
Progress of the Analytical Methods and Application of Plutonium Isotopes in the Environment
Abstract
:1. Introduction
2. Plutonium Isotope Pretreatment and Purification Separation
2.1. Sample Pretreatment Techniques
2.2. Purification and Separation Methods for Plutonium Isotopes
3. Methods for Detecting Plutonium Isotopes
3.1. Alpha Spectrometry
3.2. TIMS
3.3. AMS
3.4. ICP-MS
3.5. SIMS

{kind=link}
{kind=link}
{kind=link}
| Method | Measurable Isotope | Interference | Detection Limit (mBq) | Detect-ion Time | Cost | Advantage | Disadvantage | Ref. |
|---|---|---|---|---|---|---|---|---|
| α- spectrometry | 238Pu | 241Am, 210Po, 228Th | 0.01–0.1 | Day–weeks | High | Simple, mature, reliable, low cost, and most used. | Large sample size. Long measurement time. | [8,17,46] |
| 239+240Pu | 229Th, 231Pa, 232U, 243Am | 0.01–0.1 | ||||||
| 241Pu | 210Po, 228Th | 0.1–1.0 | ||||||
| AMS | 239Pu | 238U, molecules with the same or similar m/z as 239Pu. | 10−4–10−3 | Hour/min | High | The best detection limits. Possibility to distinguish between 239Pu and 240Pu. Small sample size and short measurement time. | Technically complex and costly. | [14,58,85,87] |
| 240Pu | Molecules with the same or similar m/z as 240Pu and | 10−4–10−3 | ||||||
| 241Pu | 241Am. Molecules with the same or similar m/z as 241Pu | / | ||||||
| TIMS | 239Pu | Matrix elements | 10−4–10−3 | Hour/min | High | High precision. Good sensitivity. | Complex sample preparation. Large sample volume and difficult operation. | [105] |
| 240Pu | 10−4–10−3 | |||||||
| 241Pu | / | |||||||
| ICP-MS | 239Pu | 238U1H,208Pb31P,206Pb33S,204Hg35Cl,205Tl34S,203Tl36Ar,202Hg37Cl,199Hg40Ar | 0.01–0.1 | Hour/min | Low | Low detection limit and high measurement accuracy. Can distinguish 239Pu from 240Pu. Small sample size and short measurement time. | Inability to effectively exclude the disturbance of homogeneous isotopes and molecular ions, and high requirements for sample preparation. Difficult to remove the system background, matrix effects. | [91,93,106,107] |
4. Plutonium in the Environment
4.1. Plutonium Released by Nuclear Weapon Tests and Spent Fuel Reprocessing Plant
4.2. Plutonium Released by Nuclear Power Plant Accidents
4.3. Global Fallout Plutonium for Environmental Application
5. Conclusions and Outlook
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Taylor, D.M. Environmental plutonium—Creation of the universe to twenty-first century mankind. In Proceedings of the 2nd International Symposium on Plutonium in the Environment, Osaka, Japan, 9–12 November 1999; pp. 1–14. [Google Scholar]
- Vintró, L.; Mitchell, P.I.; Smith, K.J.; Kershaw, P.J.; Livingston, H.D. Chapter 3 Transuranium nuclides in the world’s oceans. Radioact. Environ. 2005, 6, 79–108. [Google Scholar]
- Monty, C. UNSCEAR Report 2000: Sources and Effects of Ionizing Radiation. J. Radiol. Prot. 2001, 21, 83. [Google Scholar]
- Lindahl, P.; Lee, S.H.; Worsfold, P.; Keith-Roach, M. Plutonium isotopes as tracers for ocean processes: A review. Mar. Environ. Res. 2010, 69, 73–84. [Google Scholar] [CrossRef]
- Aarkrog, A. Input of anthropogenic radionuclides into the World Ocean. Deep. Sea Res. Part II 2003, 50, 2597–2606. [Google Scholar] [CrossRef]
- Hirose, K.; Povinec, P.P. Sources of plutonium in the atmosphere and stratosphere-troposphere mixing. Sci. Rep. 2015, 5, 15707. [Google Scholar] [CrossRef] [PubMed]
- Povinec, P.P.; Buesseler, A.A.; Delfanti, K.O.; Hirose, R.; Hong, K.; Ito, G.H.; Livingston, T.; Nies, H.D.; Noshkin, H.; Shima, V.E.; et al. 90Sr, 137Cs and 239,240Pu concentration surface water time series in the Pacific and Indian Oceans—WOMARS results. J. Environ. Radioact. 2005, 81, 63–87. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.L.; Roos, P. Critical comparison of radiometric and mass spectrometric methods for the determination of radionuclides in environmental, biological and nuclear waste samples. Anal. Chim. Acta 2008, 608, 105–139. [Google Scholar] [CrossRef]
- Hattendorf, B.; Gunther, D. Characteristics and capabilities of an ICP-MS with a dynamic reaction cell for dry aerosols and laser ablation. J. Anal. At. Spectrom. 2000, 15, 1125–1131. [Google Scholar] [CrossRef]
- Hou, X.L.; Zhang, W.C.; Wang, Y.Y. Determination of Femtogram-Level Plutonium Isotopes in Environmental and Forensic Samples with High-Level Uranium Using Chemical Separation and ICP-MS/MS Measurement. Anal. Chem. 2019, 91, 11553–11561. [Google Scholar] [CrossRef]
- Wallenius, M.; Mayer, K. Age determination of plutonium material in nuclear forensics by thermal ionisation mass spectrometry. Fresenius J. Anal. Chem. 2000, 366, 234. [Google Scholar] [CrossRef]
- Betti, M.; Tamborini, G.; Koch, L. Use of secondary ion mass spectrometry in nuclear forensic analysis for the characterization of plutonium and highly enriched uranium particles. Anal. Chem. 1999, 71, 2616–2622. [Google Scholar] [CrossRef]
- Portier, S.; Brémier, S.; Walker, C.T. Secondary ion mass spectrometry of irradiated nuclear fuel and cladding: An overview. Int. J. Mass. Spectrom. 2007, 263, 113–126. [Google Scholar] [CrossRef]
- Chamizo, E.; Enamorado, S.M.; Garcia-Leon, M.; Suter, M.; Wacker, L. Plutonium measurements on the 1 MV AMS system at the Centro Nacional de Aceleradores (CNA). Nucl. Instrum. Methods Phys. Res. B 2008, 266, 4948–4954. [Google Scholar] [CrossRef]
- Wang, Z.T.; Yang, G.S.; Zheng, J.; Cao, L.G.; Yu, H.J.; Zhu, Y.; Tagami, K.; Uchida, S. Effect of Ashing Temperature on Accurate Determination of Plutonium in Soil Samples. Anal. Chem. 2015, 87, 5511–5515. [Google Scholar] [CrossRef] [PubMed]
- Kazi, Z.H.; Cornett, J.R.; Zhao, X.L.; Kieser, L. Americium and plutonium separation by extraction chromatography for determination by accelerator mass spectrometry. Anal. Chim. Acta 2014, 829, 75–80. [Google Scholar] [CrossRef]
- Jakopic, R.; Tavcar, P.; Benedik, L. Sequential determination of Pu and Am radioisotopes in environmental samples; a comparison of two separation procedures. Appl. Radiat. Isot. 2007, 65, 504–511. [Google Scholar] [CrossRef]
- Chu, Y.N. Plutonium determination in soil by leaching and ion-exchange separation. Anal. Chem. 1971, 43, 449–452. [Google Scholar] [CrossRef]
- Sill, C.W. Some Problems in Measuring Plutonium in the Environment. Health Phys. 1975, 29, 619–626. [Google Scholar] [CrossRef]
- Lee, S.H.; Gastaud, J.; La Rosa, J.J.; Kwong, L.L.W.; Povinec, P.P.; Wyse, E.; Fifield, L.K.; Hausladen, P.A.; Di Tada, L.M.; Santos, G.M. Analysis of plutonium isotopes in marine samples by radiometric, ICP-MS and AMS techniques. J. Radioanal. Nucl. Chem. 2001, 248, 757. [Google Scholar] [CrossRef]
- Toribio, M.; Garcia, J.F.; Rauret, G.; Pilvio, R.; Bickel, M. Plutonium determination in mineral soils and sediments by a procedure involving microwave digestion and extraction chromatography. Anal. Chim. Acta 2001, 447, 179–189. [Google Scholar] [CrossRef]
- Donard, O.F.X.; Bruneau, F.; Moldovan, M.; Garraud, H.; Epov, V.N.; Boust, D. Multi-isotopic determination of plutonium (Pu-239, Pu-240, Pu-241 and Pu-242) in marine sediments using sector-field inductively coupled plasma mass spectrometry. Anal. Chim. Acta 2007, 587, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Varga, Z.; Suranyi, G.; Vajda, N.; Stefanka, Z. Determination of plutonium and americium in environmental samples by inductively coupled plasma sector field mass spectrometry and alpha spectrometry. Microchem. J. 2007, 85, 39–45. [Google Scholar] [CrossRef]
- Sill, C.W.; Puphal, K.W.; Hindman, F.D. Simultaneous Determination of Alpha-Emitting Nuclides of Radium through Californium in Soil. Anal. Chem. 1974, 46, 1725–1737. [Google Scholar] [CrossRef] [PubMed]
- Xing, S.; Luo, M.Y.; Yuan, N.; Liu, D.Q.; Yang, Y.; Dai, X.X.; Zhang, W.C.; Chen, N. Accurate Determination of Plutonium in Soil by Tandem Quadrupole ICP-MS with Different Sample Preparation Methods. At. Spectrosc. 2021, 42, 62–70. [Google Scholar] [CrossRef]
- Croudace, I.; Warwick, P.; Taylor, R.N.; Dee, S. Rapid procedure for plutonium and uranium determination in soils using a borate fusion followed by ion-exchange and extraction chromatography. Anal. Chim. Acta 1998, 371, 217–225. [Google Scholar] [CrossRef]
- Warwick, P.; Croudace, I.; Oh, J.-S. Radiochemical determination of 241Am and Pu (α) in environmental materials. Anal. Chem. 2001, 73, 3410–3416. [Google Scholar] [CrossRef]
- Luo, M.Y.; Xing, S.; Yang, Y.G.; Song, L.J.; Ma, Y.; Wang, Y.D.; Dai, X.X.; Happel, S. Sequential analyses of actinides in large-size soil and sediment samples with total sample dissolution. J. Environ. Radioact. 2018, 187, 73–80. [Google Scholar] [CrossRef]
- Maxwell, S.L.; Culligan, B.K.; Hutchison, J.B. Rapid fusion method for determination of plutonium isotopes in large rice samples. J. Radioanal. Nucl. Chem. 2013, 298, 1367–1374. [Google Scholar] [CrossRef]
- Maxwell, S.L.; Culligan, B.K.; Jones, V.D.; Nichols, S.T.; Bernard, M.A.; Noyes, G.W. Determination of 237Np and Pu isotopes in large soil samples by inductively coupled plasma mass spectrometry. Anal. Chim. Acta 2010, 682, 130–136. [Google Scholar] [CrossRef]
- Maxwell, S.L.; Culligan, B.K.; Hutchison, J.B.; Spencer, R.B. Rapid fusion method for determination of actinides in fecal samples. J. Radioanal. Nucl. Chem. 2013, 298, 1533–1542. [Google Scholar] [CrossRef]
- Maxwell, S.L.; Culligan, B.; Hutchison, J.B.; Mcalister, D.R. Rapid fusion method for the determination of Pu, Np, and Am in large soil samples. J. Radioanal. Nucl. Chem. 2015, 305, 599–608. [Google Scholar] [CrossRef]
- Hubley, N.; Brown, J.W.N.; Guthrie, J.; Robertson, J.D.; Brockman, J.D. Development of ammonium bifluoride fusion method for rapid dissolution of trinitite samples and analysis by ICP-MS. J. Radioanal. Nucl. Chem. 2016, 307, 1777–1780. [Google Scholar] [CrossRef]
- Hubley, N.T.; Brockman, J.D.; Robertson, J.D. Evaluation of ammonium bifluoride fusion for rapid dissolution in post-detonation nuclear forensic analysis. Radiochim. Acta 2017, 105, 629–635. [Google Scholar] [CrossRef]
- Yang, G.S.; Zheng, J.; Kim, E.; Zhang, S.; Seno, H.; Kowatari, M.; Aono, T.; Kurihara, O. Rapid analysis of Np-237 and Pu isotopes in small volume urine by SF- ICP-MS and ICP-MS/MS. Anal. Chim. Acta 2021, 1158, 338431. [Google Scholar] [CrossRef]
- Wang, H.; Ni, Y.; Zheng, J.; Huang, Z.; Xiao, D.; Aono, T. Low-temperature fusion using NH4HSO4 and NH4HF2 for rapid determination of Pu in soil and sediment samples. Anal. Chim. Acta 2019, 1050, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Michel, H.; Barci-Funel, G.; Dalmasso, J.; Ardisson, G. One step ion exchange process for the radiochemical separation of americium, plutonium and neptunium in sediments. J. Radioanal. Nucl. Chem. 1999, 240, 467–470. [Google Scholar] [CrossRef]
- Zheng, J.; Yamada, M. Precise determination of Pu isotopes in a seawater reference material using ID-SF-ICP-MS combined with two-stage anion-exchange chromatography. Anal. Sci. 2007, 23, 611–615. [Google Scholar] [CrossRef]
- Zoriy, M.V.; Pickhardt, C.; Ostapczuk, P.; Hille, R.; Becker, J.S. Determination of Pu in urine at ultratrace level by sector field inductively coupled plasma mass spectrometry. Int. J. Mass Spectrom. 2004, 232, 217–224. [Google Scholar] [CrossRef]
- Michel, H.; Levent, D.; Barci, V.; Barci-Funel, G.; Hurel, C. Soil and sediment sample analysis for the sequential determination of natural and anthropogenic radionuclides. Talanta 2008, 74, 1527–1533. [Google Scholar] [CrossRef]
- La Rosa, J.J.; Burnett, W.; Lee, S.H.; Levy, I.; Gastaud, J.; Povinec, P.P. Separation of actinides, cesium and strontium from marine samples using extraction chromatography and sorbents. J. Radioanal. Nucl. Chem. 2001, 248, 765. [Google Scholar] [CrossRef]
- La Rosa, J.; Gastaud, J.; Lagan, L.; Lee, S.H.; Levy-Palomo, I.; Povinec, P.P.; Wyse, E. Recent developments in the analysis of transuranics (Np, Pu, Am) in seawater. J. Radioanal. Nucl. Chem. 2005, 263, 427–436. [Google Scholar] [CrossRef]
- Sidhu, R.S. A robust procedure for the determination of plutonium and americium in seawater. J. Radioanal. Nucl. Chem. 2003, 256, 501–504. [Google Scholar] [CrossRef]
- Ageyev, V.A.; Odintsov, O.O.; Sajeniouk, A.D. Routine radiochemical method for the determination of 90 Sr, 238 Pu, 239+240 Pu, 241 Am and 244 Cm in environmental samples. J. Radioanal. Nucl. Chem. 2005, 264, 337–342. [Google Scholar]
- Kimura, T. Simultaneous Determination of Neptunium, Plutonium, Americium and Curium Using Coprecipitation with Bismuth Phosphate. JJ. Radioanal. Nucl. Chem. Ar. 1990, 139, 297–305. [Google Scholar] [CrossRef]
- Varga, Z.; Suranyi, G.; Vajda, N.; Stefanka, Z. Improved sample preparation method for environmental plutonium analysis by ICP-SFMS and alpha-spectrometry. J. Radioanal. Nucl. Chem. 2007, 274, 87–94. [Google Scholar] [CrossRef]
- Varga, Z.; Suranyi, G.; Vajda, N.; Stefanka, Z. Rapid sequential determination of americium and plutonium in sediment and soil samples by ICP-SFMS and alpha-spectrometry. Radiochim. Acta 2007, 95, 81–87. [Google Scholar] [CrossRef]
- Rameback, H.; Skalberg, M. Separation of neptunium, plutonium, americium and curium from uranium with di-(2-ethylhexyl)-phosphoric acid (HDEHP) for radiometric and ICP-MS analysis. J. Radioanal. Nucl. Chem. 1998, 235, 229–233. [Google Scholar] [CrossRef]
- Norisuye, K.; Okamura, K.; Sohrin, Y.; Hasegawa, H.; Nakanishi, T. Large volume preconcentration and purification for determining the Pu-240/Pu-239 isotopic ratio and Pu-238/Pu239+240 alpha-activity ratio in seawater. J. Radioanal. Nucl. Chem. 2006, 267, 183–193. [Google Scholar] [CrossRef]
- Rim, J.H.; Gonzales, E.R.; Armenta, C.E.; Unlu, K.; Peterson, D.S. Developing and evaluating di(2-ethylhexyl) orthophosphoric acid (HDEHP) based polymer ligand film (PLF) for plutonium extraction. J. Radioanal. Nucl. Chem. 2013, 296, 1099–1103. [Google Scholar] [CrossRef]
- Kalmykov, S.N.; Aliev, R.A.; Sapozhnikov, D.Y.; Sapozhnikov, Y.A.; Afinogenov, A.M. Determination of Np-237 by radiochemical neutron activation analysis combined with extraction chromatography. Appl. Radiat. Isot. 2004, 60, 595–599. [Google Scholar] [CrossRef]
- Tonouchi, S.; Habuki, H.; Katoh, K.; Yamazaki, K.; Hashimoto, T. Determination of plutonium by inductively coupled plasma mass spectrometry (ICP-MS). J. Radioanal. Nucl. Chem. 2002, 252, 367–371. [Google Scholar] [CrossRef]
- Momoshima, N.; Kakiuchi, H.; Maeda, Y.; Hirai, E.; Ono, T. Identification of the contamination source of plutonium in environmental samples with isotopic ratios determined by inductively coupled plasma mass spectrometry and alpha-spectrometry. J. Radioanal. Nucl. Chem. 1997, 221, 213–217. [Google Scholar] [CrossRef]
- Qiao, J.X.; Hou, X.L.; Miro, M.; Roos, P. Determination of plutonium isotopes in waters and environmental solids: A review. Anal. Chim. Acta 2009, 652, 66. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Aarkrog, A.; Nielsen, S.P.; Dahlgaard, H.; Nies, H.; Yixua, Y.; Mandrup, K. Determination of plutonium in environmental samples by controlled valence in anion exchange. J. Radioanal. Nucl. Chem. 1993, 172, 281–288. [Google Scholar] [CrossRef]
- Moreno, J.; Vajda, N.; Danesi, P.R.; Larosa, J.J.; Zeiller, E.; Sinojmeri, M. Combined procedure for the determination of90Sr,241Am and Pu radionuclides in soil samples. J. Radioanal. Nucl. Chem. 1997, 226, 279–284. [Google Scholar] [CrossRef]
- Stu¨rup, S.; Henning, D.; Steffen, C.N. High resolution inductively coupled plasma mass spectrometry for the trace determination of plutonium isotopes and isotope ratios in environmental samples. J. Anal. At. Spectrom. 1998, 13, 1321–1326. [Google Scholar] [CrossRef]
- Hrnecek, E.; Steier, P.; Wallner, A. Determination of plutonium in environmental samples by AMS and alpha spectrometry. Appl. Radiat. Isot. 2005, 63, 633–638. [Google Scholar] [CrossRef]
- Huang, Z.; Ni, Y.; Wang, H.; Zheng, J.; Yamazaki, S.; Sakaguchi, A.; Long, X.; Uchida, S. Simultaneous determination of ultra-trace level 237Np and Pu isotopes in soil and sediment samples by SF-ICP-MS with a single column chromatographic separation. Microchem. J. 2019, 148, 597–604. [Google Scholar] [CrossRef]
- Wang, Z.L.; Yamada, M. Plutonium activities and Pu-240/Pu-239 atom ratios in sediment cores from the East China Sea and Okinawa Trough: Sources and inventories. Earth Planet. Sci. Lett. 2005, 233, 441–453. [Google Scholar] [CrossRef]
- Nunnemann, M.; Erdmann, N.; Hasse, H.U.; Huber, G.; Kratz, J.V.; Kunz, P.; Mansel, A.; Passler, G.; Stetzer, O.; Trautmann, N.; et al. Trace analysis of plutonium in environmental samples by resonance ionization mass spectroscopy (RIMS). J. Alloys Compd. 1998, 271, 45–48. [Google Scholar] [CrossRef]
- Ketterer, M.E.; Hafer, K.M.; Mietelski, J.W. Resolving Chernobyl vs. global fallout contributions in soils from Poland using Plutonium atom ratios measured by inductively coupled plasma mass spectrometry. J. Environ. Radioact. 2004, 73, 183–201. [Google Scholar] [CrossRef] [PubMed]
- Kaye, J.H.; Strebin, R.S.; Orr, R.D. Rapid, Quantitative-Analysis of Americium, Curium and Plutonium Isotopes in Hanford Samples Using Extraction Chromatography and Precipitation Plating. J. Radioanal. Nucl. Chem. 1995, 194, 191–196. [Google Scholar] [CrossRef]
- Kuwabara, J.; Noguchi, H. Development of rapid bioassay method for plutonium. J. Radioanal. Nucl. Chem. 2002, 252, 273–276. [Google Scholar] [CrossRef]
- Lopez-Lora, M.; Levy, I.; Chamizo, E. Simple and fast method for the analysis of (236)U, (237)Np, (239)Pu and (240)Pu from seawater samples by Accelerator Mass Spectrometry. Talanta 2019, 200, 22–30. [Google Scholar] [CrossRef]
- Wang, Z.T.; Xie, Y.; Lin, J.X.; Li, Z.J.; Tan, Z.Y. Rapid method for sequential determination of Pu and Am in soil and sediment samples by sector-field inductively coupled plasma mass spectrometry. J. Radioanal. Nucl. Chem. 2021, 328, 137–147. [Google Scholar] [CrossRef]
- Zapata-Garcia, D.; Wershofen, H.; Seferinoglu, M.; Dirican, A.; Aslan, N.; Ozcayan, G.; Yucel, U. Comparison of two methods for the rapid radiochemical analysis of air dust samples in emergency situations. Appl. Radiat. Isot. 2019, 150, 120–126. [Google Scholar] [CrossRef]
- Luo, M.Y.; Liu, D.Q.; Yang, Y.; Dai, X.X.; Wu, Y.; Shi, K.L. Simultaneous determination of actinides and Sr-90 in large-size soil and sediment samples. J. Environ. Radioact. 2022, 247, 106854. [Google Scholar] [CrossRef]
- Rozhkova, A.K.; Kuzmenkova, N.V.; Sibirtsev, A.M.; Petrov, V.G.; Shi, K.L.; Hou, X.L.; Kalmykov, S.N. Simultaneous separation of actinides and technetium from large volumes of natural water for their determination. J. Radioanal. Nucl. Chem. 2022, 331, 2037–2044. [Google Scholar] [CrossRef]
- Metzger, S.C.; Ticknor, B.W.; Rogers, K.T.; Bostick, D.A.; McBay, E.H.; Hexel, C.R. Automated Separation of Uranium and Plutonium from Environmental Swipe Samples for Multiple Collector Inductively Coupled Plasma Mass Spectrometry. Anal. Chem. 2018, 90, 9441–9448. [Google Scholar] [CrossRef]
- Shibahara, Y.; Kubota, T.; Fujii, T.; Fukutani, S.; Takamiya, K.; Konno, M.; Mizuno, S.; Yamana, H. Determination of isotopic ratios of plutonium and uranium in soil samples by thermal ionization mass spectrometry. J. Radioanal. Nucl. Chem. 2016, 307, 2281–2287. [Google Scholar] [CrossRef]
- Habibi, A.; Boulet, B.; Gleizes, M.; Lariviere, D.; Cote, G. Rapid determination of actinides and (90)Sr in river water. Anal. Chim. Acta 2015, 883, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Dulanská, S.; Bilohuščin, J.; Remenec, B.; Galanda, D.; Mátel, L.u. Determination of 239Pu, 241Am and 90Sr in urine using pre-filter material and combined sorbents AnaLig® Pu-02, AnaLig® Sr-01, DGA® Resin. J. Radioanal. Nucl. Chem. 2015, 304, 127–132. [Google Scholar] [CrossRef]
- Dai, X.; Kramer-Tremblay, S. Five-column chromatography separation for simultaneous determination of hard-to-detect radionuclides in water and swipe samples. Anal. Chem. 2014, 86, 5441–5447. [Google Scholar] [CrossRef] [PubMed]
- Rollin, S.; Corcho-Alvarado, J.A.; Sahli, H.; Putyrskaya, V.; Klemt, E. High-resolution records of cesium, plutonium, americium, and uranium isotopes in sediment cores from Swiss lakes. Environ. Sci. Pollut. R 2022, 29, 85777–85788. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, S.K.; Saxena, M.K.; Jain, H.C. Determination of Plutonium by Alpha Spectrometry. J. Radioanal. Nucl. Chem. 1992, 156, 111–118. [Google Scholar] [CrossRef]
- Zhang, H.-T.; Zhu, F.R.; Xu, J.; Dai, Y.H.; Li, D.M.; Yi, X.W.; Zhang, L.X.; Zhao, Y.G. Age determination of plutonium material by alpha spectrometry and thermal ionization mass spectrometry. Radiochim. Acta 2008, 96, 327–331. [Google Scholar] [CrossRef]
- Maassen, J.; Inglis, J.D.; Wende, A.; Kayzar-Boggs, T.M.; Steiner, R.E.; Kara, A. Analysis of sub-picogram quantities of 238Pu by thermal ionization mass spectrometry. J. Radioanal. Nucl. Chem. 2019, 321, 1073–1080. [Google Scholar] [CrossRef]
- Burger, S.; Riciputi, L.R.; Bostick, D.A.; Turgeon, S.; McBay, E.H.; Lavelle, M. Isotope ratio analysis of actinides, fission products, and geolocators by high-efficiency multi-collector thermal ionization mass spectrometry. Int. J. Mass Spectrom. 2009, 286, 70–82. [Google Scholar] [CrossRef]
- Jakopic, R.; Richter, S.; Kuhn, H.; Benedik, L.; Pihlar, B.; Aregbe, Y. Isotope ratio measurements of pg-size plutonium samples using TIMS in combination with “multiple ion counting” and filament carburization. Int. J. Mass Spectrom. 2009, 279, 87–92. [Google Scholar] [CrossRef]
- Lee, C.G.; Suzuki, D.; Esaka, F.; Magara, M.; Song, K. Ultra-trace analysis of plutonium by thermal ionization mass spectrometry with a continuous heating technique without chemical separation. Talanta 2015, 141, 92–96. [Google Scholar] [CrossRef]
- Chamizo, E.; Christl, M.; Fifield, L.K. Measurement of U-236 on the 1 MV AMS system at the Centro Nacional de Aceleradores (CNA). Nucl. Instrum. Methods Phys. Res. Sect. B 2015, 358, 45–51. [Google Scholar] [CrossRef]
- Brown, T.A.; Marchetti, A.A.; Martinelli, R.E.; Cox, C.C.; Knezovich, J.P.; Hamilton, T.F. Actinide measurements by accelerator mass spectrometry at Lawrence Livermore National Laboratory. Nucl. Instrum. Meth. Phys. Res. B 2004, 223, 788–795. [Google Scholar] [CrossRef]
- Winkler, S.; Ahmad, I.; Golser, R.; Kutschera, W.; Orlandini, K.A.; Paul, M.; Priller, A.; Steier, P.; Valenta, A.; Vockenhuber, C. Developing a detection method of environmental Pu-244. Nucl. Instrum. Meth. Phys. Res. B 2004, 223, 817–822. [Google Scholar] [CrossRef]
- Pacesila, D.; Bishop, S.; Stanciu, I.; Enachescu, M.; Petre, A.R.; Virgolici, M.; Iancu, D.; Tugulan, L.; Done, L.; Petraglia, A.; et al. Ultrasensitive detection of Pu-244 in environmental samples by accelerator mass spectrometry. J. Anal. At. Spectrom. 2022, 37, 2581–2588. [Google Scholar] [CrossRef]
- Wacker, L.; Chamizo, E.; Fifield, L.K.; Stocker, M.; Suter, A.; Synal, H.A. Measurement of actinides on a compact AMS system working at 300 kV. Nucl. Instrum. Meth. Phys. Res. B 2005, 240, 452–457. [Google Scholar] [CrossRef]
- Kierepko, R.; Mietelski, J.W.; Ustrnul, Z.; Anczkiewicz, R.; Wershofen, H.; Holgye, Z.; Kapala, J.; Isajenko, K. Plutonium isotopes in the atmosphere of Central Europe: Isotopic composition and time evolution vs. circulation factors. Sci. Total Environ. 2016, 569, 937–947. [Google Scholar] [CrossRef] [PubMed]
- Bisinger, T.; Hippler, S.; Michel, R.; Wacker, L.; Synal, H.A. Determination of plutonium from different sources in environmental samples using alpha-spectrometry and AMS. Nucl. Instrum. Methods Phys. Res. Sect. B 2010, 268, 1269–1272. [Google Scholar] [CrossRef]
- Kim, C.S.; Kim, C.K.; Lee, J.I.; Lee, K.J. Rapid determination of Pu isotopes and atom ratios in small amounts of environmental samples by an on-line sample pre-treatment system and isotope dilution high resolution inductively coupled plasma mass spectrometry. J. Anal. At. Spectrom. 2000, 15, 247–255. [Google Scholar] [CrossRef]
- Salminen-Paatero, S.; Nygren, U.; Paatero, J. Pu-240/Pu-239 mass ratio in environmental samples in Finland. J. Environ. Radioact. 2012, 113, 163–170. [Google Scholar] [CrossRef]
- Ni, Y.Y.; Zheng, J.; Guo, Q.J.; Men, W.; Tagami, K.; Uchida, S. Rapid determination of ultra-trace plutonium isotopes (Pu-239, Pu-240 and Pu-241) in small-volume human urine bioassay using sector-field inductively coupled plasma mass spectrometry. Anal. Chim. Acta 2018, 1000, 85–92. [Google Scholar] [CrossRef]
- Shi, Y.; Collins, R.; Broome, C. Determination of uranium, thorium and plutonium isotopes by ICP-MS. J. Radioanal. Nucl. Chem. 2013, 296, 509–515. [Google Scholar] [CrossRef]
- Bu, W.T.; Gu, M.; Ding, X.T.; Ni, Y.Y.; Shao, X.P.; Liu, X.M.; Yang, C.T.; Hu, S. Exploring the ability of triple quadrupole inductively coupled plasma mass spectrometry for the determination of Pu isotopes in environmental samples. J. Anal. At. Spectrom. 2021, 36, 2330–2337. [Google Scholar] [CrossRef]
- Gao, R.Q.; Hou, X.L.; Zhang, L.Y.; Zhang, W.C.; Zhang, M.T. Determination of Ultra-Low Level Plutonium Isotopes in Large Volume Environmental Water Samples. Chin. J. Anal. Chem. 2020, 48, 765–773. [Google Scholar] [CrossRef]
- Zhang, W.C.; Lin, J.F.; Fang, S.; Li, C.; Yi, X.W.; Hou, X.L.; Chen, N.; Zhang, H.T.; Xu, Y.H.; Dang, H.J.; et al. Determination of ultra-trace level plutonium isotopes in soil samples by triple-quadrupole inductively coupled plasma-mass spectrometry with mass-shift mode combined with UTEVA chromatographic separation. Talanta 2021, 234, 122652. [Google Scholar] [CrossRef] [PubMed]
- Matsueda, M.; Kawakami, T.; Koarai, K.; Terashima, M.; Fujiwara, K.; Iijima, K.; Furukawa, M.; Takagai, Y. Using CO2 Reactions to Achieve Mass-spectrometric Discrimination in Simultaneous Plutonium-isotope Speciation with Inductively Coupled Plasma-Tandem Mass Spectrometry. Chem. Lett. 2022, 51, 678–682. [Google Scholar] [CrossRef]
- Xing, S.; Zhang, W.; Qiao, J.; Hou, X. Determination of ultra-low level plutonium isotopes (Pu-239,(240)pu) in environmental samples with high uranium. Talanta 2018, 187, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Santos, M.; Marcalo, J.; de Matos, A.P.; Gibson, J.K.; Haire, R.G. Gas-phase oxidation reactions of neptunium and plutonium ions investigated via Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. J. Phys. Chem. A 2002, 106, 7190–7194. [Google Scholar] [CrossRef]
- Tanner, S.D.; Li, C.S.; Vais, V.; Baranov, V.I.; Bandura, D.R. Chemical resolution of Pu(+) from U(+) and Am(+) using a band-pass reaction cell inductively coupled plasma mass spectrometer. Anal. Chem. 2004, 76, 3042–3048. [Google Scholar] [CrossRef]
- Gourgiotis, A.; Granet, M.; Isnard, H.; Nonell, A.; Gautier, C.; Stadelmann, G.; Aubert, M.; Durand, D.; Legand, S.; Chartier, F. Simultaneous uranium/plutonium separation and direct isotope ratio measurements by using CO2 as the gas in a collision/reaction cell based MC-ICPMS. J. Anal. At. Spectrom. 2010, 25, 1939–1945. [Google Scholar] [CrossRef]
- Xu, Y.H.; Li, C.; Yu, H.P.; Fang, F.M.; Hou, X.L.; Zhang, C.; Li, X.F.; Xing, S. Rapid determination of plutonium isotopes in small samples using single anion exchange separation and ICP-MS/MS measurement in NH3-He mode for sediment dating. Talanta 2022, 240, 123152. [Google Scholar] [CrossRef]
- Boulyga, S.F.; Tibi, M.; Heumann, K.G. Application of isotope-dilution laser ablation ICP-MS for direct determination of Pu concentrations in soils at pg g−1 levels. Anal. Bioanal. Chem. 2004, 378, 342–347. [Google Scholar] [PubMed]
- Wallenius, M.; Lutzenkirchen, K.; Mayer, K.; Ray, I.; Heras, L.A.I.D.L.; Betti, M.; Cromboom, O.; Hild, M.; Lynch, B.; Nicholl, A.; et al. Nuclear forensic investigations with a focus on plutonium. J. Alloy Compd. 2007, 444, 57–62. [Google Scholar] [CrossRef]
- Tamborini, G.; Betti, M. Characterisation of radioactive particles by SIMS. Microchim. Acta 2000, 132, 411–417. [Google Scholar] [CrossRef]
- Efurd, D.W.; Steiner, R.E.; Roensch, F.R.; Glover, S.E.; Musgrave, J.A. Determination of the Pu-240/Pu-239 atom ratio in global fallout at two locations in the Northern Hemisphere. J. Radioanal. Nucl. Chem. 2005, 263, 387–391. [Google Scholar] [CrossRef]
- Kim, C.S.; Kim, C.K.; Martin, P.; Sansone, U. Determination of Pu isotope concentrations and isotope ratio by inductively coupled plasma mass spectrometry: A review of analytical methodology. J. Anal. Atom Spectrom. 2007, 22, 827–841. [Google Scholar] [CrossRef]
- Truscott, J.B.; Jones, P.; Fairman, B.E.; Evans, E.H. Determination of actinide elements at femtogram per gram levels in environmental samples by on-line solid phase extraction and sector-field-inductively coupled plasma-mass spectrometry. Anal. Chim. Acta 2001, 433, 245–253. [Google Scholar] [CrossRef]
- Yamamoto, M.; Yamauchi, Y.; Chatani, K.; Igarashi, S.; Komura, K.; Ueno, K.; Sakanoue, M. Distribution of Global Fallout 237np, Pu Isotopes, and 241am in Lake and Sea Sediments. J. Radioanal. Nucl. Chem. 1991, 147, 165–176. [Google Scholar] [CrossRef]
- Livingston, H.D.; Schneider, D.L.; Bowen, V.T. 241Pu in the marine environment by a radiochemical procedure. Earth Planet. Sci. Lett. 1975, 25, 361–367. [Google Scholar] [CrossRef]
- Holm, E.; Rioseco, J.; Pettersson, H. Fallout of Transuranium Elements Following the Chernobyl Accident. J. Radioanal. Nucl. Chem. 1992, 156, 183. [Google Scholar] [CrossRef]
- Kelley, J.M.; Bond, L.A.; Beasley, T.M. Global distribution of Pu isotopes and Np-237. Sci. Total Environ. 1999, 238, 483–500. [Google Scholar] [CrossRef]
- Hisamatsu, S.I.; Sakanoue, M. Determination of Transuranium Elements in a So-called “Bikini Ash” Sample and in Marine Sediment Samples Collected Near Bikini Atoll. Health Phys. 1978, 35, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Koide, M.; Bertine, K.K.; Chow, T.J.; Goldberg, E.D. The 240Pu/239Pu ratio, a potential geochronometer. Earth Planet. Sci. Lett. 1985, 72, 1–8. [Google Scholar] [CrossRef]
- Muramatsu, Y.; Hamilton, T.; Uchida, S.; Tagami, K.; Yoshida, S.; Robison, W. Measurement of 240Pu/239Pu isotopic ratios in soils from the Marshall Islands using ICP-MS. Sci. Total Environ. 2001, 278, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Lujaniene, G.; Aninkevicius, V.; Lujanas, V. Artificial radionuclides in the atmosphere over Lithuania. J. Environ. Radioactiv. 2009, 100, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Tagami, K.; Watanabe, Y.; Uchida, S.; Aono, T.; Ishii, N.; Yoshida, S.; Kubota, Y.; Fuma, S.; Ihara, S. Isotopic evidence of plutonium release into the environment from the Fukushima DNPP accident. Sci. Rep. 2012, 2, 304. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Lee, S.H.; Lee, H.M.; Hong, G.H. Distribution of 239,240Pu in marine products from the seas around the Korean Peninsula after the Fukushima nuclear power plant accident. J. Environ. Radioact. 2020, 217, 106191. [Google Scholar] [CrossRef] [PubMed]
- Johansen, M.P.; Anderson, D.; Child, D.; Hotchkis, M.A.C.; Tsukada, H.; Okuda, K.; Hinton, T.G. Differentiating Fukushima and Nagasaki plutonium from global fallout using Pu-241/Pu-239 atom ratios: Pu vs. Cs uptake and dose to biota. Sci. Total Environ. 2021, 754, 141890. [Google Scholar] [CrossRef]
- Yamada, M.; Oikawa, S.; Shirotani, Y.; Kusakabe, M.; Shindo, K. Transuranic nuclides Pu, Am and Cm isotopes, and Sr-90 in seafloor sediments off the Fukushima Daiichi Nuclear Power Plant during the period from 2012 to 2019. J. Environ. Radioact. 2021, 227, 106459. [Google Scholar] [CrossRef]
- Masson, O.; Piga, D.; Gurriaran, R.; D’Amico, D. Impact of an exceptional Saharan dust outbreak in France: PM10 and artificial radionuclides concentrations in air and in dust deposit. Atmos. Environ. 2010, 44, 2478–2486. [Google Scholar] [CrossRef]
- Muramatsu, Y.; Ruhm, W.; Yoshida, S.; Tagami, K.; Uchida, S.; Wirth, E. Concentrations of Pu-239 and Pu-240 and their isotopic ratios determined by ICP-MS in soils collected from the Chernobyl 30-km zone. Environ. Sci. Technol. 2000, 34, 2913–2917. [Google Scholar] [CrossRef]
- Lindahl, P.; Roos, P.; Eriksson, M.; Holm, E. Distribution of Np and Pu in Swedish lichen samples (Cladonia stellaris) contaminated by atmospheric fallout. J. Environ. Radioact. 2004, 73, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Tagami, K.; Uchida, S. Release of Plutonium Isotopes into the Environment from the Fukushima Daiichi Nuclear Power Plant Accident: What Is Known and What Needs to Be Known. Environ. Sci. Technol. 2013, 47, 9584–9595. [Google Scholar] [CrossRef] [PubMed]
- Imanaka, T.; Endo, S.; Sugai, M.; Ozawa, S.; Shizuma, K.; Yamamoto, M. Early radiation survey of Iitate village, which was heavily contaminated by the Fukushima Daiichi accident, conducted on 28 and 29 March 2011. Health Phys. 2012, 102, 680–686. [Google Scholar] [CrossRef] [PubMed]
- Shinonaga, T.; Steier, P.; Lagos, M.; Ohkura, T. Airborne Plutonium and non-natural Uranium from the Fukushima DNPP found at 120 km distance a few days after reactor hydrogen explosions. Environ. Sci. Technol. 2014, 48, 3808–3814. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.K.; Byun, J.I.; Chae, J.S.; Choi, H.Y.; Choi, S.W.; Kim, D.J.; Kim, Y.J.; Lee, D.M.; Park, W.J.; Yim, S.A.; et al. Radiological impact in Korea following the Fukushima nuclear accident. J. Environ. Radioact. 2012, 111, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.P.; Xu, Y.H.; Pan, S.M.; Song, X.W.; Zhang, K.X.; Guo, H.T.; Gu, Z. Sources of plutonium isotopes and Cs-137 in coastal seawaters of Liaodong Bay and Bohai Strait, China and its environmental implications. Mar. Pollut. Bull. 2018, 130, 240–248. [Google Scholar] [CrossRef]
- Wang, R.R.; Lei, L.; Li, G.; Liu, Z.Y. Identification of the Distribution and Sources of Pu in the Northern South China Sea: Influences of Provenance and Scavenging. ACS Earth Space Chem. 2019, 3, 2684–2694. [Google Scholar] [CrossRef]
- Zhuang, Q.; Li, G.; Wang, F.; Tian, L.; Jiang, X.; Zhang, K.; Liu, G.; Pan, S.; Liu, Z. 137Cs and 239+ 240Pu in the Bohai Sea of China: Comparison in distribution and source identification between the inner bay and the tidal flat. Mar. Pollut. Bull. 2019, 138, 604–617. [Google Scholar] [CrossRef]
- Wang, J.; Du, J.; Zheng, J.; Bi, Q.; Ke, Y.; Qu, J. Plutonium in Southern Yellow Sea sediments and its implications for the quantification of oceanic-derived mercury and zinc. Environ. Pollut. 2020, 266, 115262. [Google Scholar] [CrossRef]
- Zhang, R.; Wang, R.; Liu, Z.; Zhang, F.; Li, T.; Guan, M.; Han, Q. Distribution and transport of plutonium in the sediments of the Yangtze River estuary and the adjacent East China Sea. Appl. Geochem. 2020, 115, 104532. [Google Scholar] [CrossRef]
- Wang, J.L.; Du, J.Z.; Qu, J.G.; Bi, Q.Q. Distribution of Pu isotopes and Pb-210 in the Bohai Sea and Yellow Sea: Implications for provenance and transportation. Chemosphere 2021, 263, 127896. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.R.; Zhu, Y.W.; Hu, F.F.; Xu, X.Y.; Sun, Z.Y.; Liu, Z.Y. Distribution and source identification of Pu in the tidal flat wetlands of northern Jiangsu Province and radial sand ridge of southern Yellow Sea-An explanation of “land-sea interaction”. Catena 2021, 206, 105506. [Google Scholar] [CrossRef]
- Wang, R.; Fu, Y.; Lei, L.; Li, G.; Liu, Z.J.A.o. Distribution and source identification of Pu in river basins in southern China. ACS Omega 2019, 4, 22646–22656. [Google Scholar] [CrossRef]
- Huang, Y.A.; Sun, X.M.; Zhang, W. Spatio-temporal distribution of Pu239+240 in sediments of the China sea and adjacent waters. J. Environ. Radioactiv. 2022, 253, 107010. [Google Scholar] [CrossRef] [PubMed]
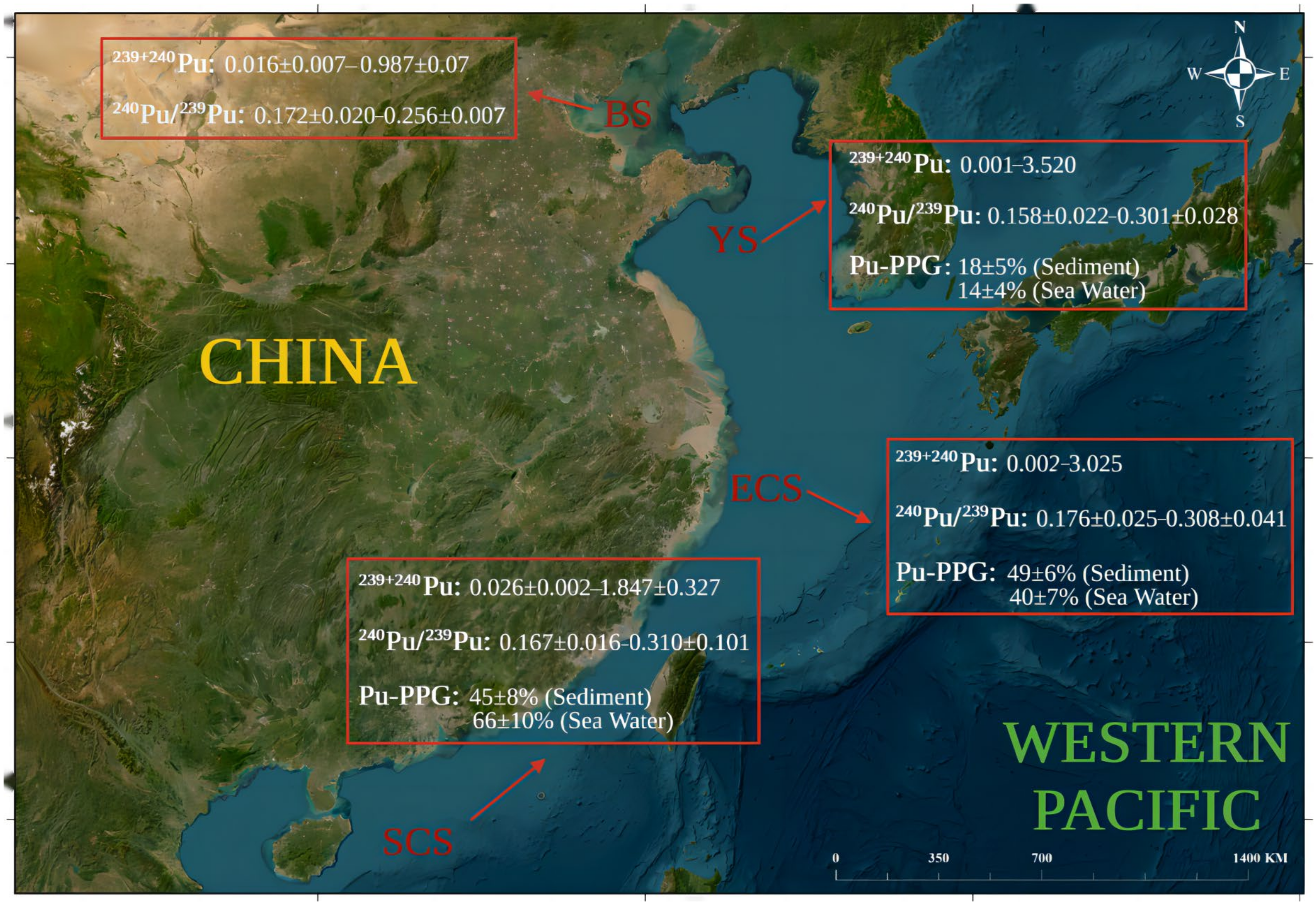
- Wu, J.; Sun, J.; Xiao, X. An overview of current knowledge concerning the inventory and sources of plutonium in the China Seas. Mar. Pollut. Bull. 2020, 150, 110599. [Google Scholar] [CrossRef] [PubMed]
- Hirose, K.; Aoyama, M.; Katsuragi, Y.; Sugimura, Y. Annual Deposition of Sr-90, Cs-137 and Pu-239, 240 from the 1961–1980 Nuclear Explosions: A Simple Model. J. Meteorol. Soc. Jpn. 2007, 65, 259–277. [Google Scholar] [CrossRef]
- Lujanienė, G.; Valiulis, D.; Byčenkienė, S.; Šakalys, J.; Povinec, P.P. Plutonium isotopes and 241Am in the atmosphere of Lithuania: A comparison of different source terms. Atmos. Environ. 2012, 61, 419–427. [Google Scholar] [CrossRef]
- Chamizo, E.; García-León, M.; Enamorado, S.M.; Jiménez-Ramos, M.C.; Wacker, L. Measurement of plutonium isotopes, 239Pu and 240Pu, in air-filter samples from Seville (2001–2002). Atmos. Environ. 2010, 44, 1851–1858. [Google Scholar] [CrossRef]
- Choi, M.S.; Lee, D.S.; Choi, J.C.; Cha, H.J.; Yi, H.I. 239+240Pu concentration and isotope ratio (240Pu/239Pu) in aerosols during high dust (Yellow Sand) period, Korea. Sci. Total Environ. 2006, 370, 262–270. [Google Scholar] [CrossRef]


| Plutonium Isotope | Half-Life | Specific Activity (Bq g−1) | Principal Decay Mode | Decay Energy (MeV) | Production Method |
|---|---|---|---|---|---|
| 238Pu | 87.7 year | 6.338 × 1011 | α | α 5.499 (70.9%) 5.456 (29.0%) | 242Cm daughter 238Np daughter |
| 239Pu | 2.411 × 104 year | 2.296 × 109 | α | α 5.157 (70.77%) 5.144 (17.11%) 5.106 (11.94%) γ 0.129 | 239Np daughter |
| 240Pu | 6.561 × 103 year | 8.401 × 109 | α | α 5.168 (72.8%) 5.124 (27.1%) | Multiple n capture |
| 241Pu | 14.35 year | 3.825 × 1012 | β− | α 4.896 (83.2%) 4.853 (12.2%) β− 0.021 γ 0.149 | Multiple n capture |
| 242Pu | 3.75 × 105 year | 1.458 × 108 | α | α 4.902 (76.49%) 4.856 (23.48%) | Multiple n capture |
| 244Pu | 8.08 × 107 year | 6.710 × 105 | α | α 4.589 (81%) 4.546 (19%) | Multiple n capture |
| Sample Type | Nuclide | Resin Column | Chemical Yield | Ref. |
|---|---|---|---|---|
| Natural water | Pu, U, Am, Tc | TEVA, TRU, DOWEX 1 × 8 | >70% | [69] |
| Dust | Pu, U, Am, Sr | UTEVA, DGA, TRU, Sr | >75% | [67] |
| Seawater | Pu, U, Np | TEVA, UTEVA | >80% | [65] |
| Soil | Pu, Np | AG MP-1M, TEVA | >72% | [59] |
| Wipe sample | Pu, U | TEVA, UTEVA | >80% | [70] |
| Soil | Pu, U | UTEVA, DOWEX 1 × 8 | / | [71] |
| Water | Pu, Sr, Am, U, Th, Np | AnaLig Pu-02/Sr-01, DGA | 90% | [72] |
| Urine | Pu, Am, Sr | AG MP-1M, TEVA, UTEVA | 84% | [73] |
| Wipe sample/Water | Pu, Np, Th, U, Am, Cm, Pm, Y, Sr, Fe | AG MP-1M, TEVA, UTEVA, DGA, Sr, TRU | 99.6 ± 4.3% | [74] |
| Core sediment | Pu, Cs, Am, U | TEVA, UTEVA, DGA | / | [75] |
| Soil/Sediment | Pu, Am, Sr, Y | AG MP-1M, DGA, TEVA, UTEVA | >75% | [68] |
| Soil/Sediment | Pu, Am | UTEVA, DGA, TEVA | 71–91% | [66] |
| Source | 238Pu/239+240Pu Activity Ratio | 241Pu/239+240Pu Activity Ratio | 240Pu/239Pu Atomic Ratio | Ref. |
|---|---|---|---|---|
| Global Settlement | 0.03 | 13–15 | 0.18 | [108,109,110,111] |
| Nuclear Test | <0.01 | 27–30 | 0.3 | [112,113,114] |
| Chernobyl Accident | 0.5 | 85 | 0.41 | [110,115] |
| Fukushima Nuclear Accident | 1.2 | 108 | 0.30–33 | [116,117,118,119] |
| Sahara Dust | 0.028 | 3.1 | 0.192 | [120] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Shao, Y.; Luo, M.; Ma, L.; Xu, G.; Wu, M. Progress of the Analytical Methods and Application of Plutonium Isotopes in the Environment. Processes 2023, 11, 1430. https://doi.org/10.3390/pr11051430
Liu X, Shao Y, Luo M, Ma L, Xu G, Wu M. Progress of the Analytical Methods and Application of Plutonium Isotopes in the Environment. Processes. 2023; 11(5):1430. https://doi.org/10.3390/pr11051430
Chicago/Turabian StyleLiu, Xidong, Yang Shao, Min Luo, Lingling Ma, Gang Xu, and Minghong Wu. 2023. "Progress of the Analytical Methods and Application of Plutonium Isotopes in the Environment" Processes 11, no. 5: 1430. https://doi.org/10.3390/pr11051430