessible surfaces, the head group charge, and the number of bonds between the collector and the kaolinite show good consistency with the actual flotation results of the three collectors, which can be used as a screening index for kaolinite flotation collectors.
1. Introduction
As a clay mineral, kaolinite is a white layered silicate product produced by natural weathering of feldspar and silicate minerals. It has good plasticity, insulation, acid and alkali resistance, and excellent adhesion. It is widely used in ceramics, paper making, textile, rubber, environmental protection, biological medicine, and refractory and other industries. Kaolinite is a very important mineral with large reserves in the world. However, kaolinite is also a gangue mineral that needs to be removed in many industries. For example, in iron ore flotation, coal flotation, and slime precipitation, kaolinite is an unfavorable mineral, which should be removed.
At present, kaolinite in mixed minerals is mainly removed by flotation, and its surface properties are changed by adding reagents so that it can be floated out. Many scholars have studied this. Liu et al. [1] used a series of tertiary amines to study the flotation behavior of kaolinite. The results showed that the maximum recovery of kaolinite flotation using N,N-dimethyl-dodecyl amine (DRN), N,N-diethyl-dodecyl amine (DEN), and N,N-dipropyl-dodecyl amine (DPN) increased by 93%, 88%, and 84% respectively, while the recovery of N,N-dibenzyl-dodecyl amine (DBN) was relatively low. Jiang et al. [2] studied the flotation behavior of dodecyl trimethyl ammonium chloride (DTAC), tetradecyl trimethyl ammonium chloride (TTAC), and hexadecyl trimethyl ammonium chloride (CTAC) on kaolinite with different particle sizes. The flotation results show that the flotation recovery of kaolinite is higher with the increase of collector concentration, and the flotation recovery of kaolinite with a 0.045–0.1 mm particle size will increase with the increase of the carbon chain of quaternary ammonium salt. However, the flotation results of a −0.045 mm particle size is just the opposite. Barani [3] floated the tailings of Iran’s kaolin by using dodecylamine (DDA) as collectors and obtained the best results under acidic conditions. Xu et al. [4] investigated the flotation behavior of different size particles when using DTAC and CTAC as the collectors. The results showed that the length of the carbon chain in the collector has significant effects on the flotation. Hu et al. [5] studied the flotation behavior of cetyltrimethylammonium bromide (CTAB) on kaolinite and found that kaolinite in the acidic pH area has better floatability and that the adsorption of CTAB on kaolinite is mainly controlled by electrostatic interaction. Jiang et al. [6] synthesized new carboxylic hydroxamic acid compounds, including 2-carboxyl-6-methylcyclohexane carboxamic acid (CMCA), 2-carboxymethyl-decylen-4-yl hydroxamic acid (CDHA), and 2-carboxymethyl-tetradecen-4-yl hydroxamic acid (CTHA) and conducted flotation tests on diaspore, kaolinite, and illite with these compounds. Bu et al. [7] studied the separation of quartz from kaolinite with DDA as the collector. Liu et al. [8] studied the flotation behavior of N, N-dipropyl dodecyl amine on kaolinite and diaspore. The flotation results showed that with the increase of collector dosage, the recoveries of both will increase. In other studies, a large number of new cationic collectors have been synthesized, and their flotation behaviors on kaolinite, pyrophyllite, illite, and other minerals have been studied [9,10,11,12]. In the above research, the collector performance in kaolinite flotation is mainly studied through large-scale experiments, and there is no clear direction for collector selection. In recent years, with the development of computer simulation technology, scholars can study the interaction mechanism between collector and mineral surface from a microscopic perspective.
Shen et al. [13] used a dodecylamine chloride/fatty acid mixture as a collector for fine kaolinite flotation and studied its related adsorption behavior through flotation tests and molecular dynamics simulation methods. The results showed that the mixed collector had higher kaolinite flotation recovery than the single DDA collector. Silva [14] et al. compared the flotation differences between ether amine and amide through molecular simulation, and the results showed that the adsorption effect of amide on a quartz surface was better than that of ether amine, mainly due to the larger size of the amide head group. Wang et al. [15] used molecular dynamics simulation to study the influence of the average molecular weight of sodium petroleum sulfonate on the flotation performance of fluorite. The results showed that, with the increase of the average molecular weight of sodium petroleum sulfonate, the flotation recovery of fluorite gradually decreased. Quezada et al. [16] used Gromacs software to study the adsorption behavior of polyacrylic acid polyelectrolyte (PAA) on the surface of quartz, kaolinite, and montmorillonite. Molecular simulation can not only be used to reveal the interaction between collectors and kaolinite [17,18,19], quartz [20,21,22], fluorite [23], and other crystal minerals [24,25] but also can simulate the interaction between coal and collectors [26]. Pradip et al. [27] proposed the design of flotation reagents based on molecular simulation and pointed out the application prospect of changing the traditional trial and error method to the molecular modeling method to guide the selection of flotation reagents.
In this study, three cationic surfactants with different head group structures were selected as collectors of kaolinite, and the influence of substituent structure changes on the properties of the reagents and the mechanism of the interaction between the reagents and kaolinite were studied using the DFT method.
2. Materials and Methods
2.1. Materials
2.1.1. Reagents
Amine and ammonium salt cationic surfactants are mainly used as cationic collectors in kaolinite flotation. The molecular structure of cationic surfactants, such as the length and structure of non-polar carbon chain and the number and structure of hydrophilic functional groups, will have an impact on the parameters of the collector molecules, such as electrification, frontier orbital energy, dipole moment, molecular atomic area and volume, and molecular energy, and thus have a greater impact on its flotation performance.
When the H atom on the -NH3 in amine cationic surfactant is replaced by different functional groups, the electronic effect of the substituent will affect the adsorption behavior of the surfactant molecules on the surface of kaolinite. Three amine surfactants with different substituent structures were selected in this paper. The chemical formulas and abbreviations of the surfactants are shown in Table 1. Due to the low solubility of dodecylamine and dodecyl diethanolamine under neutral conditions, in this study, the two reagents were mixed with concentrated hydrochloric acid in a molar ratio of 1:1 to prepare the corresponding ammonium salt. Then, distilled water was used to prepare 0.1 mol/L solution for use. All reagents were purchased from Aladdin Reagent Co., Ltd. All reagents are analytically pure.

Table 1.
Chemical formulas and abbreviations of the surfactants.
2.1.2. Materials
The kaolinite used in the test was obtained from Jinyan Kaolin Co., Ltd., Huaibei City, Anhui Province. The kaolinite sample was first crushed and grounded, and a −0.074 mm particle size was taken for flotation test. The chemical components of the kaolinite samples are shown in Table 2.
Table 2.
Chemical components of kaolinite samples.
2.2. DFT Study of Collector Properties
The change of substituent structure in collector molecules will affect the electronic distribution of molecules through an induction effect and a conjugation effect. Therefore, the influence of substituent structure changes on the quantum mechanical properties of the molecules can be accurately obtained through the density functional theory study of collector molecules.
The molecular structures of the three collectors were constructed by using the Visual module in Materials Studio 2017, as shown in Figure 1.
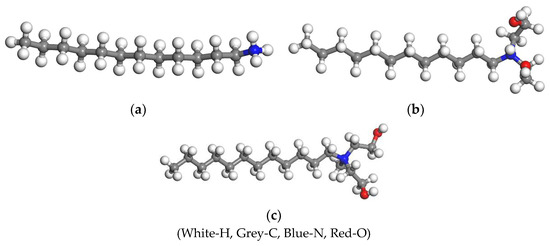
Figure 1.
Molecular structures of the three collectors. (a) DDA; (b) BHDA; (c) BHMDC.
The Dmol3 module in the simulation software was used to optimize the structure of each reagent molecule. The specific parameter settings were as follows: task is set to geometry optimization; GGA-PBE is used for the exchange correlation function; effective core potentials and DNP basis sets are used; all calculation precision is set to fine; SCF tolerance is set to 1.0 × 10−6 eV/atom; and select TS coupling method for correction. In the actual flotation process, all reagents were prepared into an ionic state, so in the calculation process, all reagents were calculated for their properties in the ionic state, and the charge was set to +1. The calculation properties were orbitals and population analysis.
2.3. DFT Study of Adsorption Process
2.3.1. Establishment of Initial Adsorption Model System
Kaolinite is a typical layered silicate mineral, with the molecular formula of Al4 (Si4O10) (OH)8. The crystal cells of kaolinite are mainly connected by aluminum oxide octahedron and silicon oxide tetrahedron. The layers are connected by hydrogen bonds with weak energy, so kaolinite is easy to be slimed into fine flake particles in water. There are mainly two different planes generated during the cleavage of kaolinite. One is 001 plane dominated by Al-OH, and the other is 001 (-) plane dominated by Si-O, as shown in Figure 2. Therefore, this paper mainly studied the adsorption behavior of the collector on these two surfaces.
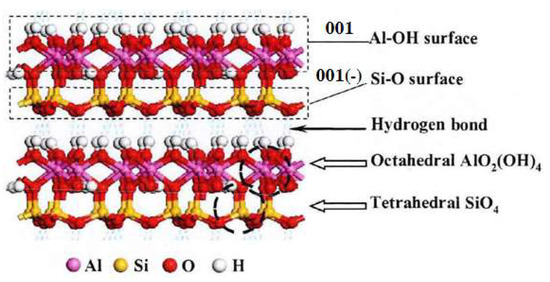
Figure 2.
Ball- stick model of crystal structure of kaolinite.
The geometric structure of the initial kaolinite unit cell was optimized using the CASTEP (Cambridge Sequential Total Energy Package) module based on the density functional theory. All calculations were performed in reciprocal space. The lattice parameters after the optimization of the kaolinite cell are shown in Table 3. It can be seen from Table 3 that the lattice parameters obtained by the calculation method in this paper are relatively close to those measured in the experiment, indicating that the selection of the simulation parameters was reasonable.
Table 3.
Lattice parameters of the kaolinite cell.
Table 3.
Lattice parameters of the kaolinite cell.
| a | b | c | α | β | γ | |
|---|---|---|---|---|---|---|
| Simulation | 5.195 | 9.006 | 7.371 | 93.027 | 105.987 | 89.867 |
| Experimental [28] | 5.153 | 8.942 | 7.391 | 91.926 | 105.046 | 89.797 |
001 and 001 (-) surfaces were obtained by using the cleave surface function in the software. A vacuum layer with a thickness of 50 Å was added on the top of the surface. The CASTEP module was still used to optimize the geometric structure of the constructed kaolinite surface. In the calculation process, the Monkhorst Pack grid k points were set, and the surface model was gamma points. The lowest atom and its connected atoms were fixed in the calculation process.
2.3.2. Calculation Method
The optimized collector molecules were added to the surface of kaolinite, and the head groups were oriented towards the surface of kaolinite, as shown in Figure 3. In order to avoid the interaction between surfactant molecules caused by periodic structure, the dimensions of the periodic cells were set as 15 Å × 15 Å × 55.5 Å. The adsorption process of each collector molecule on the surface of kaolinite was calculated to minimize the energy. The specific parameter settings were as follows: Task was set to Geometry Optimization; GGA-PBE was used for the exchange correlation function; effective core potentials and DNP basis sets were used; all calculation precision was set to fine; SCF tolerance was set to 1.0 × 10−6 eV/atom; and TS coupling method was selected for correction. The electron density difference, and population analysis of the system were calculated respectively.
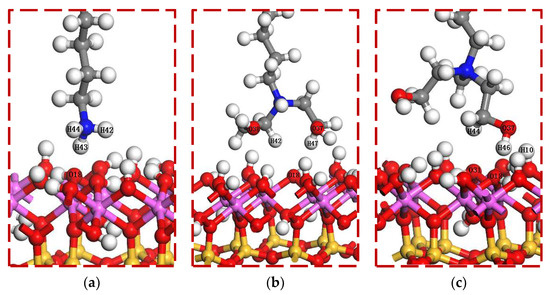
Figure 3.
Initial adsorption configuration for DFT simulation. (a) DDA/001 surface; (b) BHDA/001 surface; (c) BHMDC/001 surface.
2.4. Flotation Process
The flotation of kaolinite was carried out on an XFD flotation machine. The volume of the flotation cell was 150 mL, and the impeller speed was 1400 rpm. The flotation process was as follows. Firstly, 9 g of dried kaolinite sample was weighed and added into the flotation cell. Then, distilled water was added and stirred for 3 min to disperse the kaolinite. Then, a collector was added into the flotation cell and conditioned for 2 min. Then, the inflation valve was opened, and the foam product was collected for 3 min. collecting foam products for 3 min. Finally, the foam product was dried, weighed, and the yield of kaolinite was calculated.
3. Results and Discussion
3.1. Molecular Structure
3.1.1. Molecular Surface Area
Molecular surfaces usually have multiple representations, including van der Waals surface, Connolly surface, solvent contactable surface, etc., as shown in Figure 4. In Figure 4, blue is the surface that the center of the basketball passes through when the basketball rolls. This surface is called the accessible solvent surface.
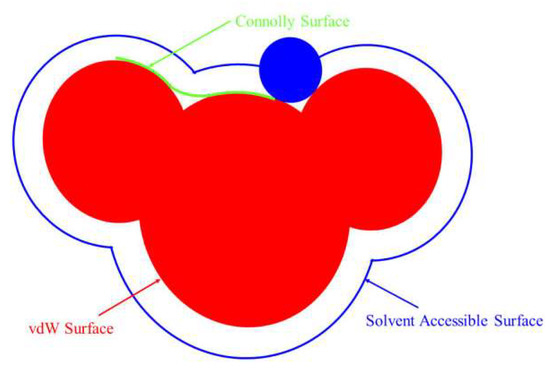
Figure 4.
Molecular surface of reagent.
Generally, solvent-accessible surfaces are useful for studying biomacromolecules. They can not only reflect the size and shape of molecules after solvation but also analyze the hydrophilic and hydrophobic sites of macromolecules. For the surfactant molecules in this study, the area occupied by single molecules when spreading at the gas–liquid interface at the critical micelle concentration can be estimated by calculating their molecular surfaces. The specific values of the van der waals surface, connolly surface, and solvent contactable surface of each agent molecule in this study are shown in Table 4. It can be seen from Table 4 that the molecular surface areas of BHDA and BHMDC were significantly larger than DDA. It can be seen from the molecular structure that the branched substituents of the head group of BHDA and BHMDC were larger. When they are adsorbed on the mineral surface, due to the steric effect, it is conducive to reducing the amount of adsorption of the agents on the molecular surface.
Table 4.
The specific values of each surface.
3.1.2. Mulliken Charge Distribution
The charge on the atom can be obtained by fitting the molecular electrostatic potential. The Mulliken charge is a method to calculate the atomic partial charge. The Mulliken charge distribution of each atom in the reagent molecule is shown in Figure 5, and the Mulliken charge values of the central N atom and the functional group are shown in Table 5.
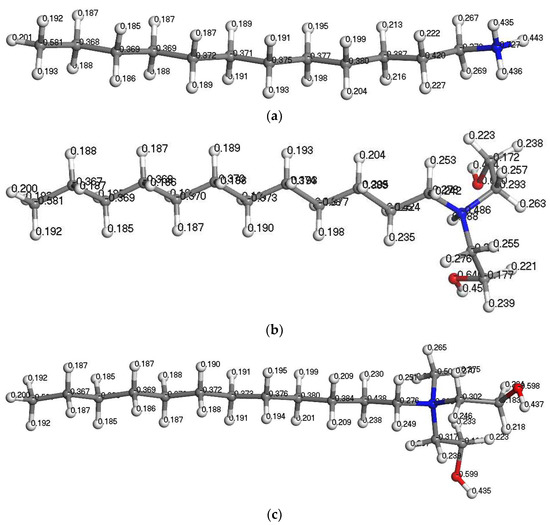
Figure 5.
Mulliken charge distribution in the collector molecules. (a) DDA; (b) BHDA; (c) BHMDC.
Table 5.
Mulliken charge distribution of collectors.
It can be seen from Table 5 that the Mulliken charge of the central N atom was negative, the positive charge of the BHDA and BHMDC functional groups was greater than DDA. BHMDC has the strongest positive electricity. The zeta potential of silicate minerals is negative at a wide range of pH values, while ammonium salt cationic surfactants are mainly adsorbed on the surface of silicate minerals by electrostatic action. Therefore, the introduction of benzyl and alcohol hydroxyl groups into the head group of amine surfactants can improve the electrostatic interaction between the reagents and kaolinite.
3.1.3. Molecule Frontier Orbital
According to the frontier orbital theory, the higher the HOMO orbital energy value of reactants, the higher the electron cloud density and the stronger the electron giving ability. The smaller the absolute value of the energy difference between the highest occupied orbit (HOMO) of one reactant and the lowest empty orbit (LUMO) of another reactant, the more conducive to the interaction between them. The highest occupied orbit and the lowest occupied orbit of each reagent and kaolinite were calculated, and the orbital energy difference between different reagent molecules and kaolinite, as well as the orbital energy difference of the reagent itself, were calculated according to Equations (1)–(3). The distribution of the LUMO orbital isosurface of each reagent molecule is shown in Figure 6 (the isosurface is 0.02 electrons/Å3), and the frontier orbital energy and energy difference are shown in Table 6. It can be seen from Table 6 that the HOMO orbital values of the three collectors are very close, indicating that they have similar electron supply capacity. However, in the process of adsorption with kaolinite, since the three collectors are cationic, their ability to attract electrons is more important. It can be seen from Table 6 that DDA has the lowest LUMO orbital energy, so DDA has the strongest electron attraction ability and the strongest interaction with kaolinite.
where ΔE1 is the energy difference between the LUMO orbit of the reagent and the HOMO orbit of the kaolinite; ΔE2 is the energy difference between the HOMO orbital of the reagent and the LUMO orbital of the kaolinite; ΔE3 is the energy difference between HOMO orbit and LUMO orbit of the agent itself; and represents the HOMO and LUMO orbital energy of the agent, respectively; and represents the HOMO and LUMO orbital energies of kaolinite, respectively.
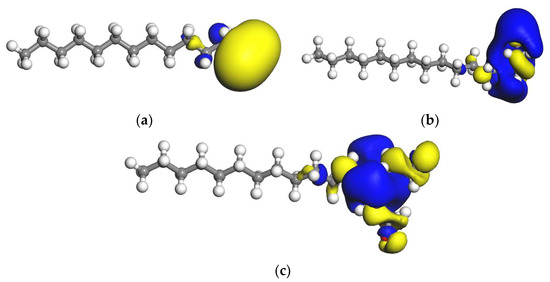
Figure 6.
LUMO orbital of collector molecule. (a) DDA; (b) BHDA; (c) BHMDC. The isosurface is 0.02 electrons/Å3.
Table 6.
Frontier orbital energy and energy difference.
It can be seen from Figure 6 that the LUMO orbitals of each reagent molecule were mainly concentrated in the head group and mainly located on the hydrogen atom. The yellow and blue regions in the figure are spintrons α and β, respectively.
When the energy level difference between the HOMO and LUMO between the two molecules is less than 0.2206 a.u., electrons can transition between their orbits. It can be seen from Table 6 that the energy level difference between the HOMO of reagent and the LUMO of kaolinite Δ E2 is greater than 0.2206 a.u., indicating that electrons cannot transition from the HOMO orbital of reagents to the LUMO orbital of kaolinite. The energy level difference between HOMO of kaolinite and LUMO of reagent Δ E1 is less than 0.2206 a.u., indicating that the LUMO orbital of the reagent can interact with the HOMO orbital of kaolinite.
3.1.4. Dipole Moment and Electronegativity
Dipole moment is a physical quantity that characterizes the size of the molecular polarity. In physics, the system composed of two electric charges with equal signs and opposite distance of d is called a dipole. The product of its electric quantity and distance is the size of the dipole moment. The molecular dipole moment can reflect the spatial configuration of the molecule, the geometry of the arrangement of atoms in the molecule, and the angle between chemical bonds. In the flotation process, the dipole moment not only affects the dispersion of the reagent in water but also affects its adsorption on the mineral surface. The larger the dipole moment of the reagent molecule, the easier it is for the reagent to interact with the mineral.
The greater the electronegativity of the reagent molecule, the stronger the ability of the reagent to obtain electrons. Therefore, the stronger the electronegativity of reagents in the flotation process, the easier it is for the collector molecules to be adsorbed on the mineral surface. The electronegativity of molecules is equal to the average value of ionization potential and electron affinity of the reagent molecules. Ionization potential refers to the energy required when the molecule loses an electron and becomes a positive ion, which is equal to the HOMO of the molecule. Electron affinity energy refers to the energy released when the molecule gets an electron and becomes a negative ion, which is equal to the LUMO of the molecule.
The calculation results of the dipole moment and the electronegativity of the reagent molecules are shown in Table 7. It can be seen from Table 7 that, compared with the other three reagents, DDA has the largest dipole moment and the largest electronegativity, indicating that DDA has a stronger interaction with the surface of kaolinite.
Table 7.
Dipole moment and electronegativity of collector.
3.2. Interaction between Collector and Kaolinite Surface
3.2.1. Mulliken Bond Populations
The Mulliken bond populations of each reagent molecule adsorbed on the 001 and 001 (-) face of kaolinite are shown in Table 8. The closer the Mulliken bond is, the larger the population value is, which indicates that there is a stronger interaction between the two atoms. From Table 8, it can be seen that the H atom in each reagent head group interacted with the O atom on the surface of kaolinite 001, such as H43 in the DDA molecule, H47 in the BHDA molecule, and H46 in the BHMDC molecule. It can also be seen from Table 8 that the hydroxyl group introduced in the reagent head functional group will increase the interaction between the reagent and the H atom on the surface of kaolinite 001 and increase the adsorption strength of the reagent, such as O37 in the BHDA molecule and O37 in the BHMDC molecule.
Table 8.
Mulliken bond populations.
From the aspect of interaction strength between reagents and the kaolinite surface, the order of the three reagents is BHMDC > DDA > BHDA. In terms of the number of atoms that can interact with each other, the order of the three reagents is BHMDC > BHDA > DDA. It can be seen from Figure 3 that BHMDC can cover more of the kaolinite surface and better improve the hydrophobicity of the kaolinite surface. In addition, because of the large head group of BHMDC, the steric hindrance effect is stronger, which can effectively reduce the consumption of the collector.
3.2.2. Electron Density Differential
The electron density differential refers to the difference between the electron density of the kaolinite collector adsorption equilibrium system and the electron density in the case of kaolinite and the collector alone. It can be calculated by the Equation (4):
is the electronic density of the kaolinite/collector system after adsorption, and and are the electronic density of kaolinite and collector before adsorption, respectively.
The electron density differential of the collector at the adsorption equilibrium of 001 and 001 (-) faces of kaolinite is shown in Figure 7. It can be seen from Figure 7 that there were electrons transferred between the three cationic collectors and kaolinite surfaces, and the interaction between the collectors and 001 faces of the kaolinite was stronger. In addition, the electron transfer between DDA and BHMDC and the kaolinite surface was stronger than BHDA, which is consistent with the analysis results of the Mulliken bond populations.
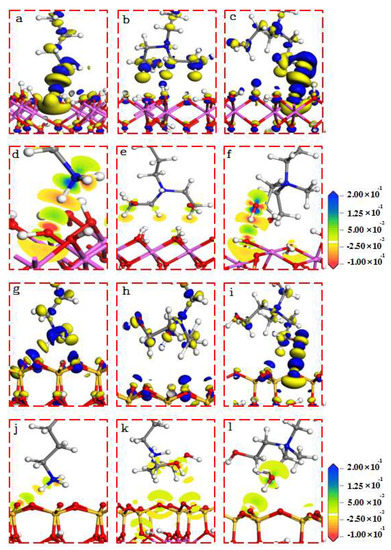
Figure 7.
Electron density differential of the collector at the adsorption equilibrium. The isosurface is 0.01 electrons/Å3 ((a–c) for 001 surface; (g–i) for 001 (-) surface).
In Figure 7, blue represents electron aggregation, and yellow represents electron consumption. The cationic collector used in this study has a strong ability to attract electrons, so the electron aggregation was mainly located near the head group of the collector, while the electron consumption was mainly located near the O of the kaolinite. And there was an area of electron aggregation between the collector and the surface of kaolinite, which was caused by the electron attraction of the collector head group.
3.2.3. Interaction Energy
The adsorption strength of the collector on the surface of kaolinite can be expressed by the interaction energy. The greater the interaction energy, the more stable the adsorption. The calculation formula for interaction energy is shown in Equation (5).
In the formula, Eads is the interaction energy, Etotal is the total energy of the system after the adsorption is stabilized, Ecollector is the energy of the collector molecule, and Esurface is the energy of the kaolinite 001 or 001 (-) surface.
The calculation results from the interaction energy between the three collectors, and the 001 and 001 (-) faces of the kaolinite are shown in Table 9. It can be seen from Table 9 that the interaction energy of the three collectors on the 001 (-) surface of the kaolinite was greater than that on the 001 surface, indicating that the collector adsorption on the 001 (-) surface of the kaolinite was more stable, which was consistent with previous studies. The interaction energy of DDA on the 001 and 001 (-) surfaces of the kaolinite was greater than that of the other two collectors, indicating that the adsorption of DDA on the surface of the kaolinite was more stable. This was consistent with the results of frontier orbital analysis, dipole moment, and electronegativity analysis.
Table 9.
Interaction energy between three collectors and kaolinite surface.
3.3. Flotation Results
The flotation results of the kaolinite at different collector concentrations are shown in Figure 8. It can be seen from Figure 8 that, with the increase of the collector concentration, the recovery rate of kaolinite increases continuously. At the same collector concentration, the recovery of kaolinite when BHMDC is used is significantly higher than DDA and BHDA, indicating that the quaternization of cationic collectors is conducive to improving their flotation performance.
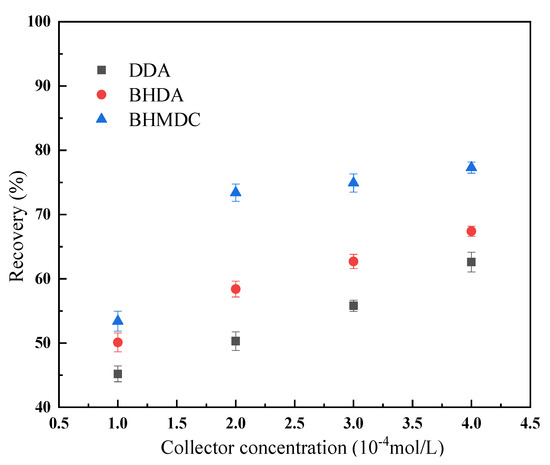
Figure 8.
Flotation results of kaolinite at different collector concentrations. (The data points represent the mean values (n = 3) ± standard deviation).
Combined with the DFT calculation results, we can find that there was a certain correlation between the properties of the collector molecules and their flotation performance with kaolinite. The calculation results of the frontier orbit, dipole moment, and electronegativity show that DDA was more easily adsorbed on the surface of the kaolinite, and the calculation results of the interaction energy between the collector and the kaolinite also show that DDA was more easily adsorbed on the surface of kaolinite. However, the flotation results showed that the flotation recovery of the kaolinite was higher when BHDMA and BHDA were used as collectors. It revealed that it is inaccurate to use the above indicators to screen cationic collectors for kaolinite flotation. The above indicators can be used to determine whether the collector can float kaolinite, but they cannot be used for the screening and optimization of the collector.
The calculation results of the molecular surface area, Mulliken charge distribution, and Mulliken bond populations showed that, compared with DDA and BHDA, BHMDC has the largest solvent-accessible surfaces (655.77 Å2), the largest head group charge (0.658e), and the largest number of bonds (10). The larger the molecular surface area of the collector molecule is, the more surface there is that can be covered by a single collector molecule when it is adsorbed on the kaolinite surface, thus reducing the consumption of the collector. A large head group charge can ensure that the BHMDC can be stably adsorbed on the surface of kaolinite. The more bonds that are formed between the collector molecules and the kaolinite surface, the more stable the final adsorption configuration of the collector and the larger the covered surface area will be, and the floatability of kaolinite is improved.
4. Conclusions
In the present work, three cationic surfactants with different head group structures were selected as collectors of kaolinite, and the influence of substituent structure changes on the properties of the reagents and the mechanism of the interaction between the reagents and kaolinite were studied using the DFT method.
Increasing the number of substituents in the dodecylamine head group can significantly increase its connolly surface, solvent-accessible surface, and head group charge, which is conducive to reducing the consumption of the collector used in the flotation process. DDA has the lowest LUMO orbital energy, so DDA has the strongest electron attraction ability and the strongest interaction with kaolinite. The calculated results of the solvent-accessible surfaces, the head group charge, and the number of bonds between the collector and kaolinite show good consistency with the actual flotation results of the three collectors, which can be used as the screening index for kaolinite flotation collectors. The research results of this paper have important guiding significance for the selection, design, and optimization of flotation collectors.
Author Contributions
Conceptualization, L.S.; methodology, L.S.; software, L.S.; validation, L.S., J.G. and Y.L.; formal analysis, J.G.; investigation, J.G.; resources, Y.L.; data curation, Y.L.; writing—original draft preparation, L.S.; writing—review and editing, E.Q.; visualization, E.Q.; supervision, E.Q.; project administration, E.Q.; funding acquisition, L.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Natural Science Foundation of China (52104241); the China Postdoctoral Science Foundation (2019M652163); and the Anhui Postdoctoral Science Foundation (2019B338).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Liu, C.; Hu, Y.; Cao, X. Substituent effects in kaolinite flotation using dodecyl tertiary amines. Miner. Eng. 2009, 22, 849–852. [Google Scholar] [CrossRef]
- Jiang, H.; Liu, G.; Hu, Y.; Xu, L.; Yu, Y.; Xie, Z.; Chen, H. Flotation and adsorption of quaternary ammonium salts collectors on kaolinite of different particle size. Int. J. Min. Sci. Technol. 2013, 23, 249–253. [Google Scholar] [CrossRef]
- Barani, K. Flotation of kaolinite from tailings of kaolin-washing plants by cationic collectors. In Proceedings of the E3S Web of Conferences, Swieradow-Zdroj, Poland, 25–28 September 2016; Volume 8, p. 01014. [Google Scholar]
- Longhua, X.; Yuehua, H.; Faqin, D.; Hao, J.; Houqin, W.; Zhen, W.; Ruohua, L. Effects of particle size and chain length on flotation of quaternary ammonium salts onto kaolinite. Mineral. Petrol. 2015, 109, 309–316. [Google Scholar] [CrossRef]
- Hu, Y.; Jiang, H.; Wang, D. Electrokinetic behavior and flotation of kaolinite in CTAB solution. Miner. Eng. 2003, 16, 1221–1223. [Google Scholar] [CrossRef]
- Yu-Ren, J.; Zhi-Gang, Y.; Yun-Lai, Y.; Xiao-Hong, Z. Synthesis and collecting properties of novel carboxyl hydroxamic acids for diaspore and aluminosilicate minerals. Miner. Eng. 2010, 23, 830–832. [Google Scholar] [CrossRef]
- Bu, X.; Evans, G.; Xie, G.; Peng, Y.; Zhang, Z.; Ni, C.; Ge, L. Removal of fine quartz from coal-series kaolin by flotation. Appl. Clay Sci. 2017, 143, 437–444. [Google Scholar] [CrossRef]
- Liu, C.; Hu, Y.; Feng, A.; Guo, Z.; Cao, X. The behavior of N,N-dipropyl dodecyl amine as a collector in the flotation of kaolinite and diaspore. Miner. Eng. 2011, 24, 737–740. [Google Scholar] [CrossRef]
- Cao, X.F.; Hu, Y.H.; Xu, J. Synthesis of γ-alkoxy-propylamines and their collecting properties on aluminosilicate minerals. J. Cent. South Univ. Technol. 2004, 11, 280–285. (In English) [Google Scholar] [CrossRef]
- Cao, X.F.; Zhang, L.M.; Hu, Y.H.; Liu, C.M.; Ouyang, K. Synthesis of N, N-diethyl dodecyl amine and its flotation properties on bauxite. J. Cent. South Univ. Technol. 2008, 15, 188–192. (In English) [Google Scholar] [CrossRef]
- Hu, Y.H.; Ouyang, K.; Cao, X.F.; Zhang, L.M. Flotation of kaolinite and diaspore with hexadecyl dimethyl benzyl ammonium chloride. J. Cent. South Univ. Technol. 2008, 15, 378–381. (In English) [Google Scholar] [CrossRef]
- Cao, X.F.; Liu, C.M.; Hu, Y.H. Flotation of kaolinite with dodecyl tertiary amines. J. Cent. South Univ. Technol. 2009, 16, 749–752. (In English) [Google Scholar] [CrossRef]
- Shen, L.; Zhu, J.; Liu, L.; Wang, H. Flotation of fine kaolinite using dodecylamine chloride/fatty acids mixture as collector. Powder Technol. 2017, 312, 159–165. [Google Scholar] [CrossRef]
- Silva, K.; Silva, L.A.; Pereira, A.M.; Bastos, L.C.; Correia, J.C.G.; Piçarra, A.; Bicalho, L.; Lima, N.; Filippova, I.V.; Filippov, L.O. Comparison between etheramine and amidoamine (N-[3-(dimethylamino)propyl] dodecanamide) collectors: Adsorption mechanisms on quartz and hematite unveiled by molecular simulations. Miner. Eng. 2022, 180, 107470. [Google Scholar] [CrossRef]
- Wang, Z.; Ren, Z.; Gao, H.; Gao, Z.; Xu, L.; Zhu, X.; Liu, Y. Effect of active substance content and molecular weight of petroleum sulfonate on fluorite flotation: Molecular dynamics simulation. Miner. Eng. 2021, 174, 107257. [Google Scholar] [CrossRef]
- Quezada, G.R.; Piceros, E.; Robles, P.; Moraga, C.; Gálvez, E.; Nieto, S.; Jeldres, R.I. Polyacrylic acid to improve flotation tailings management: Understanding the chemical interactions through molecular dynamics. Metals 2021, 11, 987. [Google Scholar] [CrossRef]
- Cao, X.F.; Lu, J.A.; Liu, R.Q. The designation of the collector in aluminosilicate minerals flotation and the study of its mechanism. Adv. Mater. Res. 2014, 997, 655–659. [Google Scholar] [CrossRef]
- Nulakani, N.V.R.; Baskar, P.; Patra, A.S.; Subramanian, V. Adsorption of guanidinium collectors on aluminosilicate minerals-a density functional study. Phys. Chem. Chem. Phys. 2015, 17, 23805–23815. [Google Scholar] [CrossRef]
- Chang, Z.; Sun, C.; Kou, J.; Fu, G.; Qi, X. Experimental and molecular dynamics simulation study on the effect of polyacrylamide on bauxite flotation. Miner. Eng. 2021, 164, 106810. [Google Scholar] [CrossRef]
- Patra, A.S.; Nulakani, N.V.R.; Pavan Kumar, Y.; Subramanian, V.; Dash, J.; Mukherjee, A.K. Design and synthesis of novel polyamine collector to recover iron values from iron ore slimes. Powder Technol. 2018, 325, 180–191. [Google Scholar] [CrossRef]
- Li, L.; Hao, H.; Yuan, Z.; Liu, J. Molecular dynamics simulation of siderite-hematite-quartz flotation with sodium oleate. Appl. Surf. Sci. 2017, 419, 557–563. [Google Scholar] [CrossRef]
- Liu, W.; Liu, W.; Zhao, Q.; Peng, X.; Wang, B.; Zhou, S.; Zhao, L. Investigating the performance of a novel polyamine derivative for separation of quartz and hematite based on theoretical prediction and experiment. Sep. Purif. Technol. 2020, 237, 116370. [Google Scholar] [CrossRef]
- Zheng, R.; Ren, Z.; Gao, H.; Chen, Z.; Qian, Y.; Li, Y. Effects of crystal chemistry on sodium oleate adsorption on fluorite surface investigated by molecular dynamics simulation. Miner. Eng. 2018, 124, 77–85. [Google Scholar] [CrossRef]
- Tang, Y.; Yao, J.; Yin, W.; Kelebek, S. Molecular dynamics simulation of cetyl phosphate adsorption in flotation of magnesite and pertinent chemical aspects. Minerals 2020, 10, 761. [Google Scholar] [CrossRef]
- Abdalla, M.A.M.; Peng, H.; Wu, D.; Abusin, L.; Mbah, T.J. Prediction of Hydrophobic Reagent for Flotation Process Using Molecular Modeling. ACS Omega 2018, 3, 6483–6496. [Google Scholar] [CrossRef]
- Xia, Y.; Zhang, R.; Cao, Y.; Xing, Y.; Gui, X. Role of molecular simulation in understanding the mechanism of low-rank coal flotation: A review. Fuel 2020, 262, 116535. [Google Scholar] [CrossRef]
- Pradip; Beena, R. Molecular modeling and rational design of flotation reagents. Int. J. Miner. Process. 2003, 72, 95–110. [Google Scholar] [CrossRef]
- Bish, D.L. Rietveld refinement of the kaolinite structure at 1.5 K. Clays Clay Miner. 1993, 41, 738–744. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).