Abstract
Coxibs are a group of non-steroidal anti-inflammatory drugs (NSAIDs), selective cyclooxygenase 2 inhibitors, characterized by a much lower gastrotoxicity compared to classic NSAIDs. They are often used in conjunction with other drugs, which greatly increases the likelihood of adverse drug interactions. The presented study analyzed the degradation rate of celecoxib and cimicoxib in solutions under the influence of other medicinal substances at different temperatures. For this purpose, triple-drug mixtures were prepared, consisting of coxib and eleven different commonly used drugs (paracetamol, ketoprofen, diclofenac, acetylsalicylic acid, ibuprofen, meloxicam, tramadol, doxycycline, bisoprolol, and caffeine). Then, the mixtures were incubated at two temperatures. Within the time specified by the research plan, further aliquots of the mixtures were subjected to a chromatographic analysis. Separation was conducted on HPTLC F254 silica gel chromatographic plates as a stationary phase, using chloroform: acetone: toluene as a mobile phase, and was detected densitometrically at wavelengths of 254 nm. The percentage changes in the tested coxibs content, depending on the time and conditions of incubation, were presented. Based on the obtained data, the basic kinetic parameters of the degradation processes were determined. The celecoxib and cimicoxib showed a relatively high durability in changing environmental conditions. It was observed that the rate of decomposition of cimicoxib and celecoxib in the tested mixtures was different and depended on the temperature and presence of other components, with cimicoxib turning out to be a more stable compound.
1. Introduction
Coxibs are a group of medicinal substances classified as non-steroidal anti-inflammatory drugs (NSAIDs). The mechanism of their action is based on the selective inhibition of type 2 cyclooxygenase (COX-2). They show a 200 times greater affinity for COX-2 than for COX-1. Clinical trials have shown that these drugs cause fewer gastrointestinal side effects. It has also been proven that, when using coxib and diclofenac, the risk of gastrointestinal bleeding is even 2–3 times lower than that when using ibuprofen or naproxen. However, existing problems with cardiovascular disorders, in particular heart attacks and strokes, led to the withdrawal of rofecoxib from the market. In addition, there are also strict guidelines for when selective COX-2 inhibitors can be taken by humans [1,2,3,4]. Currently, the most commonly used drugs in this group are celecoxib and etoricoxib. Celecoxib has anti-inflammatory, analgesic, and antipyretic properties. It has also been found to inhibit the growth of adenomas in the large intestine. It is used in rheumatoid arthritis, osteoarthritis, and as an analgesic, most often together with glucocorticoids (e.g., prednisolone), biological drugs modifying the course of a degenerative disease (e.g., infliximab), or strong painkillers (e.g., tramadol) [5,6,7,8]. In recent years, many new, selective COX-2 inhibitors have been introduced in veterinary medicine, including: deracoxib (2002), firocoxib (2007), mavacoxib (2008), robenacoxib (2009), and cimicoxib (2011). The last and newest of these drugs is cimicoxib, which is intended for use in dogs to treat the pain and inflammation associated with osteoarthritis. It is also used to treat perioperative pain after surgery, and to treat soft tissue pain [9,10,11,12,13,14,15,16,17]. Currently, many clinical trials are being conducted with coxibs in terms of evaluating their anti-cancer activity. It has been shown that their use is associated with a reduction in the incidence of episodes of cancer recurrence. In addition, anti-inflammatory substances may be used as adjuvants in conventional therapies [18,19,20,21,22,23].
Multi-drug therapy means treating one or more diseases with different drugs at the same time. Whenever the active ingredients are used together, they may react with each other. A very large number of the latest treatment regimens are based on a multi-drug system, and achieving the therapeutic goal is possible only due to the complementary mechanisms of action. This includes, e.g., blood-pressure-lowering drugs, antimicrobials, and anticancer drugs. Many more often interactions are associated by society as a side effect of therapy, i.e., undesirable interactions [24,25]. Problems associated with inappropriate use of medications are a common cause of hospitalization. In a meta-analysis, Beijer et al. wanted to determine what percentage of adverse drug reactions were associated with hospitalization. Data were collected for a year, and the study included 123,794 hospital stays, 6071 of which were drug-related, which accounted for 4.9% of the respondents [26]. A study conducted in the Netherlands analyzed data from a computer system for all hospitalizations during the year. It found that 1.8% of emergency hospital admissions were medication-related, especially with regard to anticoagulants, cytotoxics, immunosuppressants, and diuretics [27].
According to the literature, interactions are divided into: pharmaceutical, pharmacodynamic, pharmacokinetic, and mixed, based on their mechanism of formation. All kinds of incompatibilities, as well as physicochemical phenomena, are perceived as interactions in the pharmaceutical phase. Pharmacodynamic interactions arise from the combined action of drugs on the same receptor or organ, and may occur at various stages of LADME (Liberation, Absorption, Distribution, Metabolism, and Elimination) drug transformation. Drugs can affect the pH value in the body, causing a different rate of absorption of molecules. Active substances compete with each other to bind to the transporter, protein affinity, or the availability of a metabolizing enzyme. When several drugs are used simultaneously, the molecules can modify themselves in many ways that are difficult to define. Possible chemical interactions with other substances (derived from drugs or dietary supplements) are not comprehensively investigated during the registration of drugs. Safety and efficacy are key aspects of drug research and development; therefore, formulations must be designed to ensure an adequate physicochemical stability over their recommended shelf life [28].
Standard stability studies do not include analyses of drug behavior in combination with others, unless they are combination preparations containing several active substances. Therefore, addressing this issue seems to be an important aspect of further research. Maswadeh described a study of the effects of the combined use of paracetamol with a cefuroxime axetil suspension as the combination of choice for pediatric patients. An infrared spectroscopy analysis showed the influence of both paracetamol and the excipients contained in the syrup on the absorption spectrum of the antibiotic. It was found that the oral administration of both drugs together may affect the physicochemical properties, dissolution rate, solubility, absorption, and bioavailability of one or both drugs [29]. Other studies have concerned the degradation process of the drug in a combined preparation containing paracetamol and lornoxicam, indicating a significant decrease in the concentration of lornoxicam in an acidic environment [30]. Other scientists have analyzed the rate of degradation of paracetamol and ascorbic acid in a mixture of a ready-made preparation, a mixture prepared ex tempore from ingredients and individual substances. The greatest durability was shown for the mixture of the commercial product, probably due to the used preservatives [31].
Another major challenge is the problem of interaction and the influence of excipients on the degradation process of the active substance. This is determined at the formulation stage by the method of accelerated degradation. Unfortunately, the effect of an excipient from one preparation on another is unpredictable, and in many cases, may lead to a reduction in its stability. One example is the reaction of drugs with lactose, which forces some active substances into a lactose-free form. The authors showed that the main degradation product was amlodipine besylate glycosyl. Other examples of drugs that react similarly include fluoxetine, cetirizine, olanzapine, pregabalin, acyclovir, and hydrochlorothiazide [32,33,34]. The process of the increased degradation of duloxetine via hydroxypropyl methylcellulose used as a wetting agent in tablet casings and coatings is enhanced by an increased humidity and temperature [35]. Serajuddin et al. reported that sodium fosinopril forms three degradation products when tablets are coated with magnesium stearate as a lubricant. They observed two different degradation pathways, in which magnesium ion mediated the hydrolysis [36]. The phenomenon of adduct formation and simultaneous reduction in drug concentration in an oral solution of cherry-flavored famotidine were also investigated [37]. Dousa et al. examined various pharmaceutical preparations containing phenylephrine and sucrose, finding a loss of phenylephrine activity and the formation of new degradation products from the condensation of phenylephrine with aldehydes [34]. After hydrolysis, oxidation is the second most common way of pharmaceutical degradation. For example, the degradation of buprenorphine in the presence of citric acid, found in buprenorphine and naloxone sublingual tablets, has been confirmed [38,39]. In clinical practice, mixtures of drugs are often used, e.g., in infusion pumps, which allows, among others, for shortening the administration time and number of administered preparations. The most important aspect here is the stability and resistance to degradation of the individual components [40]. In one such study, scientists determined the physicochemical stability of different volumes of an infusion mixture of fosaprepitant, dexamethasone, and ondansetron or granisteron prepared ex tempore, used as an antiemetic in the premedication of chemotherapy [41]. The stability of morphine, midazolam, and levomepromazine at different doses in one infusion bag of different capacities, subjected to varying changing conditions of temperature and UV radiation, was also investigated. It was shown that levomepromazine decomposed fastest in a 0.9% NaCl solution, and the storage of morphine at 4 °C led to its precipitation [42]. Espinosa-Boch et al. evaluated the stability of mixtures of haloperidol and ondansetron stored at various temperatures, confirming their stability up to 48 h after preparation [43].
Thus, as indicated in the literature, many factors may alter the active substances in pharmaceutical formulations when used together. For this reason, it is justified to conduct continuous research in the discussed area in order to create a reliable map of possible drug interactions. The present study aimed to analyze the behavior of celecoxib and cimicoxib in mixtures. In addition to the selected coxibs, the triple mixtures included the most commonly used drugs from various therapeutic groups, i.e., antibiotics, other analgesics and anti-inflammatory drugs, antihistamines, and purine alkaloids. The research concerns an analysis of the degradation rates of celecoxib and cimicoxib in solutions under the influence of co-existing medicinal substances at various temperatures. The quantification of the coxibs in the presence of the other components was performed using thin-layer chromatography (TLC) with densitometric detection. To conduct the research, apart from celecoxib (CEL) and cimicoxib (CIM), other painkillers belonging to the NSAID group were also selected, such as: paracetamol (PAR), ketoprofen (KET), diclofenac (DIC), acetylsalicylic acid (ASA), ibuprofen (IBU), and meloxicam (MEL). Another drug included in the study was tramadol (TRA), which belongs to the group of opioid painkillers, doxycycline (DOX), an antibiotic from the tetracycline group, bisoprolol (BIS), from the β-blocker group, which is most often used in hypertension, and loratadine (LOR), a long-acting antihistamine used for the treatment of allergies. The last, most popular, and commonly used substance was caffeine (CAF), which is found in combination with other drugs, as well as in beverages such as coffee and tea. The developed methodology may be useful for studying the expected drug interactions between coxibs and other drugs commonly used by people treated for pain and/or rheumatoid diseases. The stability of the active substances used at the same time may affect the required dose of the drug, weakening or intensifying its effect or causing the formation, among other things, of harmful products.
2. Materials and Methods
2.1. Chemicals and Apparatus
Methanol, chloroform, acetone, toluene, and other organic solvents of an analytical grade were purchased from Merck (Darmstadt, Germany). A Densitometer TLC Scanner 3 with Cat4 software (CAMAG, Muttenz, Switzerland), applicator Linomat V (CAMAG, Switzerland), and analytical balance XA 52/Y (Radwag, Radom, Poland) were used. Chromatographic plates HPTLC Silica gel 60F254 (No. 1.05548) were purchased from Merck (Darmstadt, Germany).
The standard substance of caffeine was purchased from Sigma-Aldrich (Darmstadt, Germany). The analyzed pharmaceutical preparations were: Celebrex (Pfizer Europe, Tadworth, UK) containing 200 mg of celecoxib, Cimalgex (Vétoquinol SA, Lure, France) containing 80 mg of cimicoxib, Mobic (Boehringer Ingelheim, Rhein, Germany) containing 7.5 mg of meloxicam, Olfen UNO (Ratiopharm GmbH, Ulm, Germany) containing 150 mg of diclofenac, APAP (US Pharmacia, Poland) containing 500 mg of paracetamol, Bibloc (SANDOZ, Warszawa, Poland) containing 5 mg of bisoprolol, Poltram (Polpharma, Gdansk, Poland) containing 50 mg of tramadol, Clatrine (MSD, Warszawa, Poland) containing 10 mg of loratadine, Doxycyclinum TZF (Polfa, Warszawa, Poland) containing 100 mg of doxycycline, Ketonal Fotre (Sandoz, Langkampfen, Austria) containing 100 mg of ketoprofen, Acard (Polfa, Poland) containing 300 mg of acetylsalicylic acid, and Ibuprofen Pabi (Adamed, Pieńków, Poland) containing 200 mg of ibuprofen. All the preparations were purchased from the local pharmacy or veterinary office. The preparations had an expiry date of not less than one year at the time of the study.
2.2. Solutions for Analysis
Solutions of the analyzed compounds were prepared by pulverizing and weighing out a tablet mass corresponding to 10 mg of the active ingredient. Methanolic solutions of each drug were prepared in volumetric flasks by dissolving a sample in 100 mL of the solvent. The concentrations of each of the active substances were about 0.01% (w/v).
2.3. Ternary Mixtures Solutions
Ternary mixtures were prepared for analysis so that each mixture contained celecoxib or cimicoxib and two other drugs. Additionally, the remaining substances were mixed on a peer-to-peer basis. A total of 54 combinations were prepared, as shown in Table 1. A mixture of celecoxib + ibuprofen + ketoprofen was not included, due to it being the least likely composition of all the proposed combinations. All the other combinations are likely to be used in human or animal clinical practice. The mixtures were prepared in a 1:1:1 volume ratio by mixing the appropriate solutions of the tested substances in 2 mL vials. The sealed vials were placed in the incubator at 25 °C or 70 °C, and in the dark (control). During the time specified in the research plan, successive portions of the mixtures were collected and subjected to a chromatographic analysis.

Table 1.
Ternary mixtures ingested for analysis of celecoxib and cimicoxib.
2.4. Chromatographic Conditions
The method, previously developed by our team and fully validated, was used to determine the contents of celecoxib and cimicoxib [44]. Chromatography was performed on 10 × 10 cm aluminum sheets precoated with silica gel 60F254. The samples were applied to the plates as bands (5 mm wide, 10 mm apart) by a Linomat V sample applicator equipped with a 100 μL syringe (Hamilton, Bonaduz, Switzerland) with an application rate of 600 nL/s. The first application was 10 mm from the bottom and 10 mm from the left edge of the plate. The volume of the applied mixture was 30 µL. The plates were taken into a chromatographic chamber (18 × 16 × 8 cm; Sigma-Aldrich), previously saturated with mobile phase vapor for 20 min at room temperature. Well-developed peaks were obtained with a mobile phase containing chloroform: acetone: toluene (12:5:2, v/v/v). The development distance was 10 cm in 20 min. After the development, the plates were dried at room temperature for about 20 min. Densitometric detection was performed using a TLC Scanner3 with winCats4 software (CAMAG, Muttenz, Switzerland). The source of radiation was a deuterium lamp emitting a continuous UV spectrum between 200 and 400 nm. The scanning speed was 20 mm/s and the slit dimensions were 4.00 × 0.45 mm. Based on the obtained absorption spectra, an analytical wavelength of 254 nm was selected for the measurements.
3. Results
The mixtures, prepared as described above, were subjected to a chromatographic analysis under the developed and validated TLC procedure with densitometric detection [44]. Under the above conditions, the correct separation of the analyzed mixtures was found. The following retardation factors RF were obtained: CEL 0.73, CIM 0.25, PAR 0.39, KET 0.29, DIC 0.20, ASA 0.16, IBU 0.46, MEL 0.31, TRA 0.11, DOX 0.08, BIS 0.41, and LOR 0.40. Under the described conditions, the required sensitivity and symmetrical peaks were obtained, which allows for the use of this procedure for the analysis of the substances included in the test plan.
During the analysis, changes in the peak area values were observed, which were recorded using a densitometer at a wavelength of 254 nm. The obtained densitograms were analyzed for changes in the contents of celecoxib and cimicoxib in the sample (Figure 1 and Figure 2). The variable value of the peak area corresponding to the tested compound was monitored and then converted into a percentage of the content of a given coxib, relative to the original sample (immediately after the preparation of the solution). In a few cases, it was noticed that, in the recorded densitograms, apart from the three main compounds of a given mixture, there were also other additional peaks. This may have indicated the ongoing degradation process of the mixture components, and the formation of new chemical compounds with unknown properties. The following tables show the percentage changes in the active substances, CEL (Table 2, Table 3, Table 4, Table 5, Table 6 and Table 7) and CIM (Table 8 and Table 9), in each ternary mixture, taking into account the various temperatures and sample incubation times.
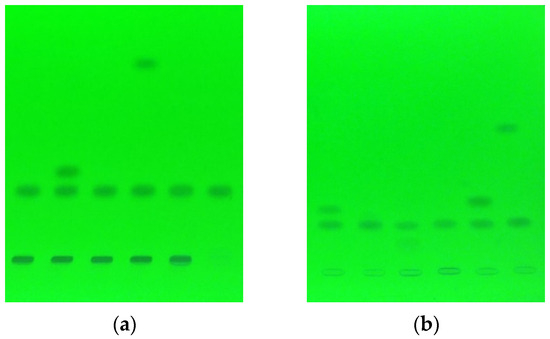
Figure 1.
An example of chromatograms for analysis of mixtures containing cimicoxib at temperature 25 °C after 42 days of incubation (a), and at 70 °C after 21 days (b).
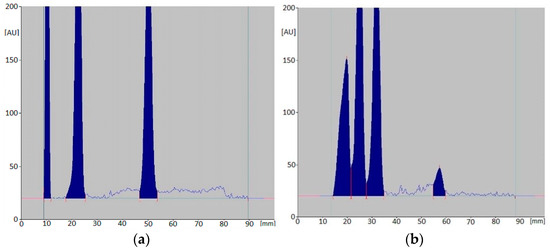
Figure 2.
An example of densitograms for mixtures: CIM, DOX, LOR (a) and CIM, CAF, LOR (b) after 56-day incubation at 25 °C.
Table 2.
Celecoxib content (%) depending on the incubation time of the mixtures (series 1) at 25 °C.
Table 3.
Celecoxib content (%) depending on the incubation time of the mixtures (series 2) at 25 °C.
Table 4.
Celecoxib content (%) depending on the incubation time of the mixtures (series 3) at 25 °C.
Table 5.
Celecoxib content (%) depending on the incubation time of the mixtures (series 1) at 70 °C.
Table 6.
Celecoxib content (%) depending on the incubation time of the mixtures (series 2) at 70 °C.
Table 7.
Celecoxib content (%) depending on the incubation time of the mixtures (series 3) at 70 °C.
Table 8.
Cimicoxib content (%) depending on the incubation time of the mixtures at 25 °C.
Table 9.
Cimicoxib content (%) depending on the incubation time of the mixtures at 70 °C.
Based on the results for the changes in the contents of the analyzed coxibs over time, calculations were made to determine the basic kinetic parameters of the ongoing degradation processes. First, the order of the reaction for each of the tested samples with the coxibs was graphically determined (Figure 3). The obtained data allowed us to determine the rate of the reaction proceeding in accordance with the first-order kinetics.
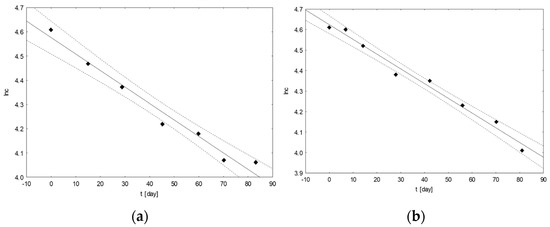
Figure 3.
An example of graphs of the ln(c[%]) = f(t[days]) dependence for mixtures of drugs with the following composition: CEL, CAF, ASA (a) and CIM, TRA, KET (b), incubated at 25 °C.
After determining the order of the reaction for each mixture at both temperatures, the basic kinetic parameters were calculated, such as: the reaction rate k and the time t0.5 and t0.1 (the period after which 50% and 10% of the original substance was degraded), using the formulas developed for the appropriate order of the reaction. The obtained results, presented in Table 10, Table 11, Table 12 and Table 13, were used to compare the durability of the tested mixtures, depending on their composition and temperature.
Table 10.
Calculated kinetic parameters for tested multi-drug mixtures containing celecoxib (series 1) stored at 25 °C and 70 °C.
Table 11.
Calculated kinetic parameters for tested multi-drug mixtures containing celecoxib (series 2) stored at 25 °C and 70 °C.
Table 12.
Calculated kinetic parameters for tested multi-drug mixtures containing celecoxib (series 3) stored at 25 °C and 70 °C.
Table 13.
Calculated kinetic parameters for tested multi-drug mixtures containing cimicoxib stored at 25 °C and 70 °C.
4. Discussion
Drug interactions are a fairly common problem, and are often responsible for the significant morbidity and mortality of patients [45,46]. For several generations, researchers have been studying the combined effects of drugs. A growing database indicates that drug interactions are a major contributor to treatment failure or avoidable medical complications. Methods for data analysis have changed over the years, but the underlying problem has not changed [47]. With clinicians’ growing understanding of the importance of drug interactions, the space for analysts who adequately analyze this problem within various medical disciplines is increasing [48,49]. Both doctors and patients can obtain relevant information about drug interactions using appropriate sources, e.g., on the internet (available publications or databases) [50].
Two of the few active substances from the coxib group were selected for the discussed study due to their fairly common use in medicine. Celecoxib is a drug used to treat osteoarthritis, rheumatoid arthritis, and ankylosing spondylitis. According to the indications, the decision to prescribe a selective COX-2 inhibitor should be made by a doctor, based on an assessment of the individual risks that may occur in a particular patient, whereas cimicoxib is the youngest representative of this group of drugs, whose properties have not yet been fully understood. It is used to treat the pain and inflammation associated with osteoarthritis in dogs. However, longer treatment requires the regular control of analytical parameters by a veterinarian. Drug stability can reduce effectiveness, increase unexpected side effects, and even increase the possible toxicity of a given drug, its metabolites, or degradation products. The analyzed coxibs can be used for both acute and chronic pain in the course of musculoskeletal disorders, which increases the possibility of potential treatment.
Due to the use of cimicoxib in veterinary medicine, only those mixtures that can be used in clinical veterinary practice were selected for further analysis. The mixtures with paracetamol, diclofenac, ibuprofen, acetylsalicylic acid, and caffeine were omitted, as the use of these drugs in animals is prohibited. The listed substances are highly toxic or cause frequent side effects in a large number of animal species, which disqualifies them from being widely used [17,51,52]. Therefore, only 14 mixtures of cimicoxib in mixtures with other drugs were developed for further research (Table 1). An analysis of the remaining cimicoxib-containing mixtures was also performed, however, for the reasons mentioned above, the results are presented as Supplementary Data. Under the conditions of the stability testing of the mixtures containing celecoxib and cimicoxib carried out in parallel, the samples were stored at the specified temperature in dark glass vials. After the maximum analysis time (at a higher temperature), there were no decreases in the contents of the analyzed substances to below 60% for celecoxib and 70% for cimicoxib.
Based on the above parameters, the degree of degradation of the tested coxibs in the tested triple-drug mixtures was determined. After analyzing the obtained results and calculated parameters, it can be seen that the degradation rates of celecoxib and cimicoxib in the tested mixtures varied depending on the composition of the mixture, but the process was always faster at elevated temperatures. The degradation rate of the tested drug substances increased (k increased), while the values of t0.5 and t0.1 decreased.
Comparing the kinetic parameters for all the mixtures at the temperature of 25 °C, celecoxib showed the greatest stability in the mixture with paracetamol and bisoprolol (t0.5 = 3807.7 h; k = 1.82 × 10−4 h−1). The next mixtures in which celecoxib was the most stable were those with paracetamol and diclofenac (t0.5 = 3447.8 h; k = 2.01 × 10−4 h−1) and with ketoprofen and loratadine (t0.5 = 3413.8 h; k = 2.03 × 10−4 h−1), while celecoxib showed the lowest stability at this temperature in the mixtures with doxycycline and loratadine (t0.5 = 1750.0 h; k = 3.96 × 10−4 h−1) and doxycycline and acetylsalicylic acid (t0.5 = 1838.2 h; k = 3.77 × 10−4 h−1). The other mixtures in which celecoxib was not very stable were those additionally containing paracetamol and ketoprofen (t0.5 = 1985.7 h; k = 3.49 × 10−4 h−1), as well as tramadol and loratadine (t0.5 = 2002.9 h; k = 3.46 × 10−4 h−1). A similar analysis of the results obtained for 70 °C indicated that celecoxib was the most stable in the mixtures with ketoprofen and diclofenac (t0.5 = 3193.6 h; k = 2.17 × 10−4 h−1) and ketoprofen and loratadine (t0.5 = 3164.4 h; k = 2.19 × 10−4 h−1). The next most stable mixtures of celecoxib were those with loratadine and acetylsalicylic acid (t0.5 = 2557.2 h; k = 2.71 × 10−4 h−1), as well as paracetamol and diclofenac (t0.5 = 2341.2 h; k = 2.96 × 10−4 h−1). Based on these parameters, it can be concluded that celecoxib was the least stable at this temperature in the mixtures with diclofenac and loratadine (t0.5 = 513.3 h; k = 1.35 × 10−3 h−1) and doxycycline with bisoprolol (t0.5 = 587.3 h; k = 1.18 × 10−3 h−1).
Similarly, comparing the results for the mixtures containing cimicoxib stored at 25 °C, it was found that the drug was the most stable in the presence of doxycycline and bisoprolol (t0.5 = 9058.8 h; k = 7.65 × 10−5 h−1) and tramadol (t0.5 = 9480.2 h; k = 7.31 × 10−5 h−1). This drug was also stable in the presence of doxycycline and loratadine (t0.5 = 6187.5 h; k = 1.12 × 10−4 h−1). In turn, the lowest durability of cimicoxib was found in the presence of ketoprofen and loratadine (t0.5 = 2132.3 h; k = 3.25 × 10−4 h−1) and meloxicam and loratadine (t0.5 = 2172.4 h; k = 3.19 × 10−4 h−1). The results obtained for triple-drug mixtures incubated at 70 °C were subjected to a similar analysis. Under the described conditions, cimicoxib showed the greatest stability in the presence of doxycycline and loratadine (t0.5 = 3609.4 h; k = 1.92 × 10−4 h−1), as well as tramadol and loratadine (t0.5 = 3026.2 h; k = 2.29 × 10−4 h−1). The lowest persistence was found in the mixture with doxycycline and bisoprolol (t0.5 = 845.1 h; k = 8.20 × 10−4 h−1), as well as in combination with tramadol and bisoprolol (t0.5 = 940.3 h; k = 7.37 × 10−4 h−1).
To sum up, it can be concluded that both drugs were characterized by a high stability under the tested stress conditions. At the same time, cimicoxib turned out to be a more stable substance, both as a result of the action of the other chemical compounds and the temperature. In a comparable incubation time of the mixtures, the lowest concentration of this drug was 20.31%, while that of celecoxib was 9.00%. The stability of cimicoxib was mainly affected by doxycycline and bisoprolol (approximately 20% remained). Similarly, for celecoxib, the greatest degradation (over 90% at 70 °C) was observed in the triple-drug mixtures containing diclofenac, loratadine, doxycycline, and bisoprolol. On the other hand, the highest concentrations of cimicoxib (>65% at 70 °C) were found in the presence of doxycycline and loratadine, celecoxib (almost 68% at 70 °C) in the presence of ketoprofen and diclofenac.
5. Conclusions
While drug–drug interactions are only a small fraction of drug-related side effects, they are important, often predictable, and therefore avoidable. The variable rate of degradation may be influenced by the presence of another drug substance or other excipient incompatible with a component of the mixture. The research results for the two coxibs presented in the paper indicated their relatively high stability in changing environmental conditions. However, as can be seen from the obtained data, the changes in the degradation rates and stability of the studied coxibs were very diverse depending on the accompanying medicinal substances. The kinetic parameters were variable and difficult to predict. It can be concluded that the creation of multi-drug mixtures (including the patient’s consumption of many drugs at the same time) may cause unfavorable chemical changes, resulting in worse therapeutic results. The presented methodology allows for a quick and simple assessment of drug stability in possible three-drug combinations of a given substance used during treatment. Multidisciplinary education on drug handling and awareness of the possible processes in multi-drug mixtures is an important element of minimizing drug interactions, especially in cases of a large number of drugs and/or dietary supplements being taken by patients.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/pr11092605/s1, Table S1. Cimicoxib content [%] depending on the incubation time of the mixtures at 25 °C; Table S2. Cimicoxib content [%] depending on the incubation time of the mixtures at 70 °C; Table S3. Calculated kinetic parameters for tested multidrug mixtures containing cimicoxib stored at 25 °C and 70 °C.
Author Contributions
Conceptualization, P.G. and M.S.; methodology, M.S.; software, P.G.; investigation, P.G.; data curation, P.G.; writing—original draft preparation, P.G. and M.S.; writing—review and editing, M.S. and M.D.; visualization, P.G.; supervision, M.S.; funding acquisition, P.G. and M.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was financed by funds of the Research Support Module as part of the Excellence Initiative—Research University at the Jagiellonian University program, project No. U1C/W42/NO/28.06, and the project No. N42/DBS/000268, financed by Jagiellonian University Medical College.
Data Availability Statement
Data sharing is not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Arias, L.H.M.; González, A.M.; Fadrique, R.S.; Vázquez, E.S. Gastrointestinal safety of coxibs: Systematic review and meta-analysis of observational studies on selective inhibitors of cyclo-oxygenase 2. Fundam. Clin. Pharmacol. 2019, 33, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Simmons, D.L.; Botting, R.M.; Hla, T. Cyclooxygenase isozymes: The biology of prostaglandin synthesis and inhibition. Pharmacol. Rev. 2002, 56, 387–437. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lee, H.; Ko, J.W.; Kim, J.R. Effects of celecoxib on the QTc interval: A thorough QT/QTc study. Clin. Ther. 2019, 41, 2204–2218. [Google Scholar] [CrossRef] [PubMed]
- Walker, C. Are all oral COX-2 selective inhibitors the same? A consideration of celecoxib, etoricoxib, and diclofenac. Int. J. Rheumatol. 2018, 2018, 1302835. [Google Scholar] [CrossRef]
- Dooner, H.; Mundin, G.; Mersmann, S.; Bennett, C.; Lorch, U.; Encabo, M.; Escriche, M.; Encina, G.; Smith, K. Pharmacokinetics of tramadol and celecoxib in Japanese and Caucasian subjects following administration of co-crystal of tramadol-celecoxib (CTC): A randomised, open-label study. Eur. J. Drug Metab. Pharmacokinet. 2019, 44, 63–75. [Google Scholar] [CrossRef]
- Del Grossi Paglia, M.; Tolentino Silva, M.; Cruz Lopes, L.; Barberato-Filho, S.; Giustti Mazzei, L.; Casale Abe, F.; de Cássia Bergamaschi, C. Use of corticoids and non-steroidal antiinflammatories in the treatment of rheumatoid arthritis: Systematic review and network meta-analysis. PLoS ONE 2021, 16, e0248866. [Google Scholar]
- Burns, K.A.; Robbins, L.M.; LeMarr, A.R.; Childress, A.L.; Morton, D.J.; Schroer, W.C.; Wilson, M.L. Celecoxib significantly reduces opioid use after shoulder arthroplasty. J. Shoulder Elb. Surg. 2021, 30, 1–8. [Google Scholar] [CrossRef]
- Cebrecos, J.; Carlson, J.D.; Encina, G.; Lahjou, M.; Sans, A.; Sust, M.; Vaqué, A.; Morte, A.; Gascón, N.; Plata-Salamán, C. Celecoxib-tramadol co-crystal: A randomized 4-way crossover comparative bioavailability study. Clin. Ther. 2021, 43, 1051–1065. [Google Scholar] [CrossRef]
- Jeunesse, E.C.; Schneider, M.; Woehrle, F.; Faucher, M.; Lefebvre, H.P.; Toutain, P.L. Pharmacokinetic/pharmacodynamic modeling for the determination of a cimicoxib dosing regimen in the dog. BMC Vet. Res. 2013, 9, 250. [Google Scholar] [CrossRef]
- Kongara, K.; Chambers, P. Robenacoxib in the treatment of pain in cats and dogs: Safety, efficacy, and place in therapy. Vet. Med. Res. Rep. 2018, 9, 53–61. [Google Scholar] [CrossRef]
- Morris, T.H.; Paine, S.W.; Zahra, P.W.; Li, E.C.; Colgan, S.A.; Karamatic, S.L. Pharmacokinetics of carprofen and firocoxib for medication control in racing greyhounds. Aust. Vet. J. 2020, 98, 578–585. [Google Scholar] [CrossRef]
- Kim, T.W.; Łebkowska-Wieruszewska, B.; Owen, H.; Yun, H.I.; Kowalski, C.J.; Giorgi, M. Pharmacokinetic profiles of the novel COX-2 selective inhibitor cimicoxib in dogs. Vet. J. 2014, 200, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.; Lees, P.; Seewald, W.; King, J.N. Analytical determination and pharmacokinetics of robenacoxib in the dog. J. Vet. Pharmacol. Ther. 2009, 32, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Cox, S.; Villarino, N.; Sommardahl, C.; Kvaternick, V.; Zarabadipour, C.; Siger, L.; Yarbrough, J.; Amicucci, A.; Reed, K.; Breeding, D.; et al. Disposition of firocoxib in equine plasma after an oral loading dose and a multiple dose regimen. Vet. J. 2013, 198, 382–385. [Google Scholar] [CrossRef] [PubMed]
- Bergh, M.S.; Budsberg, S.C. The coxib NSAIDs: Potential clinical and pharmacologic importance in veterinary medicine. J. Vet. Intern. Med. 2005, 19, 633–643. [Google Scholar] [CrossRef]
- Bustamante, R.; Daza, M.A.; Canfrán, S.; García, P.; Suárez, M.; Trobo, I.; Gómez de Segura, I.A. Comparison of the postoperative analgesic effects of cimicoxib, buprenorphine and their combination in healthy dogs undergoing ovariohysterectomy. Vet. Anaesth. Analg. 2018, 45, 545–556. [Google Scholar] [CrossRef]
- Kukanich, B.; Bidgood, T.; Knesl, O. Clinical pharmacology of nonsteroidal anti-inflammatory drugs in dogs. Vet. Anaesth. Analg. 2012, 39, 69–90. [Google Scholar] [CrossRef]
- Hashemi Goradel, N.; Najafi, M.; Salehi, E.; Farhood, B.; Mortezaee, K. Cyclooxygenase-2 in cancer: A review. J. Cell Physiol. 2019, 234, 5683–5699. [Google Scholar] [CrossRef]
- Meyerhardt, J.A.; Shi, Q.; Fuchs, C.S.; Meyer, J.; Niedzwiecki, D.; Zemla, T.; Kumthekar, P.; Guthrie, K.A.; Couture, F.; Kuebler, P.; et al. Effect of celecoxib vs placebo added to standard adjuvant therapy on disease-free survival among patients with stage III colon cancer: The CALGB/SWOG 80702 (Alliance) randomized clinical trial. JAMA J. Am. Med. Assoc. 2021, 325, 1277–1286. [Google Scholar] [CrossRef]
- Zappavigna, S.; Cossu, A.M.; Grimaldi, A.; Bocchetti, M.; Ferraro, G.A.; Nicoletti, G.F.; Filosa, R.; Caraglia, M. Anti-inflammatory drugs as anticancer agents. Int. J. Mol. Sci. 2020, 21, 2605. [Google Scholar] [CrossRef]
- Li, S.; Jiang, M.; Wang, L.; Yu, S. Combined chemotherapy with cyclooxygenase-2 (COX-2) inhibitors in treating human cancers: Recent advancement. Biomed. Pharmacother. 2020, 129, 110389. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.-K.; Madhurapantula, S.V.; He, G.; Wang, K.-Y.; Song, C.-H.; Zhang, J.-Y.; Wang, K.-J. Genetic variant of cyclooxygenase-2 in gastric cancer: More inflammation and susceptibility. World J. Gastroenterol. 2021, 27, 4653–4666. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.; Sun, H.; Yu, F.B.; Li, B.; Zhang, Y.; Zhu, Y.T. The role of cyclooxygenase-2 in colorectal cancer. Int. J. Med. Sci. 2020, 17, 1095–1101. [Google Scholar] [CrossRef] [PubMed]
- Corsonello, A.; Abbatecola, A.M.; Fusco, S.; Luciani, F.; Marino, A.; Catalano, S.; Maggio, M.G.; Lattanzio, F. The impact of drug interactions and polypharmacy on antimicrobial therapy in the elderly. Clin. Microbiol. Infect. 2015, 21, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Arnoldo, L.; Cattani, G.; Cojutti, P.; Pea, F.; Brusaferro, S. Monitoring polypharmacy in healthcare systems through a multi-setting survey: Should we put more attention on long term care facilities? J. Public. Health Res. 2016, 5, 104–108. [Google Scholar] [CrossRef]
- Beijer, H.J.M.; de Blaey, C.J. Hospitalisations caused by adverse drug reactions (ADR): A meta-analysis of observational studies. Pharm. World Sci. 2002, 24, 46–54. [Google Scholar] [CrossRef]
- Van Der Hooft, C.S.; Sturkenboom, M.C.J.M.; Van Grootheest, K.; Kingma, H.J.; Stricker, B.H.C. Adverse drug reaction-related hospitalisations: A nationwide study in The Netherlands. Drug Saf. 2006, 29, 161–168. [Google Scholar] [CrossRef]
- Bajaj, S.; Singla, D.; Sakhuja, N. Stability testing of pharmaceutical products. J. Appl. Pharm. Sci. 2012, 2, 129–138. [Google Scholar]
- Maswadeh, H. Incompatibility of paracetamol with pediatric suspensions containing amoxicillin, azithromycin and cefuroxime axetil. Pharmacol. Pharm. 2017, 8, 355–368. [Google Scholar] [CrossRef]
- Shah, D.A.; Patel, N.J.; Baldania, S.L.; Chhalotiya, U.K.; Bhatt, K.K. Stability indicating LC-method for estimation of paracetamol and lornoxicam in combined dosage form. Sci. Pharm. 2011, 79, 113–122. [Google Scholar] [CrossRef]
- Golonka, I.; Kawacki, A.; Musial, W. Stability studies of a mixture of paracetamol and ascorbic acid, prepared extempore, at elevated temperature and humidity conditions. Trop. J. Pharm. Res. 2015, 14, 1315–1321. [Google Scholar] [CrossRef]
- Abdoh, A.; Al-Omari, M.M.; Badwan, A.A.; Jaber, A.M.Y. Amlodipine besylate-excipients interaction in solid dosage form. Pharm. Dev. Technol. 2004, 9, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Stowell, J.G.; Cao, W.; Morris, K.R.; Byrn, S.R.; Carvajal, M.T. Effect of milling and compression on the solid-state Maillard reaction. J. Pharm. Sci. 2005, 94, 2568–2580. [Google Scholar] [CrossRef] [PubMed]
- Douša, M.; Gibala, P.; Havlíček, J.; Plaček, L.; Tkadlecová, M.; Břicháč, J. Drug-excipient compatibility testing-identification and characterization of degradation products of phenylephrine in several pharmaceutical formulations against the common cold. J. Pharm. Biomed. Anal. 2011, 55, 949–956. [Google Scholar] [CrossRef]
- Jansen, P.J.; Oren, P.L.; Kemp, C.A.; Maple, S.R.; Baertschi, S.W. Characterization of impurities formed by interaction of duloxetine HCl with enteric polymers hydroxypropyl methylcellulose acetate succinate and hydroxypropyl methylcellulose phthalate. J. Pharm. Sci. 1998, 87, 81–85. [Google Scholar] [CrossRef]
- Serajuddin, A.T.M.; Thakur, A.B.; Ghoshal, R.N.; Fakes, M.G.; Ranadive, S.A.; Morris, K.R.; Varia, S.A. Selection of solid dosage form composition through drug-excipient compatibility testing. J. Pharm. Sci. 1999, 88, 696–704. [Google Scholar] [CrossRef]
- Hotha, K.K.; Patel, T.; Roychowdhury, S.; Subramanian, V. Identification, synthesis, and characterization of unknown impurity in the famotidine powder for oral suspension due to excipient interaction by UPLC-MS/MS and NMR. J. Liq. Chromatogr. Rel. Technol. 2015, 38, 977–985. [Google Scholar] [CrossRef]
- Gabrič, A.; Hodnik, Ž.; Pajk, S. Oxidation of drugs during drug product development: Problems and solutions. Pharmaceutics 2022, 14, 325. [Google Scholar] [CrossRef]
- Mostafavi, A.; Abedi, G.; Jamshidi, A.; Afzali, D.; Talebi, M. Development and validation of a HPLC method for the determination of buprenorphine hydrochloride, naloxone hydrochloride and noroxymorphone in a tablet formulation. Talanta 2009, 77, 1415–1419. [Google Scholar] [CrossRef]
- Espinosa Bosch, M.; Sánchez Rojas, F.; Bosch Ojeda, C. Compatibility and stability of drug mixtures: An overview. Biol. Pharm. Sci. 2022, 18, 295–325. [Google Scholar] [CrossRef]
- Gil, A.M.; Gómez, M.A.M.; Colomer, E.G.; Oltra, B.P.; Martí, M.C. Stability studies of ternary mixtures containing fosaprepitant, dexamethasone, ondansetron and granisetron used in clinical practice. J. Anal. Bioanal. Technol. 2016, 7, 307. [Google Scholar]
- Fernandez-Campos, F.; Mallandrich, M.; Calpena, A.C.; Ayestarán, A.; Lacasa, C. Stability studies of binary and ternary mixtures containing morphine, midazolam, levomepromazine and hyoscine butylbromide for parenteral administration. J. Pharm. Pharmacol. 2013, 65, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Espinosa-Bosch, M.; Sanchez-Rojas, F.; Bosch-Ojeda, C. Stability of mixtures of ondansetron and haloperidol stored in infusors at different temperatures. Eur. J. Hosp. Pharm. 2018, 25, e134–e138. [Google Scholar] [CrossRef] [PubMed]
- Gumułka, P.; Dąbrowska, M.; Starek, M. TLC-densitometric determination of five coxibs in pharmaceutical preparations. Processes 2020, 8, 620. [Google Scholar] [CrossRef]
- Juurlink, D.N.; Mamdani, M.; Kopp, A.; Laupacis, A.; Redelmeier, D.A. Drug-drug interactions among elderly patients hospitalized for drug toxicity. JAMA 2003, 289, 1652–1658. [Google Scholar] [CrossRef]
- Alfaro, C.L. Emerging role of drug interaction studies in drug development: The good, the bad, and the unknown. Psychopharmacol. Bull. 2001, 35, 80–93. [Google Scholar]
- Shad, M.U.; Marsh, C.; Preskorn, S.H. The economic consequences of a drug-drug interaction. J. Clin. Psychopharmacol. 2001, 21, 119–120. [Google Scholar] [CrossRef] [PubMed]
- Glassman, P.A.; Simon, B.; Belperio, P.; Lanto, A. Improving recognition of drug interactions: Benefits and barriers to using automated drug alerts. Med. Care 2002, 40, 1161–1171. [Google Scholar] [CrossRef]
- Hazlet, T.K.; Lee, T.A.; Hansten, P.D.; Horn, J.R. Performance of community pharmacy drug interaction software. J. Am. Pharm. Assoc. 2001, 41, 200–204. [Google Scholar] [CrossRef]
- Drug Interactions Checker. Available online: www.drugs.com/interaction/list/ (accessed on 15 July 2023).
- Monteiro, B.; Steagall, P.V. Antiinflammatory drugs. Vet. Clin. N. Am. Small Anim. Pract. 2019, 49, 993–1011. [Google Scholar] [CrossRef]
- McLean, M.K.; Khan, S.A. Toxicology of frequently encountered nonsteroidal anti-inflammatory drugs in dogs and cats: An update. Vet. Clin. N. Am. Small Anim. Pract. 2018, 48, 969–984. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).