
Optimizing Recombinant Baculovirus Vector Design for Protein Production in Insect Cells

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells, Viruses and Infections
2.2. Plasmids and Recombinant Viruses
2.3. Protein Synthesis Analysis
2.3.1. Detection of eGFP Fluorescence
2.3.2. Detection of β-Galactosidase
2.3.3. SDS-PAGE, Coomassie Blue Staining, Western Blotting
2.4. Statistics
3. Results
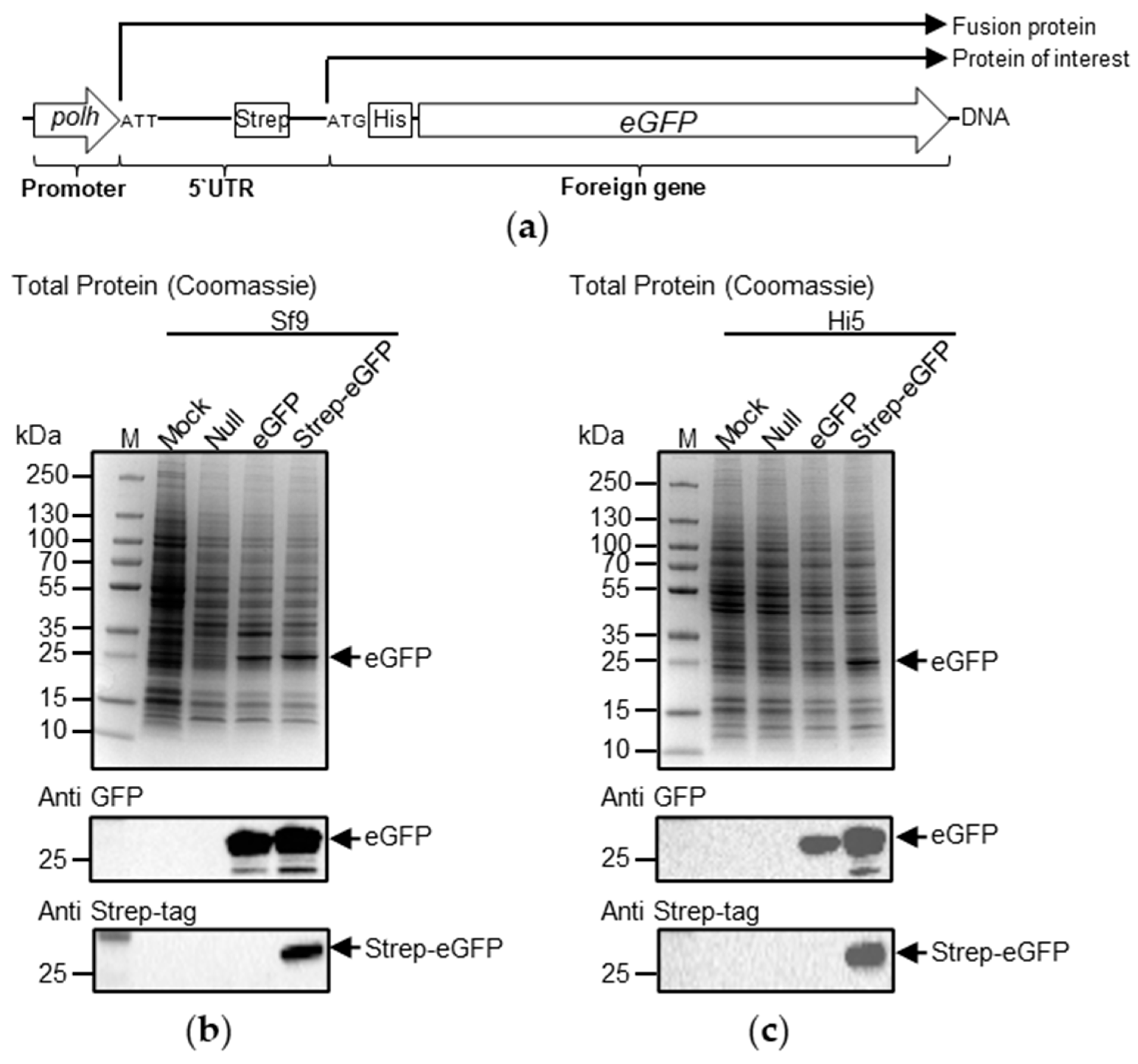
3.1. Protein Synthesis Is Initiated at AUU in Polh 5′ UTR
3.2. Protein Translation Is Initiated at AUU, AUC, or AUA in the polh 5′ UTR
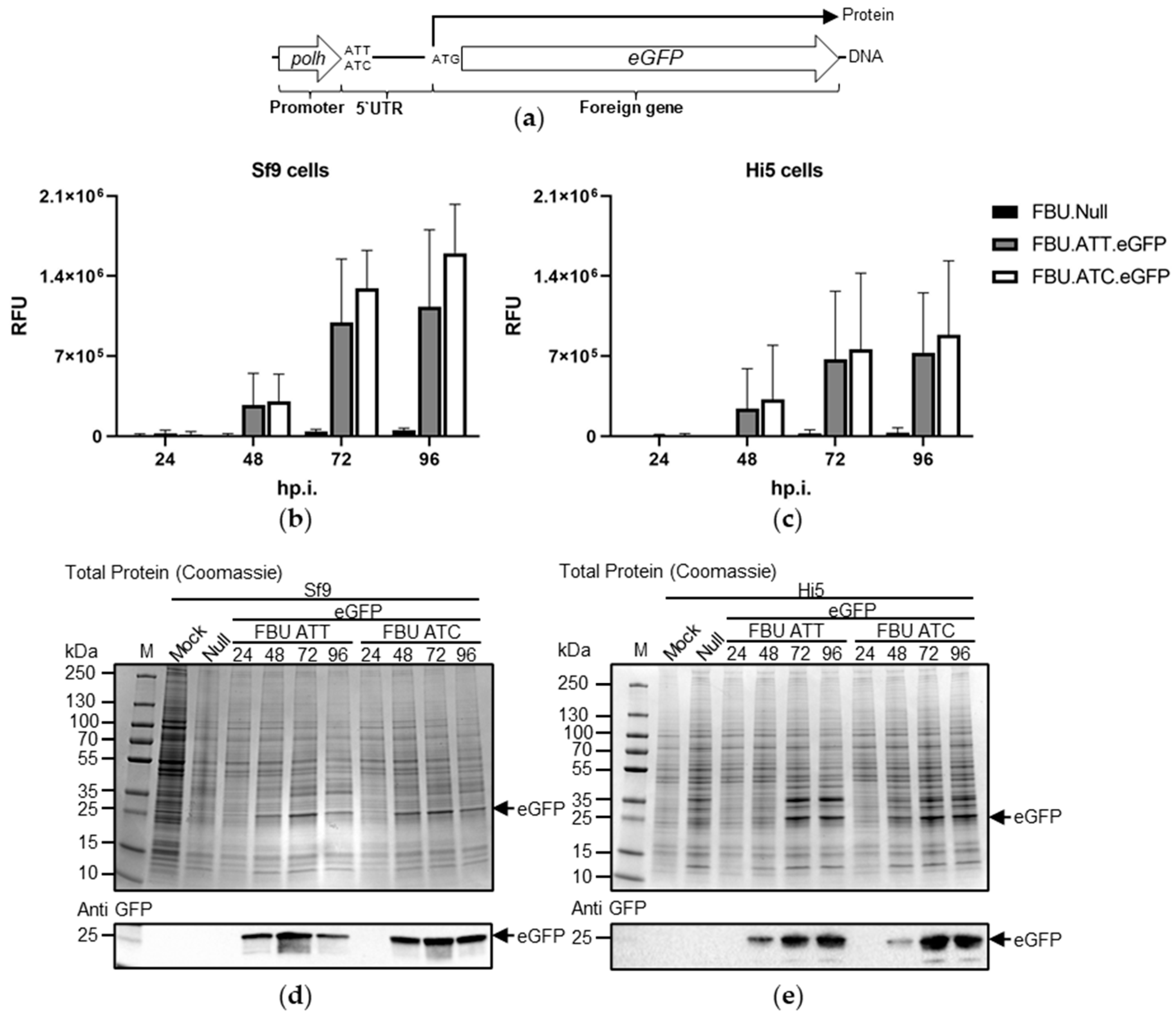
3.3. The Effect of Substituting AUC for AUU on Recombinant Protein Production
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- van Oers, M.M.; Pijlman, G.P.; Vlak, J.M. Thirty years of baculovirus-insect cell protein expression: From dark horse to mainstream technology. J. Gen. Virol. 2015, 96, 6–23. [Google Scholar] [CrossRef] [PubMed]
- Chambers, A.C.; Aksular, M.; Graves, L.P.; Irons, S.L.; Possee, R.D.; King, L.A. Overview of the Baculovirus Expression System. Curr. Protoc. Protein Sci. 2018, 91, 5.4.1–5.4.6. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.E.; Summers, M.D.; Fraser, M.J. Production of human beta interferon in insect cells infected with a baculovirus expression vector. Mol. Cell. Biol. 1983, 3, 2156–2165. [Google Scholar] [CrossRef] [PubMed]
- Pennock, G.D.; Shoemaker, C.; Miller, L.K. Strong and regulated expression of Escherichia coli beta-galactosidase in insect cells with a baculovirus vector. Mol. Cell. Biol. 1984, 4, 399–406. [Google Scholar] [CrossRef]
- Kitts, P.A.; Ayres, M.D.; Possee, R.D. Linearization of baculovirus DNA enhances the recovery of recombinant virus expression vectors. Nucleic Acids Res. 1990, 18, 5667–5672. [Google Scholar] [CrossRef] [Green Version]
- Kitts, P.A.; Possee, R.D. A method for producing recombinant baculovirus expression vectors at high frequency. Biotechniques 1993, 14, 810–817. [Google Scholar]
- Possee, R.D.; Hitchman, R.B.; Richards, K.S.; Mann, S.G.; Siaterli, E.; Nixon, C.P.; Irving, H.; Assenberg, R.; Alderton, D.; Owens, R.J.; et al. Generation of baculovirus vectors for the high-throughput production of proteins in insect cells. Biotechnol. Bioeng. 2008, 101, 1115–1122. [Google Scholar] [CrossRef]
- Hitchman, R.; Possee, R.; King, L. Baculovirus Expression Systems for Recombinant Protein Production in Insect Cells. Recent Pat. Biotechnol. 2009, 3, 46–54. [Google Scholar] [CrossRef]
- Luckow, V.A.; Lee, S.C.; Barry, G.F.; Olins, P.O. Efficient generation of infectious recombinant baculoviruses by site-specific transposon-mediated insertion of foreign genes into a baculovirus genome propagated in Escherichia coli. J. Virol. 1993, 67, 4566–4579. [Google Scholar] [CrossRef] [Green Version]
- Howard, S.C.; Ayres, M.D.; Possee, R.D. Mapping the 5′ and 3′ ends of Autographa californica nuclear polyhedrosis virus polyhedrin mRNA. Virus Res. 1986, 5, 109–119. [Google Scholar] [CrossRef]
- Matsuura, Y.; Possee, R.D.; Overton, H.A.; Bishop, D.H.L. Baculovirus Expression Vectors: The Requirements for High Level Expression of Proteins, Including Glycoproteins. J. Gen. Virol. 1987, 68, 1233–1250. [Google Scholar] [CrossRef] [PubMed]
- Rankin, C.; Ooi, B.G.; Miller, L.K. Eight base pairs encompassing the transcriptional start point are the major determinant for baculovirus polyhedrin gene expression. Gene 1988, 70, 39–49. [Google Scholar] [CrossRef]
- Possee, R.D.; Howard, S.C. Analysis of the polyhedrin gene promoter of the Autographa californica nuclear polyhedrosis virus. Nucleic Acids Res. 1987, 15, 10233–10248. [Google Scholar] [CrossRef] [Green Version]
- Luckow, V.A.; Summers, M.D. High level expression of nonfused foreign genes with Autographa californica nuclear polyhedrosis virus expression vectors. Virology 1989, 170, 31–39. [Google Scholar] [CrossRef]
- Luckow, V.A.; Summers, M.D. Signals important for high-level expression of foreign genes in Autographa californica nuclear polyhedrosis virus expression vectors. Virology 1988, 167, 56–71. [Google Scholar] [CrossRef]
- Beames, B.; Braunagel, S.; Summers, M.D.; Lanford, R.E. Polyhedrin initiator codon altered to AUU yields unexpected fusion protein from a baculovirus vector. Biotechniques 1991, 11, 378–383. [Google Scholar] [PubMed]
- Luckow, V.A.; Summers, M.D. Trends in the Development of Baculovirus Expression Vectors. Nat. Biotechnol. 1988, 6, 47–55. [Google Scholar] [CrossRef]
- Granados, R.R.; Guoxun, L.; Derksen, A.C.G.; McKenna, K.A. A new insect cell line from Trichoplusia ni (BTI-Tn-5B1-4) susceptible to Trichoplusia ni single enveloped nuclear polyhedrosis virus. J. Invertebr. Pathol. 1994, 64, 260–266. [Google Scholar] [CrossRef]
- King, L.A.; Possee, R.D. The Baculovirus System, A Laboratory Guide; London Chapman Hall: London, UK, 1992. [Google Scholar]
- Hitchman, R.B.; Siaterli, E.A.; Nixon, C.P.; King, L.A. Quantitative real-time PCR for rapid and accurate titration of recombinant baculovirus particles. Biotechnol. Bioeng. 2007, 96, 810–814. [Google Scholar] [CrossRef]
- Sambrook, J.; Russell, D. Molecular Cloning: A Laboratory Manual, 4th ed.; CSHL Press: Cold Spring Harbor, NY, USA, 2012. [Google Scholar]
- Matsuura, Y.; Possee, R.D.; Bishop, D.H.L. Expression of the S-coded Genes of Lymphocytic Choriomeningitis Arenavirus Using a Baculovirus Vector. J. Gen. Virol. 1986, 67, 1515–1529. [Google Scholar] [CrossRef]
- Drummond, D.A.; Wilke, C.O. The evolutionary consequences of erroneous protein synthesis. Nat. Rev. Genet. 2009, 10, 715–724. [Google Scholar] [CrossRef] [Green Version]
- Zitomer, R.S.; Walthall, D.A.; Rymond, B.C.; Hollenberg, C.P. Saccharomyces cerevisiae ribosomes recognize non-AUG initiation codons. Mol. Cell. Biol. 1984, 4, 1191–1197. [Google Scholar] [CrossRef]
- Peabody, D.S. Translation initiation at an ACG triplet in mammalian cells. J. Biol. Chem. 1987, 262, 11847–11851. [Google Scholar] [CrossRef]
- Peabody, D.S. Translation initiation at non-AUG triplets in mammalian cells. J. Biol. Chem. 1989, 264, 5031–5035. [Google Scholar] [CrossRef]
- Clements, J.M.; Laz, T.M.; Sherman, F. Efficiency of translation initiation by non-AUG codons in Saccharomyces cerevisiae. Mol. Cell. Biol. 1988, 8, 4533–4536. [Google Scholar] [CrossRef]
- Hann, S.R.; King, M.W.; Bentley, D.L.; Anderson, C.W.; Eisenman, R.N. A non-AUG translational initiation in c-myc exon 1 generates an N-terminally distinct protein whose synthesis is disrupted in Burkitt’s lymphomas. Cell 1988, 52, 185–195. [Google Scholar] [CrossRef]
- Kozak, M. Compilation and analysis of sequences upstream from the translational start site in eukaryotic mRNAs. Nucleic Acids Res. 1984, 12, 857–872. [Google Scholar] [CrossRef] [PubMed]
- Kozak, M. Context effects and inefficient initiation at non-AUG codons in eucaryotic cell-free translation systems. Mol. Cell. Biol. 1989, 9, 5073–5080. [Google Scholar] [CrossRef] [PubMed]
- Ayres, M.D.; Howard, S.C.; Kuzio, J.; Lopez-Ferber, M.; Possee, R.D. The complete DNA sequence of Autographa californica nuclear polyhedrosis virus. Virology 1994, 202, 586–605. [Google Scholar] [CrossRef]
- Ingolia, N.T.; Ghaemmaghami, S.; Newman, J.R.S.; Weissman, J.S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 2009, 324, 218–223. [Google Scholar] [CrossRef] [Green Version]
- Ingolia, N.T.; Lareau, L.F.; Weissman, J.S. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell 2011, 147, 789–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kearse, M.G.; Wilusz, J.E. Non-AUG translation: A new start for protein synthesis in eukaryotes. Genes Dev. 2017, 31, 1717–1731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fütterer, J.; Potrykus, I.; Bao, Y.; Li, L.; Burns, T.M.; Hull, R.; Hohn, T. Position-dependent ATT initiation during plant pararetrovirus rice tungro bacilliform virus translation. J. Virol. 1996, 70, 2999–3010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirako, Y. Non-AUG Translation Initiation in a Plant RNA Virus: A Forty-Amino-Acid Extension Is Added to the N Terminus of the Soil-Borne Wheat Mosaic Virus Capsid Protein. J. Virol. 1998, 72, 1677–1682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozak, M. Comparison of initiation of protein synthesis in procaryotes, eucaryotes, and organelles. Microbiol. Rev. 1983, 47, 74. [Google Scholar] [CrossRef]
- Kozak, M. Initiation of translation in prokaryotes and eukaryotes. Gene 1999, 234, 187–208. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sf9 eGFP | ||||
| FBU.ATG ΔATG.eGFP | FBU.ATT ΔATG.eGFP | FBU.ATC ΔATG.eGFP | FBU.ATA ΔATG.eGFP. | |
| 24 hp.i | 100% | 117.2% | 70.1% | 13.8% |
| 48 hp.i. | 100% | 6.2% | 5.4% | 6.8% |
| 72 hp.i. | 100% | 4.5% | 3.3% | 5.0% |
| 96 hp.i. | 100% | 4.3% | 3.2% | 5.1% |
| Hi5 eGFP | ||||
| FBU.ATG ΔATG.eGFP | FBU.ATT ΔATG.eGFP | FBU.ATC ΔATG.eGFP | FBU.ATA ΔATG.eGFP | |
| 24 hp.i | 100% | 80.2% | 56.5% | 49.0% |
| 48 hp.i. | 100% | 15.3% | 11.1% | 9.1% |
| 72 hp.i. | 100% | 11.2% | 5.6% | 8.5% |
| 96 hp.i. | 100% | 10.3% | 5.2% | 7.2% |
| Sf9 β-Galactosidase | ||||
| FBU.ATG ΔATG.lacZ | FBU.ATT ΔATG.lacZ | FBU.ATC ΔATG.lacZ | FBU.ATA ΔATG.lacZ | |
| 24 hp.i | 100% | 69.6% | 77.1% | 88.7% |
| 48 hp.i. | 100% | 1.0% | 0.7% | 1.4% |
| 72 hp.i. | 100% | 0.3% | 0.4% | 0.7% |
| 96 hp.i. | 100% | 0.1% | 0.0% | 0.2% |
| Hi5 β-Galactosidase | ||||
| FBU.ATG ΔATG.lacZ | FBU.ATT ΔATG.lacZ | FBU.ATC ΔATG.lacZ | FBU.ATA ΔATG.lacZ | |
| 24 hp.i | 100% | 0.0% | 0.0% | 17.2% |
| 48 hp.i. | 100% | 2.8% | 0.6% | 5.3% |
| 72 hp.i. | 100% | 7.1% | 4.3% | 5.8% |
| 96 hp.i. | 100% | 6.4% | 4.2% | 6.2% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bannach, C.; Buck, D.R.; Bobby, G.; Graves, L.P.; Li, S.; Chambers, A.C.; Gan, E.; Arinto-Garcia, R.; Possee, R.D.; King, L.A. Optimizing Recombinant Baculovirus Vector Design for Protein Production in Insect Cells. Processes 2021, 9, 2118. https://doi.org/10.3390/pr9122118
Bannach C, Buck DR, Bobby G, Graves LP, Li S, Chambers AC, Gan E, Arinto-Garcia R, Possee RD, King LA. Optimizing Recombinant Baculovirus Vector Design for Protein Production in Insect Cells. Processes. 2021; 9(12):2118. https://doi.org/10.3390/pr9122118
Chicago/Turabian StyleBannach, Carina, Daniel Ruiz Buck, Genna Bobby, Leo P. Graves, Sainan Li, Adam C. Chambers, Elizabeth Gan, Raquel Arinto-Garcia, Robert D. Possee, and Linda A. King. 2021. "Optimizing Recombinant Baculovirus Vector Design for Protein Production in Insect Cells" Processes 9, no. 12: 2118. https://doi.org/10.3390/pr9122118
APA StyleBannach, C., Buck, D. R., Bobby, G., Graves, L. P., Li, S., Chambers, A. C., Gan, E., Arinto-Garcia, R., Possee, R. D., & King, L. A. (2021). Optimizing Recombinant Baculovirus Vector Design for Protein Production in Insect Cells. Processes, 9(12), 2118. https://doi.org/10.3390/pr9122118