Abstract
CDK1 (cyclin dependent kinase 1) is a key regulator of the cell cycle and is frequently dysregulated in cancer, making it a promising target for anticancer therapy. Securigera securidaca L. (S. securidaca) seeds, traditionally used in folk medicine for various ailments including cancer, were examined for their potential as CDK1/Cks2 inhibitors using in silico approaches. A total of 14 phytocompounds was identified in the GC/MS chromatogram, with gingerone being the most abundant at 25.67% and hippeastrine the least at 2%. Major constituents of the essential extract, including gingerol, eugenol, α-curcumene, and gingerol, showed high values and made up 52% of the total content of the volatile extract. Molecular docking and ADMET studies suggested that hippeastrine and naringenin are potential hit candidates against CDK1, exhibiting good drug-like properties and molecular interactions with desirable pharmacokinetic and toxicological characteristics close to dinaciclib. Furthermore, molecular dynamics (MD) simulations showed that both compounds exhibited stable conformations inside the binding site over the 100 ns MD simulation, suggesting they may stabilize the protein structure by reducing the flexibility of the CDK1 backbone. Additionally, MM-PBSA calculations further supported the stability of hippeastrine and naringenin in CDK1 complexes. Overall, these findings suggest that hippeastrine and naringenin are potential hit candidates for CDK1 inhibition, providing valuable insight into their binding and stability within the active site of CDK1. Further investigation of these compounds with in vitro and in vivo assays is warranted to assess their potential as CDK1 inhibitors for cancer therapy.
1. Introduction
Cancer is currently one of the most significant public health challenges worldwide and a leading cause of death [1]. According to the latest GLOBOCAN 2020 data, there were nearly 19.3 million new cancer cases (excluding non-melanoma skin cancer) and almost 10.0 million cancer-related deaths globally [1]. Despite remarkable progress in diagnostics and therapeutics, cancer incidence and mortality rates remain high, highlighting the pressing need for innovative strategies to manage cancer more effectively [2,3]. One of the fundamental hallmarks of cancer is the overactivity of the cell cycle [2,4]. The cell cycle is a tightly regulated and highly conserved process governed by various mechanisms including checkpoints, cyclin-dependent kinases (CDKs), and cyclins [5,6]. Dysregulation of these regulatory mechanisms leads to the loss of control over the cell cycle, resulting in uncontrolled cell proliferation and tumor progression [7,8]. Targeting cell cycle regulatory proteins, particularly CDKs, represents a promising approach for cancer therapy, as cell proliferation is a crucial step in tumor development [9,10]. CDK1, a crucial member of the cell cycle kinase family, plays a vital role in regulating cell cycle progression [11,12]. Studies have found that dysregulation of CDK1 is affiliated with aggressive tumor development, chromosomal instability, and increased cell proliferation in cancer [11,12,13,14]. The universal master kinase nature of CDK1, which is conserved from yeast to humans, highlights the significant impact of CDK1 dysregulation in cancer [13]. Moreover, CDK1 overexpression has been identified in different cancer types, along with breast cancer, gastric cancer, ovarian cancer, liver cancer, colorectal cancer, esophageal adenocarcinoma, and oral squamous cell carcinoma, indicating the potential of CDK1 inhibitors to have pan-cancer applications [11,12,13,14].
CDK1, also known as CDC2, CDC28A, cell division cycle 2 homolog A, p34 protein kinase, and p34, is capable of forming a complex with cyclins A, B, D, and E [15,16]. This kinase primarily regulates the transition between G1/S and G2/M phases while also promoting M phase progression [16,17]. Its pivotal role in cell cycle regulation stems from the fact that other CDKs, including the closely related CDK2, cannot compensate for its functions. CDK1, however, can compensate for the functions of other CDKs and can drive mammalian cell cycle progression in their absence [18]. Apart from its primary function of regulating mitosis, CDK1 can also regulate G1 phase progression and the G1/S transition by forming a complex with other cyclins, such as D1, E, and A [16]. During the late G2 phase, increased levels of cyclin B allow for stable CDK1/cyclin B complex formation [16]. However, the complex remains inactive due to Wee1- and Myt1-dependent inhibitory phosphorylation of the tyrosine 14 and 15 residues in the CDK1 subunit, which interferes with ATP alignment [16,19]. Once the inhibitory phosphates are removed by CDC25, the complex becomes activated, leading to a G2/M phase transition [20]. To exit the M phase, CDK1/cyclin B complex activity needs to be downregulated again, which is achieved through cyclin B proteolysis [21]. With the ability to phosphorylate over 100 different proteins, the activated CDK1/cyclin B complex can target not only cell cycle-related proteins but also proteins involved in cell migration and cytoskeleton regulation [21]. Recent studies have identified CDK1 as a key regulator of self-renewal and differentiation of human embryonic stem cells and human-induced pluripotent stem cells [18,22]. Although knowledge of CDK1 has increased over the past decade, not all processes in which it is involved are fully understood [16]. CDK1 overexpression likely leads to pancreatic ductal adenocarcinoma tumorigenesis by stimulating checkpoint evasion and inducing stemness properties [16]. Therefore, the design and development of CDK1 inhibitors represents a promising approach for effectively managing cancer [11,12,13,14,23].
Cancer research has increasingly recognized the potential of natural products as a source of compounds for developing anticancer drugs [24]. Among these, plant-derived compounds have shown great promise in cancer drug discovery, as many of the currently available anticancer drugs are of natural origin or based on natural product scaffolds [25]. Numerous studies have reported the anticancer properties of plants and their extracts, emphasizing the relevance of natural products in the improvement of cancer therapeutics [25,26,27].
One plant that has gained attention for its potential as a source of natural anticancer agents is S. securidaca, a member of the Fabaceae family that has been used in folk medicine for various ailments, including cancer [28,29,30,31,32,33]. Previous research has revealed the presence of several bioactive compounds in different parts of the plant, including the seeds [28,29,30,31,32,33]. Therefore, investigating the chemical composition of Securigera securidaca seeds and identifying their potential as inhibitors of CDK1/Cks2 represents a promising strategy for exploring novel and effective anticancer agents. In this study, we aimed to investigate the chemical constituents of S. securidaca seeds and evaluate their potential as CDK1 inhibitors using in silico approaches such as molecular docking, ADMET, molecular dynamic simulations, and MM-PPBSA.
2. Results and Discussion
2.1. GC/MS Analysis
GC/MS was used to calculate the percentages of each element in the essential extract. Table 1 and Figure 1 show the calculated and tabulated relative percentages of each component. The following substances were found in the GC/MS chromatogram: cyclopentane, 8-nonynoic acid, 5-hydroxymethylfurfural, 4-vinylguaiacol, eugenol, hippeastrine, vanillin, α-curcumene, α-bergamotene, carbanilic acid, gingerone, didodecyl phthalate, cholestan-3-ol, and gingerol. Hippeastrine had the lowest percentage (2%), while gingerone had the highest (25.67%). The primary components of the essential extract (gingerol, eugenol, gingerol, and α-curcumene) demonstrated higher values (25.67, 18.44, 4.02, and 3.89, respectively) and accounted for 52% of the volatile extract’s total content. These compounds are efficient against a diversity of cancers, including breast, lung, skin, and prostate cancer [34,35,36,37].

Table 1.
GC/MS analysis of S. securidaca seeds phytoconstituents.
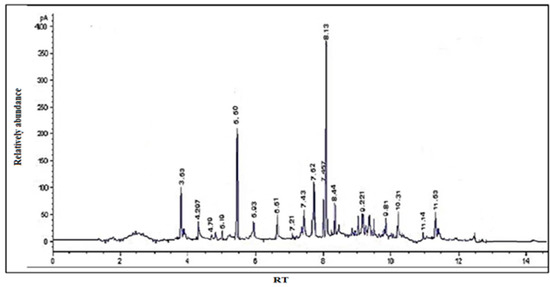
Figure 1.
GC/MS chromatogram of S. securidaca.
2.2. HPLC Analysis
Both standard and sample solutions of S. securidaca seeds were injected three times for their flavonoid content analysis, and the average of the readings was obtained. The major component was kaempferol, which appeared at RT 26.483 min, and the minor component was centaureidin, which appeared at RT 32.447 min, and their percentages in the formulation were calculated as 0.9903 mg/g and 0.551 mg/g, respectively (Figure 2). Kaempferol, a well-known natural flavanol, is found in 80% of plant-based foods such as beans, broccoli, tomato, apples, grapes, and strawberries, as well as some plant seeds or fruits. As a result, it has significance in terms of anti-angiogenesic, anti-proliferative, and anti-metastatic properties [38,39,40].
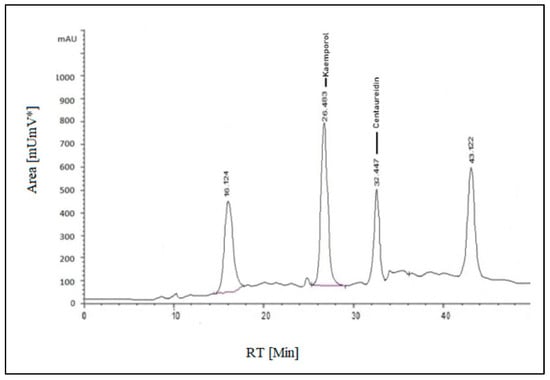
Figure 2.
HPLC-(UV-VIS) chromatogram of S. securidaca seed extract.
Following the methods used, the observed conditions were used to proceed with the analysis of steroids. The overall average readings for each sample and the standard solution were determined after being injected three times. Peak area versus the concentration (mg/mL) was used to measure diosgenin. At an RT of 8.866 min, the diosgenin concentration (3.087 mg/mL) was visible.
2.3. LC-MS/MS
To indicate the existence of phytochemical components, LC-ESI-MS was utilized to analyze the alcoholic extract made from S. securidaca (seed material). Electrospray ionization was utilized in the LC-ESI-MS analysis with a relatively low fragmentation energy of 30 eV. The analysis was conducted using positive and negative ions, and the energy value was used to have a good investigation into the analysis, which helped to identify flavonoids and phenolic acids, as well as their glycosides. The highly complex existence of the extract was reflected by the mass spectra, which were derived from the analysis and permitted the preliminary attribution of about 20 substances from a total of 156 unknown peaks that were found. The identified substances could be categorized as phenolic compounds and their derivatives. Peaks were observed at m/z 279.3, 337.3, and 443.5, corresponding to kaempferol; 278.3 and 220.2, relating to gallic acid; 313.3, 331.4, 792, 800.4, 534.4, 301.3, and 315, corresponding to kaempferol-3-O-glucuronide; 239.3, 240.3, 507.7, and 179.2, corresponding to quercetin; and 281.3, 413.4, and 221.3, corresponding to naringenin. Additionally, the low energy negative-ion mode [171–479.1] was associated with compounds called polyphenols (rutin, luteolin, apigenin, diosgenin, and centaureidin).
2.4. Molecular Docking Studies
Molecular docking has become a computational technique popularly used in structure-based drug models to anticipate the preferred orientation of even one molecule to another while a small molecule (ligand) and a protein (target) are linked [41,42,43]. It plays a crucial role in natural drug discovery, as it helps to identify potential binding sites, evaluate the affinity of the ligand–protein interaction, and predict the most beneficial ligand binding mode [44,45,46]. Such information is crucial for the choice among the most appropriate hit candidates for further drug development and the optimization of new drug molecule design [41].
In the present study, molecular docking was conducted with CDK1 (6GU7.PDB) as the protein target and 24 isolated phytochemicals from Securigera securidaca seeds as the ligands. In addition, two reference drugs, AZD5438 (co-crystallized ligand of 6GU7.PDB) and dinaciclib (experimental control), were included for comparison. The findings of the molecular docking scores are displayed in Table 2 as binding free energy (ΔG) values in kcal/mol.
Table 2.
Binding free energy (ΔG) of isolated phytochemicals 1–24 and reference controls against the cyclin-dependent kinase 1 (CDK1) (6GU7.PDB) target.
A negative ΔG value in molecular docking signifies a strong binding affinity between the protein and ligand [47]. It is used to assess the stability of any protein–ligand interaction and is an indicator of stronger binding [47,48]. The study found that 6 phytochemicals out of 24 exhibited binding energies comparable to those of reference drugs AZD5438 (−8.44 kcal/mol) and dinaciclib (−10.38 kcal/mol), namely, cholestan-3-ol (−8.69 kcal/mol), diosgenin (−9.31 kcal/mol), hippeastrine (−9.42 kcal/mol), naringenin (−10.41 kcal/mol), quercetin (−10.63 kcal/mol), and rutin (−11.92 kcal/mol). Figure 3 presented an analysis of the binding interactions between the six phytochemicals (cholestan-3-ol, diosgenin, hippeastrine, naringenin, quercetin, and rutin) and the critical amino acids inside the active binding site of CDK1. The results were also compared to AZD5438 and dinaciclib.
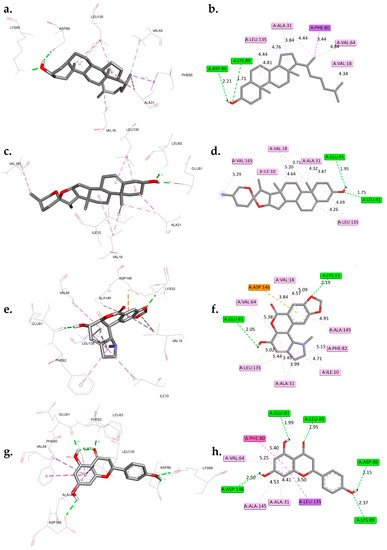
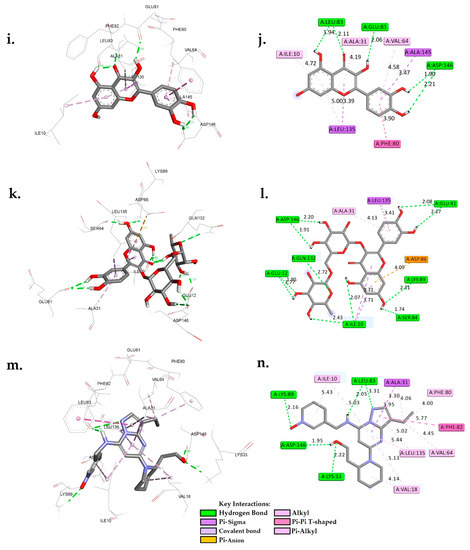
Figure 3.
A 3D and 2D interaction analysis of cholestan-3-ol (a,b), diosgenin (c,d), hippeastrine (e,f), naringenin (g,h), quercetin (i,j), rutin (k,l), and dinaciclib (m,n) with the CDK1 active binding site (6GU7.PDB).
The interactions between six phytochemicals, namely, cholestan-3-ol, diosgenin, hippeastrine, naringenin, quercetin, and rutin, with critical amino acids inside the active binding site of CDK1 were analyzed in detail, and the results are presented in Figure 3. To provide a contextual comparison, the results were also compared with reference controls, the CDK1 inhibitors AZD5438 and dinaciclib. The interactions were categorized into hydrogen bond interactions, Pi-Sigma/Pi-anion interactions, and hydrophobic interactions.
The control AZD5438 displayed hydrogen bond interactions with residues LEU83 and LYS89, with distances of 2.31 Å and 2.33 Å, respectively (Figure S1). It also had hydrophobic interactions with residues ILE10, VAL18, ALA31, LYS33, PHE80, and LEU135. The binding free energy of AZD5438 was found to be −8.44 kcal/mol.
Dinaciclib, the other control, displayed hydrogen bond interactions with residues LYS33, LEU83, LEU83, LYS89, and ASP146 with distances of 2.22 Å, 2.05 Å, 3.31 Å, 2.16 Å, and 1.95 Å, respectively. It also had a Pi-Sigma interaction with ALA31, as well as hydrophobic interactions with residues ILE10, ALA31, VAL18, VAL64, PHE82, PHE80, and LEU135. The binding free energy of dinaciclib was found to be −10.38 kcal/mol. When comparing the phytocompounds to the controls, we observed that cholestan-3-ol exhibited hydrogen bond interactions with ASP86 and LYS89 at distances of 2.21 Å and 1.71 Å, respectively. Additionally, it displayed a Pi-Sigma interaction with PHE80 and hydrophobic interactions with residues VAL18, ALA31, VAL64, and LEU135. The calculated binding free energy of cholestan-3-ol was −8.69 kcal/mol. Similarly, diosgenin showed hydrogen bond interactions with GLU81 and LEU83 at distances of 1.95 Å and 1.75 Å, respectively. It also displayed hydrophobic interactions with ILE10, VAL18, ALA31, LEU135, and VAL165, with a calculated binding free energy of −9.31 kcal/mol. Hippeastrine exhibited hydrogen bond interactions with LYS33 and GLU81, with distances of 2.19 Å and 2.05 Å, respectively. Furthermore, it displayed a Pi-anion interaction with ASP146 and hydrophobic interactions with ILE10, ALA31, VAL18, VAL64, LEU135, PHE82, and ALA145, with a binding free energy of −9.42 kcal/mol. Naringenin showed hydrogen bond interactions with GLU81, LEU83, ASP86, LYS89, and ASP146, with distances of 1.99 Å, 1.95 Å, 2.15 Å, 2.37 Å, and 2.50 Å, respectively. It also displayed a Pi-Sigma interaction with LEU135 and hydrophobic interactions with ALA31, PHE80, VAL64, and ALA145. The calculated binding free energy of naringenin was −10.41 kcal/mol.
The analysis of molecular interactions revealed that quercetin formed hydrogen bonds with GLU81, LEU83, ASP146, and ASP146 at distances of 2.06 Å, 1.94 Å, 2.11 Å, 1.90 Å, and 2.21 Å, respectively. Furthermore, it exhibited Pi-Sigma interactions with LEU135 and ALA145, as well as hydrophobic interactions with ALA31, ILE10, PHE80, VAL64, and LEU135. These findings provide valuable insights into the interactions between the hit phytocompounds, namely, cholestan-3-ol, diosgenin, hippeastrine, naringenin, quercetin, and rutin, with the target cyclin-dependent kinase 1 (CDK1). Notably, the data presented in Figure 3 indicated that the two controls, AZD5438 and dinaciclib, had more extensive interactions compared to the phytocompounds. For instance, dinaciclib formed hydrogen bonds with five residues (LYS33, LEU83, LEU83, LYS89, and ASP146), whereas quercetin interacted with only two residues (GLU81 and LEU83). Comparing each phytochemical with the two controls, it was observed that diosgenin had fewer hydrogen bond interactions compared to both controls, but its binding free energy was similar to that of AZD5438. Cholestan-3-ol had similar hydrogen bond interactions as AZD5438, but its binding free energy was slightly lower. On the other hand, quercetin had Pi-Sigma interactions with residues LEU135 and ALA145 in addition to its hydrogen bond interactions, which made it closer to dinaciclib in terms of molecular interactions.
Hippeastrine had Pi-anion interactions with ASP146, which is unique among the phytochemicals and may suggest that it is a promising candidate for further investigation. Naringenin had Pi-Sigma interactions with LEU135 and hydrophobic interactions with residues ALA31, VAL64, PHE80, and ALA145, which are similar to dinaciclib, making it another candidate for further investigation. Rutin had many hydrogen bond interactions, which were comparable to dinaciclib, but its binding free energy was slightly lower.
Molecular interaction analysis between hit phytochemicals and the CDK1 target suggested that quercetin, naringenin, and hippeastrine are promising candidates for further investigation, as they had the closest molecular interactions to the two controls. Although it is essential to point out that this assessment was conducted according to a single target, more research is required to validate these outcomes. These phytochemicals may be potential CDK1 inhibitors, as indicated by their high binding interactions and binding free energy. The fact that these phytochemicals have demonstrated anticancer properties in experimental studies may support our findings [49,50,51].
To fully evaluate the potential of these compounds as therapeutic agents, their pharmacological properties and safety must be considered. In silico ADMET analysis, which examines absorption, distribution, metabolism, excretion, and toxicity, can provide valuable insights into their pharmacological profiles and predict their potential as hit candidates. In addition, conducting molecular dynamics studies can provide a more detailed understanding of the binding mechanisms of these ligands. Therefore, combining in silico ADMET analysis and molecular dynamics studies can provide a comprehensive evaluation of these phytochemicals’ suitability as therapeutic agents.
2.5. ADMET Prediction
Lipinski’s Principle of Five (RO5), also known as Pfizer’s Principle of Five, or simply the Rule of Five (RO5), is often a commonly accepted standard for determining the oral bioavailability of bioactive molecules [52]. The rule postulates that a compound’s poor absorption, as well as permeation, is more probable when its properties exceed the following thresholds: upwards of 5 donors of hydrogen bonds, greater than 10 hydrogen bond acceptors, a molecular weight exceeding 500 Da, and a computed logP greater than 5 [53]. The RO5 has been an influential tool in the drug discovery process, with approximately 50% of new chemical entities designed for oral administration adhering to these guidelines [53]. It is important to note that this rule is not a foolproof predictor of oral bioavailability [53]. This is because some compounds with higher molecular weights or logP values can still exhibit oral absorption [53].
The Ghose filter is another commonly used drug-like filter in drug discovery, which defines drug-likeness through constraints on calculated logP (−0.4–5.6), as well as molecular weight (160–480 Da) and hydrogen bond acceptors (0–10) [54]. The Veber rule is a criterion applied to examine the drug-likeness of compounds and stipulates that the number of rotatable bonds should not be greater than 10, and the topological polar surface area (TPSA) ought to be lower than (should be less than) 140 Å2 [55]. This rule is often used in conjunction with Lipinski’s Principle of Five, which highlights the correlation between drug oral bioavailability and small, moderately lipophilic compounds [56]. In addition, the Veber rule has been utilized in terahertz light research to identify rotating bonds in drug candidate molecules [57].
The Egan rule is another criterion applied to evaluate the drug-likeness of compounds [58,59]. It considers compounds along with 0 ≥ TPSA ≤ 132 Å2 and −1 ≥ logP ≤ 6 to have good bioavailability [58,59]. The Muegge rule is another criterion used to appraise the drug-likeness of compounds, which specifies that the molecular weight should be no greater than 500 Da, the total number of hydrogen bond donors should be no more than 5, and the total number of hydrogen bond acceptors should be no more than 10 [60].
The Egan and Muegge rules are often used in conjunction with other rules such as Lipinski’s and Ghose filters to predict drug absorption [58,59,60]. It is important to note that these filters should be viewed as guidelines rather than absolute measures when evaluating drug-likeness [61]. While these rules have proven useful in predicting drug absorption, some compounds may still exhibit oral bioavailability despite exceeding certain thresholds [61].
The results shown in Table 3 present the estimated physicochemical and drug-likeness properties of the six hit phytochemicals using the Swiss-ADME webserver. Properties such as molecular weight, TPSA, molar refractivity, and octanol–water partition coefficient (LogP) are crucial for evaluating the potential of a compound for drug development. Dinaciclib, the control drug, has a molecular weight of 396.49 g/mol, a TPSA of 91.15 Å2, a molar refractivity of 117.94, and a LogP of 1.55. Its water solubility is categorized as soluble.
Table 3.
The estimated physicochemical and drug-likeness properties using the Swiss-ADME webserver for the hit phytochemicals.
Among the six hit phytochemicals, hippeastrine is the closest in terms of drug-likeness properties to dinaciclib. Its molecular weight is 315.32 g/mol, its molar refractivity is 83.56, its TPSA is 68.23 Å2, its LogP is 0.66, and its water solubility is very soluble. The drug-likeness properties as indicated by the Lipinski, Ghose, Veber, and Egan rules, as well as the Muegge filters suggest that hippeastrine is a potentially promising candidate for drug development.
Naringenin and quercetin also have drug-likeness properties that are relatively similar to dinaciclib. Naringenin has a molecular weight of 272.25 g/mol, a TPSA of 86.99 Å2, a molar refractivity of 71.57, a LogP of 2.19, and a water solubility of soluble. Quercetin has a molecular weight of 302.24 g/mol, a TPSA of 131.36 Å2, a molar refractivity of 78.03, a LogP of 1.99, and a water solubility of soluble.
Cholestan-3-ol, diosgenin, and rutin, the remaining three hit phytochemicals, have drug-likeness properties that are less similar to dinaciclib. Cholestan-3-ol and diosgenin are poorly soluble in water and do not meet the criteria set by the Lipinski, Ghose, Veber, and Egan rules and the Muegge filters. Rutin is moderately soluble in water but also does not meet the criteria set by the filters.
Pharmacokinetics is the investigation of how drugs access the body, circulate, and exit the body [23]. The effects of a drug on an individual are determined by its pharmacological characteristics at the site of action [23]. Table 4 shows the estimated pharmacokinetics and toxicological properties of six hit phytochemicals compared to dinaciclib as a control using the Swiss-ADME and pkCSM webservers. Hippeastrine and naringenin are the two best phytochemicals that are closest to the control in terms of properties. Both hippeastrine and naringenin have high GI absorption and do not cross the BBB, making them promising candidates for oral drug delivery. They are also Pgp substrates and do not inhibit CYP2C19, indicating a low potential for drug–drug interactions. However, hippeastrine is potentially toxic, with a greater tolerated dose of −0.459 log mg/kg/day, while naringenin has a higher greater tolerated dose of −0.176 log mg/kg/day. Both compounds also have relatively low rat acute toxicity LD50 effects of 2.492 and 1.791 mol/kg, respectively.
Table 4.
The estimated pharmacokinetics and toxicological properties using the Swiss-ADME and pkCSM webservers for the hit phytochemicals.
The results presented in Table 3 and Table 4 suggest that hippeastrine and naringenin exhibit drug-like properties and desirable pharmacokinetic and toxicological characteristics, which are comparable to the reference compound dinaciclib. These findings highlight the potential of these phytochemicals as promising hit candidates for further investigation, such as molecular dynamics simulations, to assess their dynamic behavior and stability. However, it is essential to conduct additional experiments and studies to validate these results and determine the safety and efficacy of these compounds.
2.6. Molecular Dynamic Simulations
To further investigate the binding of hippeastrine and naringenin to the active binding site of CDK1, we performed 100 ns in molecular dynamics (MD) simulations of the four ligand–protein complexes, including AZD5438 and dinaciclib, using GROMACS 2016. The stability of the docked complexes was analyzed by calculating the average root-mean-square deviation (RMSD), the average root-mean-square fluctuation (RMSF) of the protein and ligand backbone atoms, and the radius of gyration (ROG).
The RMSD of the Cα-backbone of CDK1 bound to the AZD5438 and dinaciclib molecules exhibited a deviation of approximately 3.5 Å and 3.3 Å, respectively, more than the course of the 100 ns MD simulation (Figure 4A). Interestingly, the Cα-backbone RMSD of CDK1 bound to hippeastrine and naringenin showed slightly smaller deviations than the AZD5438 system and were close to the dinaciclib system, with deviations of approximately 3.2 Å and 3.3 Å, respectively (Figure 4A).
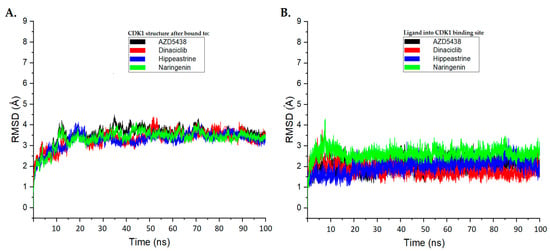
Figure 4.
The root-mean-square deviation (RMSD) analysis plots for the molecular dynamics (MD) simulation trajectories were conducted over a 100 ns time scale. (A) RMSD plots for the protein backbone of CDK1, exhibiting molecular vibrations after binding with AZD5438 (black), dinaciclib (red), hippeastrine (blue), and naringenin (green). (B) RMSD plots highlighting the conformational changes of AZD5438 (black), dinaciclib (red), hippeastrine (blue), and naringenin (green) upon binding with CDK1.
We also examined the conformational changes and stability of the ligands inside the active binding site of CDK1, as displayed in Figure 4B. Our results suggest that hippeastrine and naringenin exhibited good convergence and stable conformations within the binding site throughout the 100 ns MD simulation, with conformational changes less than 2 Å. Based on these findings, we conclude that the CDK1-bound hippeastrine and naringenin molecules are quite stable because of their higher affinity for the protein and can be considered promising candidates for further study.
In molecular dynamics simulations, RMSF and ROG assessments are valuable tools for evaluating protein stability and flexibility [62,63,64]. RMSF analysis measures the mean fluctuation of residues that occurs during simulation, while the ROG assessment determines the mass-weighted root mean square separation of the protein from its center of mass [51,52,53]. By using these methods, we can gain important insights into the structural stability and compactness of proteins [62,63,64].
In this study, RMSF and ROG analyses were performed on the MD simulation trajectories of CDK1 complexed with AZD5438, dinaciclib, hippeastrine, and naringenin over a 100 ns time scale to determine their structural stability and compactness changes. The RMSF plots in Figure 5A display the average residual fluctuations of CDK1 after binding to the different ligands. Interestingly, the RMSF values for CDK1 bound to hippeastrine and naringenin were found to be similar to those of AZD5438 and dinaciclib; on average, residual fluctuation variations in the systems were less than 2 Å. This suggests that hippeastrine and naringenin may stabilize the protein structure by reducing the flexibility of the CDK1 backbone as the controls.
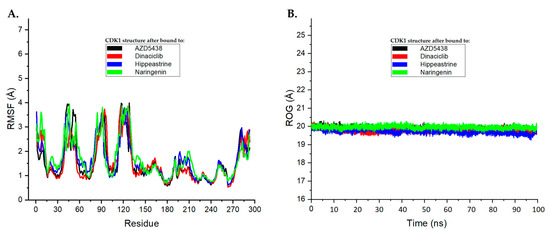
Figure 5.
The root-mean-square fluctuation (RMSF) and radius of gyration analysis plots for the molecular dynamics (MD) simulation trajectories were conducted over a 100 ns time scale. (A) RMSF plots of CDK1’s average residual fluctuations after binding to each ligand. (B) ROG plots demonstrating the compactness of the CDK1 protein structure induced by binding to each ligand. These plots are shown with AZD5438 in black, dinaciclib in red, hippeastrine in blue, and naringenin in green.
Panel B in Figure 5 shows the ROG plots, which highlight the compactness of the CDK1 protein structure and its conformational changes upon binding to the different ligands. The black, red, blue, and green curves represent the ROG plots for CDK1 bound to AZD5438, dinaciclib, hippeastrine, and naringenin, respectively. To gain a deeper understanding of protein compactness following ligand binding, we calculated the ROG values over the last 10 ns of the MD simulation. The average ROG values for AZD5438, dinaciclib, hippeastrine, and naringenin were found to be approximately 19.92 Å, 19.71 Å, 19.93 Å, and 19.56 Å, respectively. The average ROG values of CDK1 suggested that hippeastrine and naringenin may not significantly affect the compactness structure of the protein, as compared to reference drugs AZD5438 and dinaciclib. Overall, the results showed that hippeastrine and naringenin exhibited good convergence and stable conformations within the CDK1 active binding site and reduced the flexibility of the protein backbone, suggesting they may have potential as CDK1 inhibitors.
The widely applied Molecular Mechanics Poisson–Boltzmann Surface Area (MM-PBSA) strategy provides good convergence to experimental outcomes when computing the binding free energy of protein–ligand complexes [65]. This approach combines free energy estimates based on implicit solvent models with energetic calculations that utilize molecular mechanics [66]. For modelling molecular recognition, including such protein–ligand binding interactions, the MM-PBSA technique has been proven to be a reliable and effective free energy simulation strategy [66,67].
It is possible to gain further insight into CDK1–ligand binding affinity by applying MM-PBSA to CDK1–ligand complexes and potentially confirm the suitability of hippeastrine and naringenin as hit candidates by applying MM-PBSA in molecular dynamics simulations.
This study delivers valuable insights into the binding energies of ligands to the active binding site of CDK1. The outcomes demonstrate that all the ligands possess a relatively strong binding affinity for the receptor, along with binding energies ranging from −52.03 to −59.41 kJ/mol. By examining the energy contributions from different interactions, we can learn more about the molecular mechanisms underlying the interactions.
Table 5 presents data that reveal van der Waals interactions as the primary factor contributing to the binding energy for all systems, ranging from −28.63 to −31.24 kJ/mol. This finding indicates that the physical interactions among the atoms of the ligand and the receptor are the primary driving force behind ligand binding. Additionally, electrostatic interactions, ranging from −31.15 to −37.16 kJ/mol, also played a significant role in the binding process, emphasizing their importance.
Table 5.
MM-PBSA energies (ΔGBinding) for ligand binding (hippeastrine and naringenin) at the active binding site of CDK1 (6GU7.PDB). AZD5438 and dinaciclib were used for comparison. All the energy units in kJ/mol.
As expected, the polar and non-polar salvation solvation energy values for the ligands were relatively low, ranging from −16.49 to 26.94 kJ/mol and −15.22 to −16.33 kJ/mol, respectively. This implies that the solvent does not play a significant role in the binding process of the ligands to the receptor.
The MM-PBSA calculations were consistent with the RMSD and RMSF results, indicating that hippeastrine and naringenin have strong binding interactions with CDK1. The binding free energy values for these ligands were similar to those of reference ligands AZD5438 and dinaciclib, suggesting that they possess comparable binding affinities to the receptor. The electrostatic energy values for hippeastrine and naringenin were also comparable to those of AZD5438 and dinaciclib, suggesting that these ligands interact similarly with the electrostatic potential of the receptor. However, the van der Waals energy values for hippeastrine and naringenin were slightly greater than those of AZD5438 and dinaciclib, indicating that they may have slightly different interactions with the van der Waals forces of the receptor.
Overall, our findings suggest that hippeastrine and naringenin may have potential as CDK1 inhibitors. However, additional studies such as in vitro and in vivo assays are necessary to verify these results and assess the binding affinity and efficacy of these compounds as CDK1 inhibitors. This study highlights the importance of using computational methods such as RMSD, RMSF, and MM-PBSA in the early phases of drug inventions to estimate the structural stability and the potential of protein–ligand complexes.
3. Materials and Methods
3.1. Collection and Preparation of S. securidaca Seed Extracts
S. securidaca seeds were obtained from a local market in Hafr Al Batin, Saudi Arabia. The seeds were verified by comparing them to a reference herbarium sample.
3.2. Preparation of S. securidaca Seed Extracts
3.2.1. GC/MS
The samples were ground up, stored desiccated in plastic containers, and dried at 60 °C for one hour to a constant weight. For 24 h at room temperature, 500 mL of mixing ethanol with water (1:1 v/v) was continuously infused with 50 g of dried sample powder. Under reduced pressure at 45 °C, the extracted material was filtered before being concentrated to dryness. Then 100 mg of the cleaned extract was partitioned into chloroform with water (1:1 v/v) for GC/MS examine. Under lower pressure at 45 °C, the eventual findings crude organic phase was evaporated, then filtered, and concentrated to dryness. Then gas chromatography-mass was used to analyze the fraction obtained [68].
3.2.2. Glycine Test
One gram of powdered plant seeds was set in 100 mL of hexane and evaporated for one hour. Then 0.5 g of the flesh was extracted for 1 h by ultrasound waves utilizing 5 mL of an extracting remedy that contained ethanol and 0.2% metaphoric acid by volume. The solution was centrifuged at 10,000 rpm and 4 °C for 5 min. Using a concentrator, 0.8 mL of supernatant was dried for 4 h. A 0.22 m nylon syringe filter was utilized to filter the samples before they were placed in an HPLC glass vial and securely closed. After that, 0.5 L of the samples was added to the HPLC instrument [68].
3.2.3. Glycoside Test
A conical flask was filled with ground seed powder (0.5 g) (90 mL). Then 20 mL of ethanol was added, extractions were conducted, and the water bath was shaken at the same temperature of 37 °C that was used for water extraction. A rotary evaporator was used to completely fade away the ethanol from the filtrate after the extracts were thoroughly filtered, similar to the water extraction. The dried sample was mixed with 5 mL of diethyl ether, and then the mixture was vortexed for 1 min at room temperature (20 ± 2 °C) to initiate precipitation of the major phytoconstituents. To prepare it for the HPLC method, diethyl ether was evaporated after being allowed to dry up overnight inside a fume hood.
3.2.4. Flavonoids Test
Five grams of powder seeds was immersed in a solvent consisting of aqueous methanol (99%) and 1% hydrochloric acid, and effective integration of microwave (MAE) and ultrasound (UAE) guided extraction was used. One gram of the lyophilized dehydrated powders was combined with 4 mL of such extraction solvent, which contained methanol and hydrochloric acid (99:1, v/v), and then was microwaved for 15 s at 450 W. Following that, each mixture was put in a light-protected vial and sonicated in an ultrasonicator at 258 °C for 15 min at a steady frequency of 35 kHz. Having been sonicated, the lyophilized seed solutions were centrifuged for 20 min at 3500 rpm, after which each extract of the seeds was gathered in a lab flask for HPLC analysis [68].
3.2.5. Steroids Test
While using a magnetic stirrer, 100 mg of powdered seeds was extracted in 200 mL of 70% methanol for 3 h at 60 °C. Before HPLC analysis, the achieved extract was lyophilized, diluted in methanol (1:1), and filtered by a 0.22-micron nylon syringe filter.
3.3. Instrumentation and MS Parameters
3.3.1. GC/MS
A gas chromatograph with an Agilent 6890 mass spectrometer and an Agilent mass spectrometric detector, direct capillary interface, and PAS-5ms column made of fused silica (30 m, 0.32 mm, and 0.25 m in film thickness) were used. The following circumstances existed when the extract under investigation was injected. About 1.0 mL/min of helium was utilized as the carrier gas in a pulsed split-less mode. The injection volume should be 1.0 µL, with a 3 min solvent delay. The mass spectrometric detector was run in electron influence ionization mode, scanning between 50 and 500 m/z, with an ionization energy of 70 e.v. The multiplier voltage (EM 5 voltage) was kept at 1250 v out of auto-tune, while the ion source temperature was 230 °C [58]. To perform the glycine test, a 2.1 mm × 1.7 m C18 column that was thermostated at 16 °C was used in conjunction with the chromatography method. Chemstation, an analytical tool for chromatography data, was used to analyze the data (Agilent, Santa Clara, CA, USA). The Mobile Phase System was applied with a flow rate of 0.4 mL/min as described:
| Mobile Phase Grades | Time | |
| A (10% methanol with 0.1% formic acid) | 10–30 | 0–6.5 |
| 30–100 | 6.5–7 | |
| 100–10 | 8–8.5 | |
| 10 | 8.5–12.5 | |
| B (50% methanol with 0.1% formic acid) | 90–70 | 0–6.5 |
| 70–0 | 6.5–7 | |
| 0–90 | 8–8.5 | |
| 90 | 8.5–12.5 | |
The detector was a fluorescence detector with emissions at 440 and 335 nm wavelengths. Retention was used to identify amino acids [45].
3.3.2. HPLC
Chromatography was conducted using a 5 μm OmniSpher C18 column with a 250 × 4.6 mm internal diameter (Varian, Palo Alto, CA, USA). The flowing mobile phases (A consisting of 0.1% phosphoric acid mixed with water, B consisting of 100% methanol HPLC grade) were used (Table 6). The flow rate should be 0.8 mL per minute. For samples and standards, the injection volume was 20 µL, and the compounds were all isolated at room temperature. Between independent runs, a 10 min re-equilibration duration was utilized. The detection wavelength for UV-vis spectra was 280 nm [46].
Table 6.
Mobile phase grades of HPLC for analysis of S. securidaca seed phytoconstituents.
To test for flavonoids, a reverse-phase SupelcosilTM LC-18 HPLC column with a 15 cm length and 4 mm inner diameter, and 5 µm diameter octadecyl silane particles were used, as previously described [69].
In the steroids test, the HPLC system—Agilent 1200—composed of two LC-20AD pumps with a DGU-26A7 degasser was connected to a micromixer (0.5–2.6 mL for HPLC-ELSD), CTO-20AC thermostat, CBM-20A control system, and SIL 20ACYR autosampler with an evaporative light scattering detector with an ELSD 3300 N2 generator (Alltech Associates, Deerfield, IL, USA). Chemstation software was used to collect and process the data (version 3.b32). Formic acid and water (A, 99.9:0.1, V/V), as well as acetonitrile (B, 99.9:0.1, V/V), made up the mobile phase. On an Invention C-18 column (150 mm × 2.1 mm, 3 µm), isolation was first carried out by the following gradient programs: 0 min—20% B, 27 min—33.5% B, and 45 min—100% B. The flow rate was 0.2 mL min–1, and the column temperature was 20 °C. The injection volume was 1 µL.
3.4. Molecular Docking
3.4.1. Protein Structure Preparation
The 3D structure of the CDK1 complex with the co-crystallized ligand AZD5438 (PDB ID: 6GU7) [70], derived from the Protein Data Bank (PDB) [70] in PDB format, was selected as the protein target for further analysis. Dinaciclib, a novel CDK inhibitor with promising antitumor activity in clinical trials, was chosen as the reference drug for a comparative study [71,72,73]. The protein targets were preprocessed for molecular docking studies using the Biovia Discovery Studio Visualizer (Biovia, 2020) [74] to remove heteroatoms and non-essential water molecules and the YASARA web-server tool to add missing amino acids [75]. The H++ web-server tool was utilized to calculate the ionization states of titratable amino acid groups at pH 7.4 [76]. The resulting output was then transformed to PDBQT format via AutoDock Tools version 1.5.6 software [77], including adding polar hydrogen atoms and Kollman charges. The active binding site coordinate for the docking studies was recognized from the potential ligand binding domain region of the crystal structure.
3.4.2. Ligand Preparation
In this study, the reference drug dinaciclib’s 2D structure (PubChem ID: 46926350) was collected from the PubChem database (“https://pubchem.ncbi.nlm.nih.gov, accessed on 18 December 2022”) to conduct a molecular docking experiment. The protein structures for AZD5438 were taken from the Protein Data Bank (PDB ID: 6GU7), and any changes in conformation were considered when determining the structure of the drug. ChemDraw JS can be found at https://chemdrawdirect.perkinelmer.cloud/js/sample/index.html, accessed on 18 December 2022, and twenty-four phytochemical compounds were illustrated and stored in the Structural Data File (SDF) format. To ensure the stability of each ligand, an energy minimization process was initiated, incorporating the Universal Force Field (UFF) and a Conjugate Gradient (CG) optimization algorithm that runs for a thousand steps. This optimization process was conducted through the Open Babel software, and the resulting structures were saved in PDB format. The Gasteiger charges were allocated to the ligands utilizing AutoDock Tools version 1.5.6, and the ligands were stored in PDBQT format, all set for molecular docking simulations.
3.4.3. Molecular Docking Preparation
To assess the binding affinity and molecular interactions of the twenty-four phytochemical compounds along with CDK1, a molecular docking investigation was carried out. The AutoDock 4.2 Release 4.2.6 software [77,78] was utilized for the docking calculations, and the phytochemicals (ligands) were docked against co-crystallized AZD5438 and dinaciclib reference controls. To optimize the binding poses of the compounds, the Lamarckian genetic algorithm was used, with flexible ligands and a rigid CDK1 [79]. The active binding site of CDK1 was utilized for docking, and the grid box was centered on the ligand of the original CDK1 structures. The grid box size was 404,040 along the X-, Y-, and Z-axes, with the central grid point of the map located at 32.61, 16.15, and 11.25 (XYZ-coordinates). To ensure consistency in the calculations, the default values were used for all parameters except the number of runs, which was set to 150, and the greater number of assessments, which was set to 25,000,000. Finally, the binding energies and properties of the various phytochemical compounds were evaluated and examined for their potential as therapeutic hit candidates.
3.5. ADMET Prediction
The use of in silico tools to predict the ADMET impact of ligands, as well as their impurities, is increasingly important for the quality monitoring of medicines [80]. To this end, we utilized the SwissADME server [81] to predict the physicochemical, drug-like, and pharmacokinetic properties of our selected ligands, while the pkCSM server [82] was utilized to predict the toxicological impacts. To validate these predictions, we conducted a comparative study with dinaciclib as a control. The resulting predictions were used to find potential hit candidates for further in silico investigations, such as molecular dynamic simulations, based on their predicted safety and drug-likeness properties.
3.6. Molecular Dynamics
The priorities of this study were to assess the binding behavior and stability of specific phytocompounds in the active binding sites of the protein CDK1 by using molecular dynamics (MD) simulations. The crystal structure of CDK1 in combination with AZD5438 was employed as a point of origin for the simulations. MD simulations were carried out via the GROMACS software package (version 2016.3) and the Gromos96 54a7 force field [83] for 100 nanoseconds. Prior to the simulation, the topology files for the ligand and protein were produced via suitable tools, namely, the GROMACS tool, pdb2gmx, and the PRODRG server that was accessed on 5 February 2023 (“http://davapc1.bioch.dundee.ac.uk/cgi-bin/prodrg”), respectively. The systems were then solvated by the Transferable Intermolecular Potential along with the 3 Points (TIP3P) water model, and counterions were introduced to offset the charge. To complete the preparation for the simulation, the potential energy of systems was lowered with the steepest descent integrator for 50,000 greater minimization steps and an energy step size of 0.01. Thereafter, the systems were equilibrated in the NVT ensemble for 100 ps at 310 K by the v-rescale coupling method, accompanied by another 100 ps of equilibration along with the NPT ensemble at 1.0 bar utilizing the Berendsen pressure coupling procedure [68,84].
Following the temperature and pressure equilibrations, we carried out MD simulation runs on the models for a duration of 100 ns each at 1 bar and 310 K. Short-range non-bonded interactions were limited to 1.2 nm, whereas long-range electrostatic interactions were handled using the Particle Mesh Ewald (PME) algorithm [85]. The bonds and hydrogen atoms were constrained via the LINCS algorithm [86]. All simulations had a time step of 2 fs, and MD data were analyzed by recording the coordinates every 5000 steps (10 ps) and using various techniques such as RMSD and RMSF to assess the stability and strength of the interactions between the phytocompounds and the protein. For visualization of the results, we used various tools and techniques, such as Biovia Discovery Studio Visualizer (Biovia, 2020) to create molecular graphics images and OriginPro 2021 (version 9.8.0.200) to produce graphs. These images and graphs provided a detailed representation of the systems and the interactions taking place, offering a more profound comprehension of the simulation results.
3.7. Binding Free Energy Calculation Using MM/PBSA
Our study used the molecular mechanics Poisson–Boltzmann surface area (MM-PBSA) procedure [87], a commonly used procedure for measuring the binding free energy from snapshots of molecular dynamics (MD) trajectories. We examined the binding free energies of the chosen phytochemical hits and reference controls (AZD5438 and dinaciclib) against CDK1 by taking snapshots between 90 and 100 ns at intervals of 100 ps during the MD simulations equilibrium processes. This assessment was conducted by the g_mmpbsa software of Gromacs [88,89]. To compute the binding free energy of the ligand–protein complex inside the solvent, we subtracted the sum of the total energies of the protein (Gprotein) and ligand (Gligand) in the solvent when separated from the total free energy of the protein–ligand complex (Gcomplex). This was done by the following equation [75]:
ΔGbind = [Gcomplex − Gprotein + Gligand]
The energy that is free for every separate state, including the complex, protein, and ligand, was approximated by adding up the mean potential energy of molecular mechanics in a vacuum (EMM) and the energy that is free from solvation (Gsolvation) [89]:
Gx = EMM + Gsolvation
The molecules’ potential energy was calculated in a vacuum environment by adding the energy from both bonded (Ebonded) and non-bonded (Enon-bonded) interactions. Ebonded covers interactions such as bonding, angles, and dihedral, as well as improper interactions, while Enon-bonded includes electrostatic (Eelec) and van der Waals (Evdw) interactions [89]:
EMM = Ebonded + Enon_bonded = Ebonded + [Evdw + Eelec]
To obtain the free energy of solvation (Gsolvation), we added the non-polar solvation free energy (Gnon-polar) and the free energy of electrostatic solvation (Gpolar) [89]:
Gsolvation = Gpolar + Gnon-polar
The Poisson–Boltzmann equation [78] was applied to compute the electrostatic solvation free energy (Gpolar), whereas the solvent-accessible surface area (SASA) was utilized to estimate the non-polar solvation free energy (Gnon-polar) according to the following equation:
Gnon-polar = γSASA + b
In this study, two sets of values were provided for the coefficients γ and b used to estimate the solvation free energy in Equation (5). If the simulation is carried out using units of kcal/mol, then the values γ = 0.0054 kcal/mol·Å2 and b = 0.916 kcal/mol should be used. On the other hand, if the simulation is carried out using units of kJ/mol, then the values γ = 0.02267 kJ/mol·Å2 and b = 3.849 kJ/mol should be used. The choice of which set of values to use depends on the unit system being used in the simulation.
It is important to point out that the value of ΔEbonded was considered to be zero in this computation [90]. These results of the calculation of binding free energy could offer valuable information regarding the energetics of interactions among the protein and ligand molecules, which may help in recognizing the potential hit compounds.
4. Conclusions
In conclusion, our study has successfully identified hippeastrine and naringenin as promising hit candidates for CDK1 inhibition, which may offer new opportunities for the improvement of effective anti-cancer agents. The fact that these compounds were isolated from Securigera securidaca seeds, a plant that has been used for ailments in conventional medicine for a long time, highlights the possibilities of natural products being used as valuable sources for drug discovery.
Our in silico studies suggest that hippeastrine and naringenin exhibit favorable molecular interactions and drug-like properties, as well as desirable pharmacokinetic and toxicological profiles, which make them attractive candidates for further development. The good convergence and stable conformations observed in the molecular dynamic (MD) simulations moreover support the prospects of these compounds as effective CDK1 inhibitors for cancer therapy.
Moving forward, future research should prioritize validating the CDK1 inhibitory activity of hippeastrine and naringenin in vitro and in vivo, as well as exploring their mechanisms of action and potential synergistic effects with other anti-cancer agents. Additionally, efforts should be made to optimize the chemical structures of these compounds for improved potency, selectivity, and bioavailability, and to develop formulations suitable for clinical use.
Overall, our findings offer insightful information about how natural products are evolving as potential sources for novel anti-cancer medications and underscore the importance of interdisciplinary approaches to drug discovery. We hope that this research will inspire further studies in this area and contribute to the improvement of more effective and less toxic treatments for cancer patients.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/pr11051478/s1, Figure S1. (A) Superimposition and (B) 2D interaction analysis of the original co-crystallized ligand AZD5438 (depicted in blue for carbon (C), red for oxygen (O), navy for nitrogen (N), and orange for sulphur (S)). (C) Re-docked ligand (depicted in cyan for carbon (C), red for oxygen (O), navy for nitrogen (N), and orange for sulphur (S)) in the crystal structure of human CDK1/CKS2 complex with AZD5438 (PDB ID: 6GU7). The root mean square deviation between the original co-crystallized ligand of AZD5438 and the re-docked ligand was 0.92 Å.
Author Contributions
Conceptualization, M.E.M.A., G.M.A.-M. and A.E.A.; methodology, M.E.M.A., G.M.A.-M., F.A.-R., F.W. and H.A.S.; writing—original draft preparation, M.E.M.A., G.M.A.-M., F.A.A., F.W. and A.E.A.; writing—review and editing, M.E.M.A., G.M.A.-M., F.W. and A.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research work was funded by Institutional Fund Projects under no. IFP-A-2022-2-5-25. Therefore, the authors gratefully acknowledge technical and financial support from the Ministry of Education and University of Hafr Al Batin, Saudi Arabia.
Data Availability Statement
Data is contained within the article and Supplementary Material.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Mehraj, U.; Dar, A.H.; Wani, N.A.; Mir, M.A. Tumor microenvironment promotes breast cancer chemoresistance. Cancer Chemother. Pharmacol. 2021, 87, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Matthews, H.K.; Bertoli, C.; de Bruin, R.A. Cell cycle control in cancer. Nat. Rev. Mol. Cell Biol. 2022, 23, 74–88. [Google Scholar] [CrossRef]
- Panagopoulos, A.; Altmeyer, M. The Hammer and the Dance of Cell Cycle Control. Trends Biochem. Sci. 2020, 46, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Mehraj, U.; Sofi, S.; Alshehri, B.; Mir, M.A. Expression pattern and prognostic significance of CDKs in breast cancer: An integrated bioinformatic study. Cancer Biomark. 2022, 34, 505–519. [Google Scholar] [CrossRef]
- Dang, F.; Nie, L.; Wei, W. Ubiquitin signaling in cell cycle control and tumorigenesis. Cell Death Differ. 2020, 28, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Mehraj, U.; Aisha, S.; Sofi, S.; Mir, M.A. Expression pattern and prognostic significance of baculoviral inhibitor of apoptosis repeat-containing 5 (BIRC5) in breast cancer: A comprehensive analysis. Adv. Cancer Biol. Metastasis 2022, 4, 100037. [Google Scholar] [CrossRef]
- Bury, M.; Le Calvé, B.; Ferbeyre, G.; Blank, V.; Lessard, F. New Insights into CDK Regulators: Novel Opportunities for Cancer Therapy. Trends Cell Biol. 2021, 31, 331–344. [Google Scholar] [CrossRef]
- Qayoom, H.; Wani, A.; Alshehri, N.; Mir, M. An insight into the cancer stem cell survival pathways involved in chemoresistance in triple-negative breast cancer. Futur. Oncol. 2021, 17, 4185–4206. [Google Scholar] [CrossRef]
- Izadi, S.; Nikkhoo, A.; Hojjat-Farsangi, M.; Namdar, A.; Azizi, G.; Mohammadi, H.; Yousefi, M.; Jadidi-Niaragh, F. CDK1 in Breast Cancer: Implications for Theranostic Potential. Anticancer Agents Med. Chem. 2020, 20, 758–767. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; He, F.; Zhang, Z.; Xiang, Z.; Hu, D. CDK1 serves as a potential prognostic biomarker and target for lung cancer. J. Int. Med. Res. 2020, 48. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: A changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Sofi, S.; Mehraj, U.; Qayoom, H.; Aisha, S.; Almilaibary, A.; Alkhanani, M.; Mir, M.A. Targeting cyclin-dependent kinase 1 (CDK1) in cancer: Molecular docking and dynamic simulations of potential CDK1 inhibitors. Med. Oncol. 2022, 39, 1–15. [Google Scholar] [CrossRef] [PubMed]
- García-Reyes, B.; Kretz, A.-L.; Ruff, J.-P.; von Karstedt, S.; Hillenbrand, A.; Knippschild, U.; Henne-Bruns, D.; Lemke, J. The Emerging Role of Cyclin-Dependent Kinases (CDKs) in Pancreatic Ductal Adenocarcinoma. Int. J. Mol. Sci. 2018, 19, 3219. [Google Scholar] [CrossRef]
- Wijnen, R.; Pecoraro, C.; Carbone, D.; Fiuji, H.; Avan, A.; Peters, G.J.; Giovannetti, E.; Diana, P. Cyclin Dependent Kinase-1 (CDK-1) Inhibition as a Novel Therapeutic Strategy against Pancreatic Ductal Adenocarcinoma (PDAC). Cancers 2021, 13, 4389. [Google Scholar] [CrossRef] [PubMed]
- Sung, W.-W.; Lin, Y.-M.; Wu, P.-R.; Yen, H.-H.; Lai, H.-W.; Su, T.-C.; Huang, R.-H.; Wen, C.-K.; Chen, C.-Y.; Chen, C.-J.; et al. High nuclear/cytoplasmic ratio of CDK1 expression predicts poor prognosis in colorectal cancer patients. BMC Cancer 2014, 14, 951. [Google Scholar] [CrossRef]
- Neganova, I.; Tilgner, K.; Buskin, A.; Paraskevopoulou, I.; Atkinson, S.P.; Peberdy, D.; Passos, J.F.; Lako, M. CDK1 plays an important role in the maintenance of pluripotency and genomic stability in human pluripotent stem cells. Cell Death Dis. 2014, 5, e1508. [Google Scholar] [CrossRef] [PubMed]
- Novak, B.; Tyson, J.J.; Gyorffy, B.; Csikasz-Nagy, A. Irreversible cell-cycle transitions are due to systems-level feedback. Nature 2007, 9, 724–728. [Google Scholar] [CrossRef] [PubMed]
- Nigg, E.A. Mitotic kinases as regulators of cell division and its checkpoints. Nat. Rev. Mol. Cell Biol. 2001, 2, 21–32. [Google Scholar] [CrossRef]
- Prevo, R.; Pirovano, G.; Puliyadi, R.; Herbert, K.J.; Rodriguez-Berriguete, G.; O’docherty, A.; Greaves, W.; McKenna, W.G.; Higgins, G.S. CDK1 inhibition sensitizes normal cells to DNA damage in a cell cycle dependent manner. Cell Cycle 2018, 17, 1513–1523. [Google Scholar] [CrossRef] [PubMed]
- Bendris, N.; Lemmers, B.; Blanchard, J.M. Cell cycle, cytoskeleton dynamics and beyond: The many functions of cyclins and CDK inhibitors. Cell Cycle 2015, 14, 1786–1798. [Google Scholar] [CrossRef] [PubMed]
- Saikat, A.S.M. An In Silico Approach for Potential Natural Compounds as Inhibitors of Protein CDK1/Cks2. Chem. Proc. 2021, 8, 5. [Google Scholar]
- Dias, D.A.; Urban, S.; Roessner, U. A Historical Overview of Natural Products in Drug Discovery. Metabolites 2012, 2, 303–336. [Google Scholar] [CrossRef]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; Supuran, C.T. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Lu, J.-J.; Ding, J. Natural Products in Cancer Therapy: Past, Present and Future. Nat. Prod. Bioprospect. 2021, 11, 5–13. [Google Scholar] [CrossRef]
- Cragg, G.M.; Pezzuto, J.M. Natural Products as a Vital Source for the Discovery of Cancer Chemotherapeutic and Chemopreventive Agents. Med. Princ. Pract. 2015, 25, 41–59. [Google Scholar] [CrossRef]
- Ahmadi, A.; Khalili, M.; Salimi, M.; Mirsistani, N.; Niksirat, A.; Nazirzadeh, S. The Effects of Solvent Polarity on Analgesic and Anti-Inflammatory Activities of Securigera Securidaca (L.), Achillea Eriophora DC, and Portulaca Oleracea Extracts. Pharm. Chem. J. 2019, 53, 248–263. [Google Scholar] [CrossRef]
- Ahmadipour, B. Securigera securidaca seed medicinal herb supplementation of diets improves pulmonary hypertensive response in broiler chickens reared at high altitude. J. Anim. Physiol. Anim. Nutr. 2018, 102, 1601–1607. [Google Scholar] [CrossRef]
- Aldal’in, H.K.; Al-Mazaideh, G.; Al-Nadaf, A.H.; Al-Rimawi, F.; Afaneh, A.T.; Marashdeh, A.; Jamhour, R.M. Phytochemical Constituents of Securigera securidaca Seed Extract Using GS-MS and HPLC. Trop. J. Nat. Prod. Res. 2020, 4, 540–544. [Google Scholar]
- Alizadeh-Fanalou, S.; Babaei, M.; Bahreini, E. Effects of Securigera Securidaca Seed Extract, Alone and in combination with Glibenclamide, on Circulating Levels of Pro-Angiogenic/Anti-Angiogenic Biomarkers in Hyperglycemic Rats. 2020. Available online: https://doi.org/10.21203/rs.3.rs-52508/v1 (accessed on 18 December 2022).
- Al Mazaideh, G.M.; Al Quran, S.A. Effect of Methanolic Extract of Securigera securidaca as Antioxidant and Antibacterial Activities. J. Pharm. Res. Int. 2020, 10–17. [Google Scholar] [CrossRef]
- Garjani, A.; Fathiazad, F.; Zakheri, A.; Akbari, N.A.; Azarmie, Y.; Fakhrjoo, A.; Andalib, S.; Maleki-Dizaji, N. The effect of total extract of Securigera securidaca L. seeds on serum lipid profiles, antioxidant status, and vascular function in hypercholesterolemic rats. J. Ethnopharmacol. 2009, 126, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Yu, H.; Ma, Q.; Shen, S.; Das, U.N. Linoleic acid suppresses colorectal cancer cell growth by inducing oxidant stress and mitochondrial dysfunction. Lipids Health Dis. 2010, 9, 106. [Google Scholar] [CrossRef]
- Amelio, I.; Cutruzzolá, F.; Antonov, A.; Agostini, M.; Melino, G. Serine and glycine metabolism in cancer. Trends Biochem. Sci. 2014, 39, 191–198. [Google Scholar] [CrossRef]
- Fukuda, T.; Ito, H.; Mukainaka, T.; Tokuda, H.; Nishino, H.; Yoshida, T. Anti-tumor Promoting Effect of Glycosides from Prunus persica Seeds. Biol. Pharm. Bull. 2003, 26, 271–273. [Google Scholar] [CrossRef]
- Newmark, J.; Brady, R.O.; Grimley, P.M.; Gal, A.E.; Waller, S.G.; Thistlethwaite, J.R. Amygdalin (Laetrile) and prunasin beta-glucosidases: Distribution in germ-free rat and in human tumor tissue. Proc. Natl. Acad. Sci. USA 1981, 78, 6513–6516. [Google Scholar] [CrossRef]
- Kashyap, D.; Sharma, A.; Tuli, H.S.; Sak, K.; Punia, S.; Mukherjee, T.K. Kaempferol—A dietary anticancer molecule with multiple mechanisms of action: Recent trends and advancements. J. Funct. Foods 2017, 30, 203–219. [Google Scholar] [CrossRef]
- Arya, P.; Kumar, P. Diosgenin a steroidal compound: An emerging way to cancer management. J. Food Biochem. 2021, 45, e14005. [Google Scholar] [CrossRef] [PubMed]
- El Sayed, A.M.; Basam, S.M.; El-Naggar, E.-M.B.A.; Marzouk, H.S.; El-Hawary, S. LC–MS/MS and GC–MS profiling as well as the antimicrobial effect of leaves of selected Yucca species introduced to Egypt. Sci. Rep. 2020, 10, 1–15. [Google Scholar] [CrossRef]
- Meng, X.-Y.; Zhang, H.-X.; Mezei, M.; Cui, M. Molecular Docking: A Powerful Approach for Structure-Based Drug Discovery. Curr. Comput. Aided-Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Shalayel, M.H.; Al-Mazaideh, G.M.; Swailmi, F.K.A.; Aladaileh, S.; Nour, S.; Afaneh, A.T.; Marashdeh, A. Molecular docking evaluation of syzygium aromaticum isolated compounds against Exo-β-(1, 3)-glucanases of Candida albicans. J. Pharm. Res. Int. 2021, 32, 34–44. [Google Scholar] [CrossRef]
- Shalayel, M.H.; Al-Mazaideh, G.M.; Aladaileh, S.H.; Al-Swailmi, F.K.; Al-Thiabat, M.G. Vitamin D is a potential inhibitor of COVID-19: In silico molecular docking to the binding site of SARS-CoV-2 endoribonuclease Nsp15. Pak. J. Pharm. Sci. 2020, 33. [Google Scholar] [CrossRef]
- GC-MS/Phytolab-0322/2008; National Organization for Drug Control and Research (NODCAR): Cairo, Egypt, 2008.
- Rodriguez-Garcia, M.; Surman, A.J.; Cooper, G.J.; Suárez-Marina, I.; Hosni, Z.; Lee, M.P.; Cronin, L. Formation of oligopeptides in high yield under simple programmable conditions. Nat. Commun. 2015, 6, 8385. [Google Scholar] [CrossRef] [PubMed]
- Bolarinwa, I.F.; Orfila, C.; Morgan, M.R. Amygdalin content of seeds, kernels and food products commercially—Available in the UK. Food Chem. 2014, 152, 133–139. [Google Scholar] [CrossRef]
- Du, X.; Li, Y.; Xia, Y.-L.; Ai, S.-M.; Liang, J.; Sang, P.; Ji, X.-L.; Liu, S.-Q. Insights into protein–ligand interactions: Mechanisms, models, and methods. Int. J. Mol. Sci. 2016, 17, 144. [Google Scholar] [CrossRef]
- Holderbach, S.; Adam, L.; Jayaram, B.; Wade, R.C.; Mukherjee, G. RASPD+: Fast Protein-Ligand Binding Free Energy Prediction Using Simplified Physicochemical Features. Front. Mol. Biosci. 2020, 7. [Google Scholar] [CrossRef] [PubMed]
- Kopustinskiene, D.M.; Jakstas, V.; Savickas, A.; Bernatoniene, J. Flavonoids as anticancer agents. Nutrients 2020, 12, 457. [Google Scholar] [CrossRef]
- Stabrauskiene, J.; Kopustinskiene, D.M.; Lazauskas, R.; Bernatoniene, J. Naringin and Naringenin: Their Mechanisms of Action and the Potential Anticancer Activities. Biomedicines 2022, 10, 1686. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.-L.; Tian, Y.-Q.; Wu, J.-L.; Li, N.; Guo, M.-Q. Antiproliferative activities of Amaryllidaceae alkaloids from Lycoris radiata targeting DNA topoisomerase I. Sci. Rep. 2016, 6, 38284. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Leeson, P.D.; Springthorpe, B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discov. 2007, 6, 881–890. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. 1. A Qualitative and Quantitative Characterization of Known Drug Databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Segall, M. Rules for drug discovery: Can simple property criteria help you to find a drug? Drug Discov. 2014, 15, 19–24. [Google Scholar]
- Plinski, E.F.; Plinska, S. Veber’s Rules in Terahertz Light. 2020. Available online: https://www.researchsquare.com/article/rs-12857/v1 (accessed on 18 December 2022).
- Craciun, D.; Modra, D.; Isvoran, A. ADME-Tox profiles of some food additives and pesticides. In Proceedings of the AIP Conference Proceedings; AIP Publishing: Melville, NY, USA, 2015; Volume 1694. [Google Scholar] [CrossRef]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of Drug Absorption Using Multivariate Statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef] [PubMed]
- Vélez, L.A.; Delgado, Y.; Ferrer-Acosta, Y.; Suárez-Arroyo, I.J.; Rodríguez, P.; Pérez, D. Theoretical Prediction of Gastrointestinal Absorption of Phytochemicals. Int. J. Plant Biol. 2022, 13, 163–179. [Google Scholar] [CrossRef]
- Muegge, I. Selection criteria for drug-like compounds. Med. Res. Rev. 2003, 23, 302–321. [Google Scholar] [CrossRef]
- Ishak, S.N.H.; Aris, S.N.A.M.; Halim, K.B.A.; Ali, M.S.M.; Leow, T.C.; Kamarudin, N.H.A.; Masomian, M.; Rahman, R.N.Z.R.A. Molecular Dynamic Simulation of Space and Earth-Grown Crystal Structures of Thermostable T1 Lipase Geobacillus zalihae Revealed a Better Structure. Molecules 2017, 22, 1574. [Google Scholar] [CrossRef]
- Barazorda-Ccahuana, H.L.; Valencia, D.E.; Aguilar-Pineda, J.A.; Gómez, B. Art v 4 Protein Structure as a Representative Template for Allergen Profilins: Homology Modeling and Molecular Dynamics. ACS Omega 2018, 3, 17254–17260. [Google Scholar] [CrossRef]
- Boroujeni, M.B.; Dastjerdeh, M.S.; Shokrgozar, M.; Rahimi, H.; Omidinia, E. Computational driven molecular dynamics simulation of keratinocyte growth factor behavior at different pH conditions. Inf. Med. Unlocked 2021, 23. [Google Scholar] [CrossRef]
- Poli, G.; Granchi, C.; Rizzolio, F.; Tuccinardi, T. Application of MM-PBSA Methods in Virtual Screening. Molecules 2020, 25, 1971. [Google Scholar] [CrossRef]
- Wang, C.; Greene, D.; Xiao, L.; Qi, R.; Luo, R. Recent Developments and Applications of the MMPBSA Method. Front. Mol. Biosci. 2018, 4, 87. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Nguyen, P.H.; Pham, K.; Huynh, D.; Le, T.-B.N.; Wang, H.; Ren, P.; Luo, R. Calculating protein-ligand binding affinities with MMPBSA: Method and error analysis. J. Comput. Chem. 2016, 37, 2436–2446. [Google Scholar] [CrossRef]
- Rühle, V. Pressure coupling/barostats. J. Club 2008, 1–5. [Google Scholar]
- Andreotti, C.; Ravaglia, D.; Ragaini, A.; Costa, G. Phenolic compounds in peach (Prunus persica) cultivars at harvest and during fruit maturation. Ann. Appl. Biol. 2008, 153, 11–23. [Google Scholar] [CrossRef]
- Wood, D.; Korolchuk, S.; Tatum, N.; Wang, L.-Z.; Endicott, J.; Noble, M.E.; Martin, M.P. Differences in the Conformational Energy Landscape of CDK1 and CDK2 Suggest a Mechanism for Achieving Selective CDK Inhibition. Cell Chem. Biol. 2018, 26, 121–130.e5. [Google Scholar] [CrossRef] [PubMed]
- Westbrook, J.; Feng, Z.; Chen, L.; Yang, H.; Berman, H.M. The Protein Data Bank and structural genomics. Nucleic Acids Res. 2003, 31, 489–491. [Google Scholar] [CrossRef]
- Kumar, S.K.; LaPlant, B.; Chng, W.J.; Zonder, J.; Callander, N.; Fonseca, R.; Fruth, B.; Roy, V.; Erlichman, C.; Stewart, A.K. Dinaciclib, a novel CDK inhibitor, demonstrates encouraging single-agent activity in patients with relapsed multiple myeloma. Blood 2015, 125, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Saqub, H.; Proetsch-Gugerbauer, H.; Bezrookove, V.; Nosrati, M.; Vaquero, E.M.; de Semir, D.; Ice, R.J.; McAllister, S.; Soroceanu, L.; Kashani-Sabet, M.; et al. Dinaciclib, a cyclin-dependent kinase inhibitor, suppresses cholangiocarcinoma growth by targeting CDK2/5/9. Sci. Rep. 2020, 10, 18489. [Google Scholar] [CrossRef]
- Systèmes, D. BIOVIA, Discovery Studio Visualizer, Release 2019; Dassault Systèmes: San Diego, CA, USA, 2020. [Google Scholar]
- Land, H.; Humble, M.S. YASARA: A Tool to Obtain Structural Guidance in Biocatalytic Investigations. In Protein Engineering; Springer: Berlin, Germany, 2018; pp. 43–67. [Google Scholar] [CrossRef]
- Gordon, J.C.; Myers, J.B.; Folta, T.; Shoja, V.; Heath, L.S.; Onufriev, A. H++: A server for estimating p K as and adding missing hydrogens to macromolecules. Nucleic Acids Res. 2005, 33, W368–W371. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Forli, S.; Olson, A.J. A Force Field with Discrete Displaceable Waters and Desolvation Entropy for Hydrated Ligand Docking. J. Med. Chem. 2011, 55, 623–638. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Valerio, L.G., Jr. In silico toxicology for the pharmaceutical sciences. Toxicol. Appl. Pharm. 2009, 241, 356–370. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- Schmid, N.; Eichenberger, A.P.; Choutko, A.; Riniker, S.; Winger, M.; Mark, A.; Van Gunsteren, W.F. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur. Biophys. J. 2011, 40, 843–856. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; Van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Petersen, H.G. Accuracy and efficiency of the particle mesh Ewald method. J. Chem. Phys. 1995, 103, 3668–3679. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.; Fraaije, J.G. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. Accounts Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Open Source Drug Discovery Consortium; Lynn, A. g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Grover, S.; Tyagi, C.; Goyal, S.; Jamal, S.; Singh, A.; Grover, A. Hydrophobic Interactions Are a Key to MDM2 Inhibition by Polyphenols as Revealed by Molecular Dynamics Simulations and MM/PBSA Free Energy Calculations. PLoS ONE 2016, 11, e0149014. [Google Scholar] [CrossRef] [PubMed]
- Homeyer, N.; Gohlke, H. Free Energy Calculations by the Molecular Mechanics Poisson−Boltzmann Surface Area Method. Mol. Inf. 2012, 31, 114–122. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).