
A New Symmetrical Thiazolidinedione Derivative: In Silico Design, Synthesis, and In Vivo Evaluation on a Streptozotocin-Induced Rat Model of Diabetes

 ,
,  ,
,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
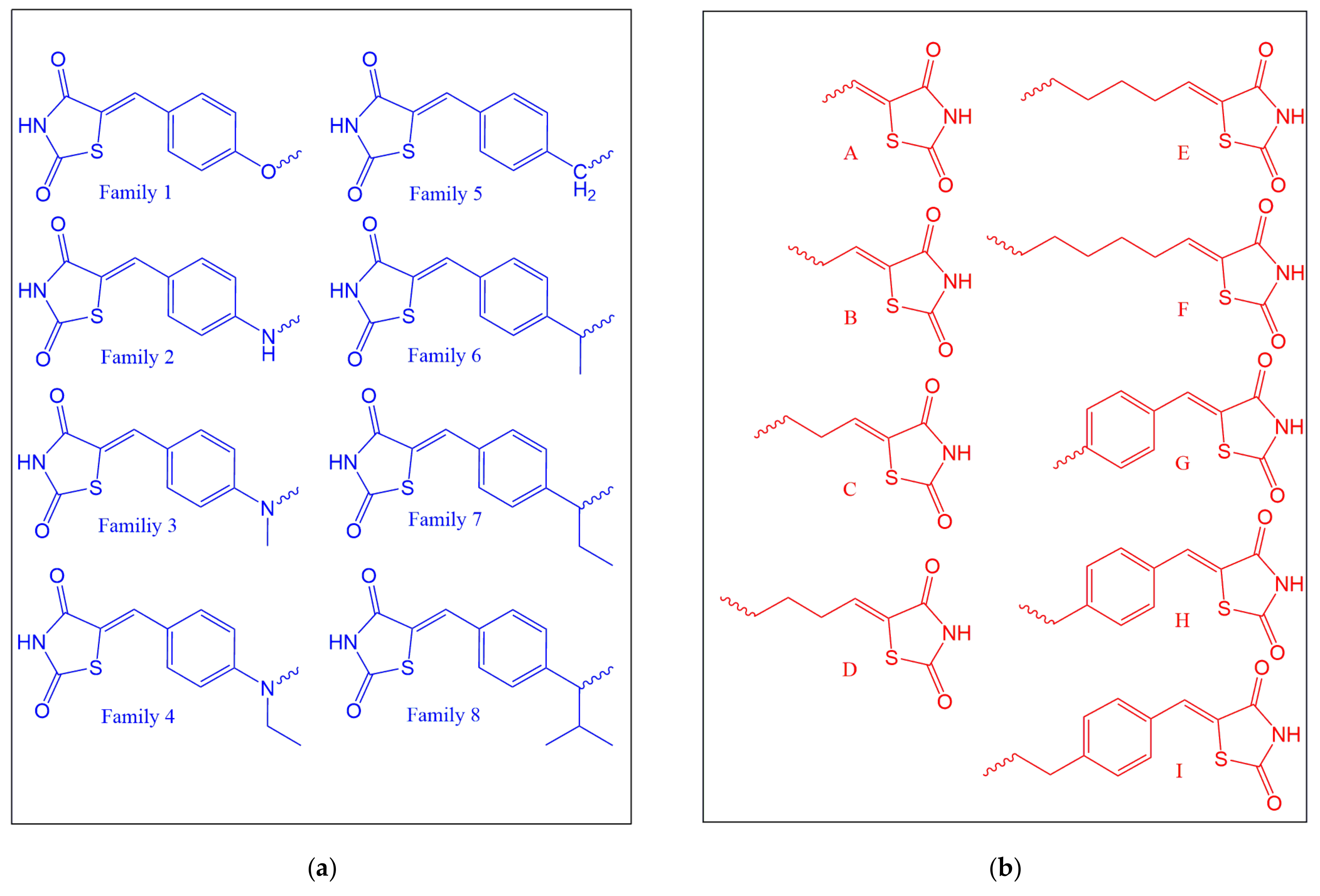
2.1. Ligand Design
2.2. Theoretical Determination of Physicochemical and Biological Properties
2.3. Docking Studies
2.4. Visualization of Interactions
2.5. Selection of the Compound to Be Synthesized
2.6. Prediction of Pharmacokinetic/Toxicological Properties and Protein Targets of Compound 1G
2.7. Chemical Synthesis of Compound 1G
2.8. Animals
2.9. Blood Sample Collection and Processing
2.10. Evaluation of Acute Oral Toxicity
2.11. Rat Model of Diabetes Induced by Streptozotocin
2.12. Administration of Treatments
2.13. The Glucose Tolerance Curve
2.14. Liver and Blood Parameters
2.15. Determination of ALT and AST Activity
2.16. Statistical Analysis
3. Results and Discussion
3.1. Docking
3.2. Prediction of the Phisicochemical Properties of 1G
3.3. Toxicityriskassessment
3.4. Prediction of Bioavailability and Toxicity
3.5. Toxicity Prediction
3.6. Synthesis and Characterization
3.7. Accute Oral Toxicity Assay
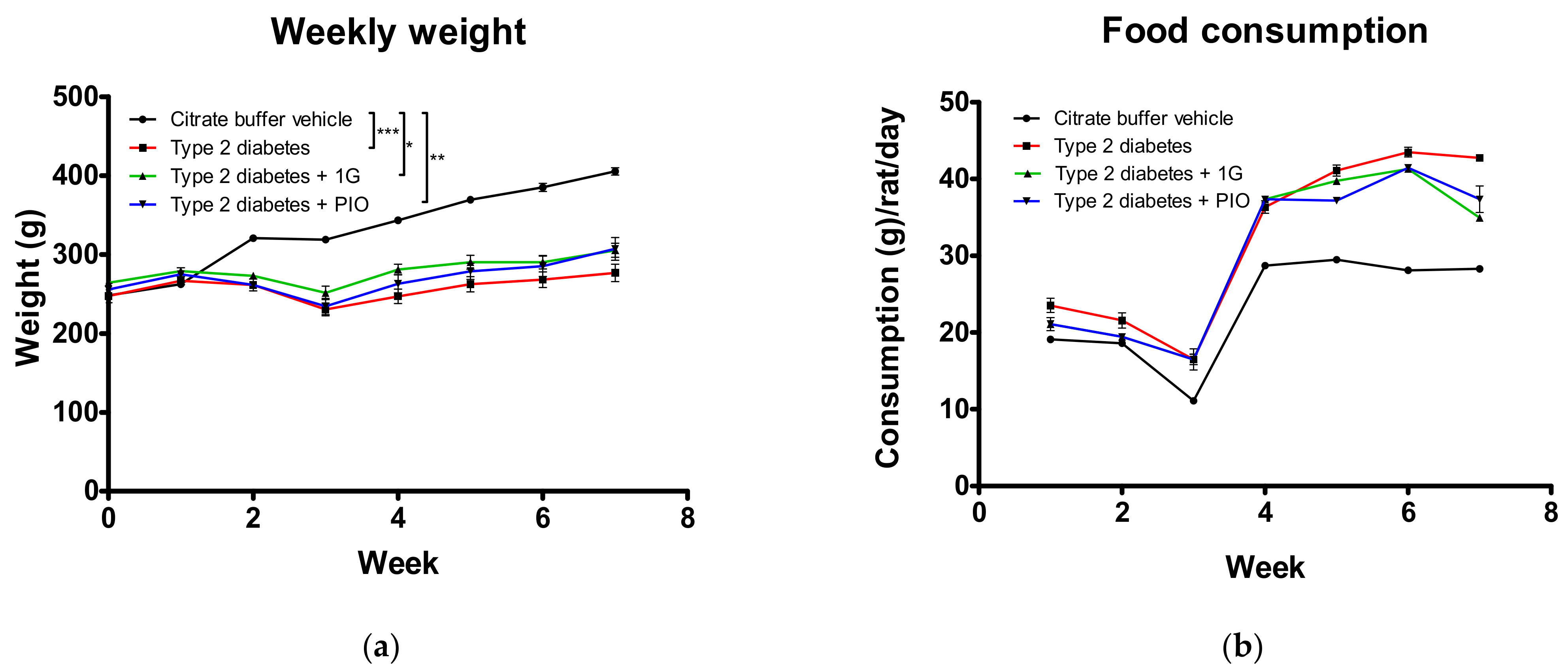
3.8. In Vivo Administration of 1G in a Rat Model of Diabetes
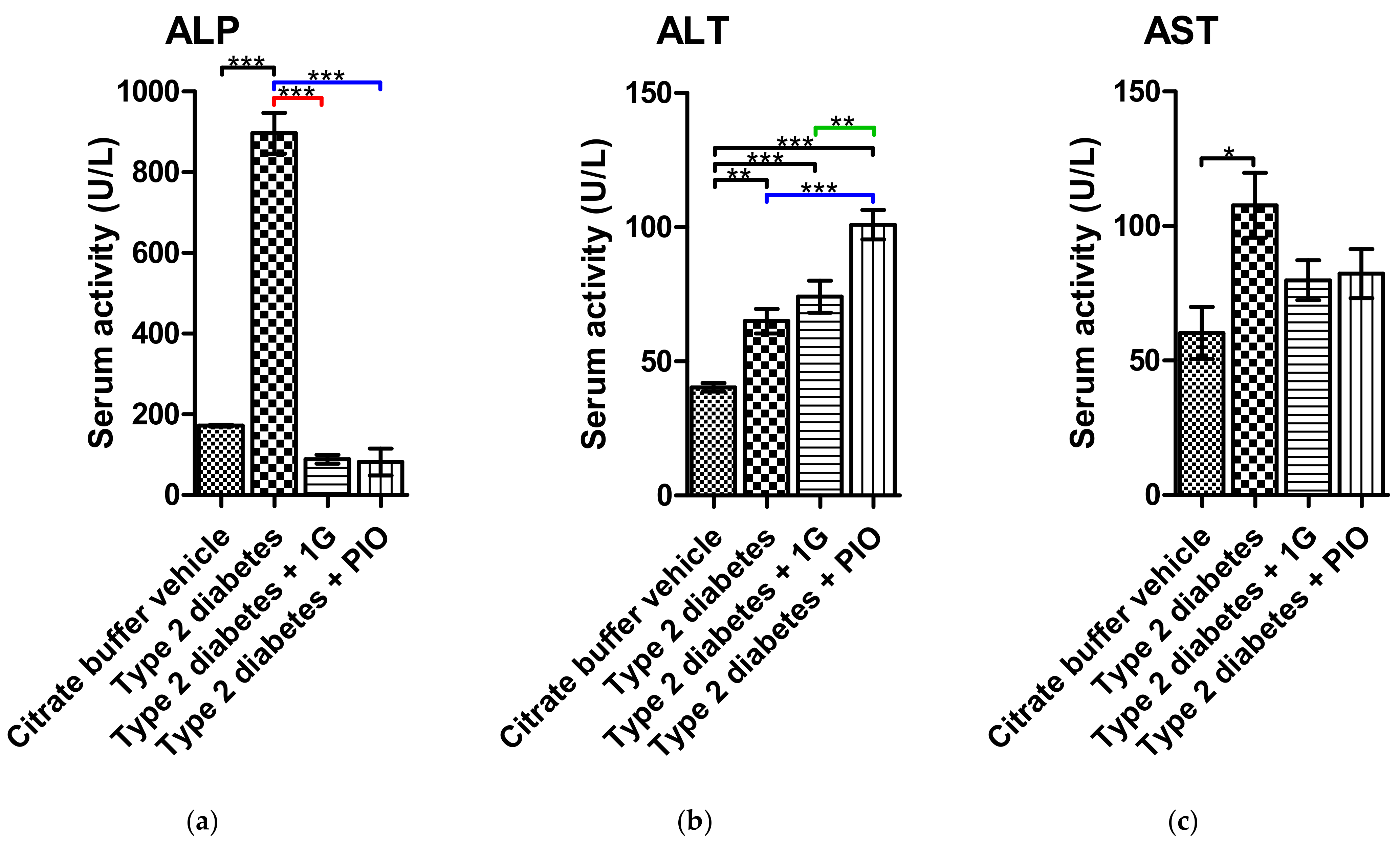
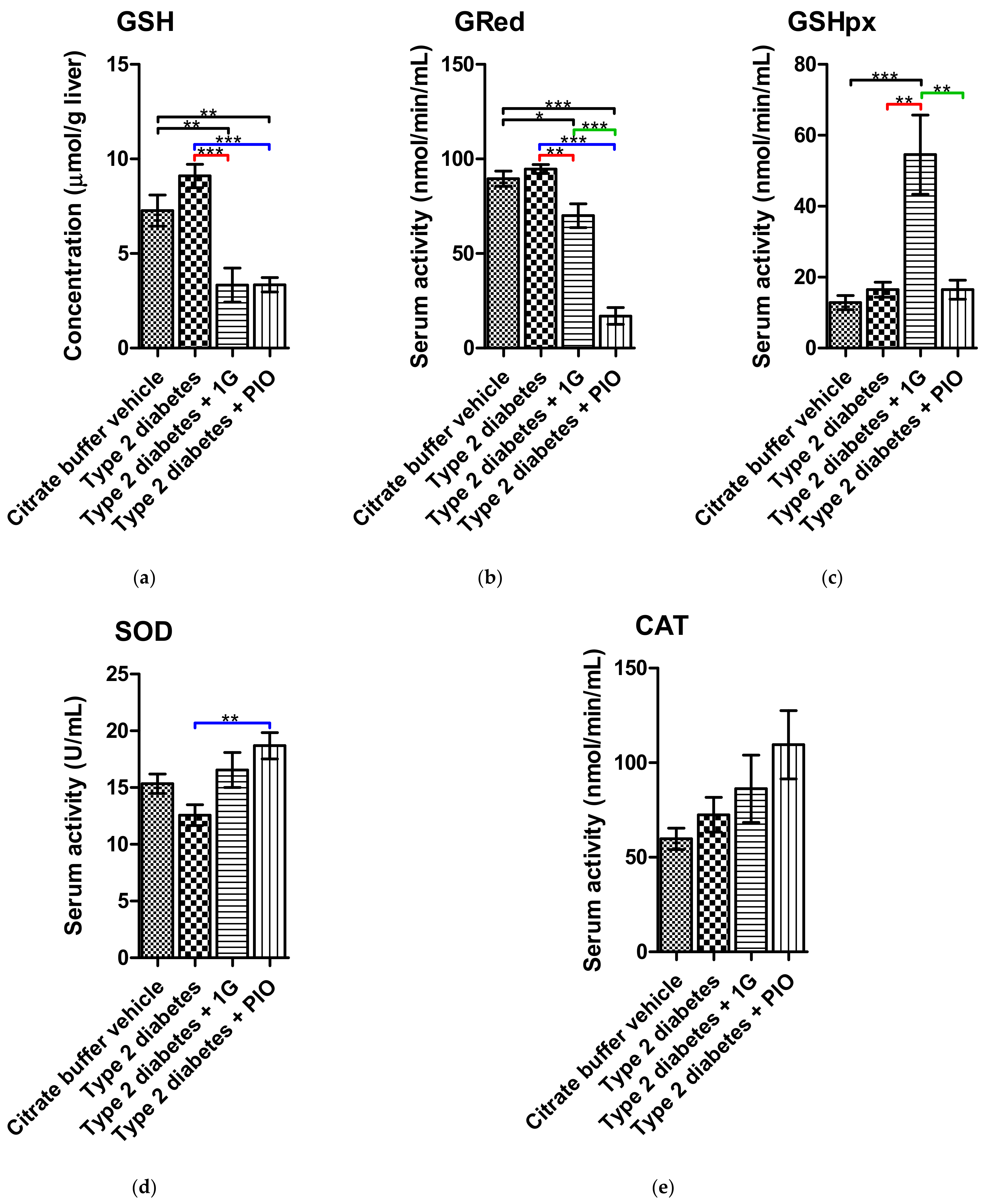
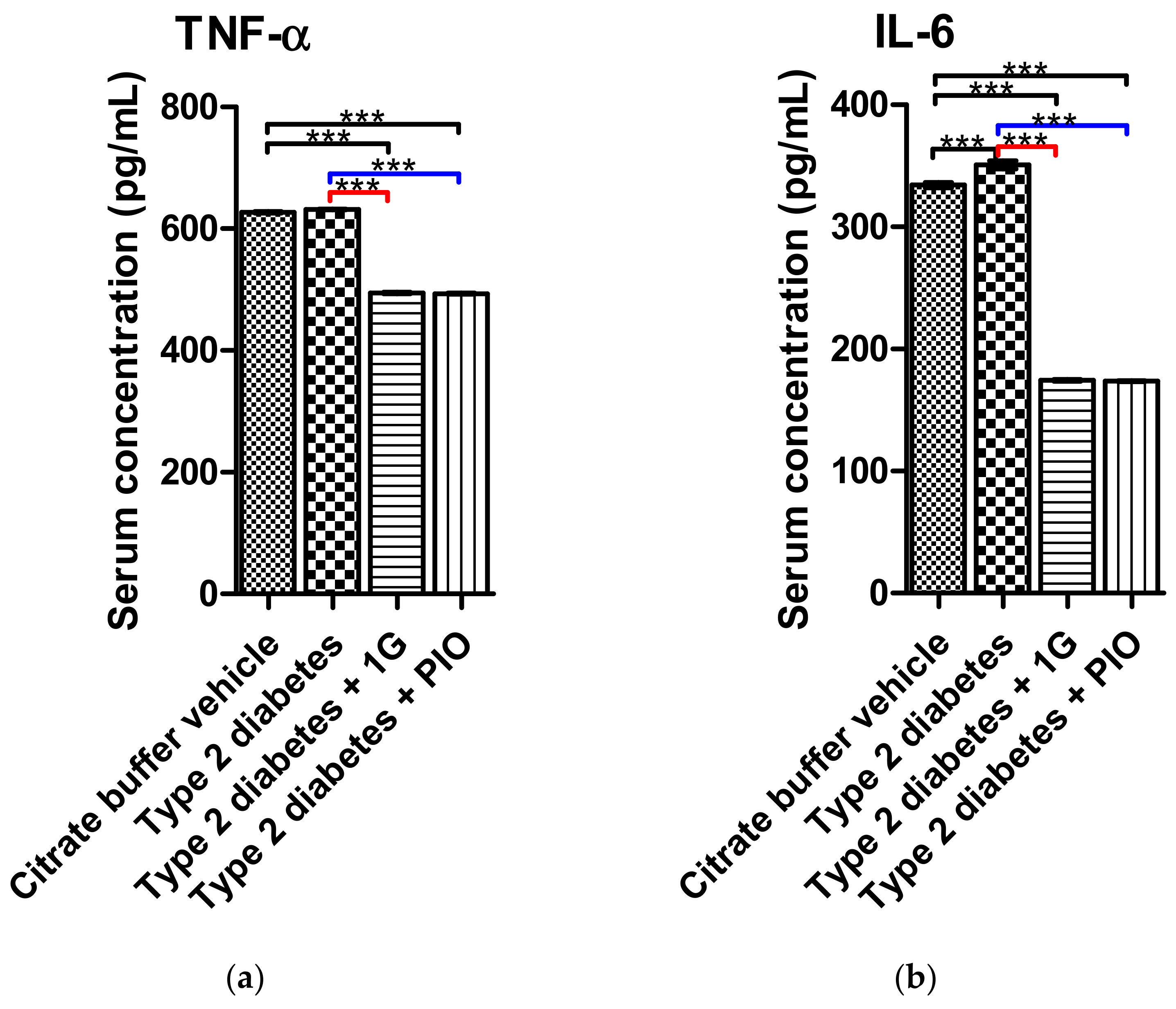
3.9. Ex Vivo Results of 1G Administration
3.10. Prediction of the Targets of 1G
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Álvarez-Almazán, S.; Filisola-Villaseñor, J.G.; Alemán-González-Duhart, D.; Tamay-Cach, F.; Mendieta-Wejebe, J.E. Current molecular aspects in the development and treatment of diabetes. J. Physiol. Biochem. 2020, 76, 13–35. [Google Scholar] [CrossRef]
- Mahmoud, A.M.; Ashour, M.B.; Abdel-Moneim, A.; Ahmed, O.M. Hesperidin and naringin attenuate hyperglycemia-mediated oxidative stress and proinflammatory cytokine production in high fat fed/streptozotocin-induced type 2 diabetic rats. J. Diabetes Complicat. 2012, 26, 483–490. [Google Scholar] [CrossRef]
- De Jesús Sandoval-Muñiz, R.; Vargas-Guerrero, B.; Flores-Alvarado, L.J.; Gurrola-Díaz, C.M. Glucotransportadores (GLUT): Aspectos clínicos, moleculares y genéticos. Gac. Med. Mex. 2016, 152, 547–557. [Google Scholar]
- Mansour, M. The roles of peroxisome proliferator-activated receptors in the metabolic syndrome. In Progress in Molecular Biology and Translational Science; Elsevier Inc.: Amsterdam, The Netherlands, 2014; Volume 121, pp. 217–266. ISBN 9780128001011. [Google Scholar]
- Navarrete-Vázquez, G.; Torres-Gómez, H.; Hidalgo-Figueroa, S.; Ramírez-Espinosa, J.J.; Estrada-Soto, S.; Medina-Franco, J.L.; León-Rivera, I.; Alarcón-Aguilar, F.J.; Almanza-Pérez, J.C. Synthesis, in vitro and in silico studies of a PPARγ and GLUT-4 modulator with hypoglycemic effect. Bioorg. Med. Chem. Lett. 2014, 24, 4575–4579. [Google Scholar] [CrossRef] [PubMed]
- Gutierréz-Hernández, A.; Galván-Ciprés, Y.; Domínguez-Mendoza, E.A.; Aguirre-Vidal, Y.; Estrada-Soto, S.; Almanza-Pérez, J.C.; Navarrete-Vázquez, G. Design, Synthesis, Antihyperglycemic Studies, and Docking Simulations of Benzimidazole-Thiazolidinedione Hybrids. J. Chem. 2019, 2019, 1650145. [Google Scholar] [CrossRef]
- Álvarez-Almazán, S.; Bello, M.; Tamay-Cach, F.; Martínez-Archundia, M.; Alemán-González-Duhart, D.; Correa-Basurto, J.; Mendieta-Wejebe, J.E. Study of new interactions of glitazone’s stereoisomers and the endogenous ligand 15d-PGJ2 on six different PPAR gamma proteins. Biochem. Pharmacol. 2017, 142, 168–193. [Google Scholar] [CrossRef]
- Hidalgo-Figueroa, S.; Ramírez-Espinosa, J.J.; Estrada-Soto, S.; Almanza-Pérez, J.C.; Román-Ramos, R.; Alarcón-Aguilar, F.J.; Hernández-Rosado, J.V.; Moreno-Díaz, H.; Díaz-Coutiño, D.; Navarrete-Vázquez, G. Discovery of Thiazolidine-2,4-Dione/Biphenylcarbonitrile Hybrid as Dual PPAR α/γ Modulator with Antidiabetic Effect: In vitro, In Silico and In Vivo Approaches. Chem. Biol. Drug Des. 2013, 81, 474–483. [Google Scholar] [CrossRef]
- Parmenon, C.; Guillard, J.; Caignard, D.H.; Hennuyer, N.; Staels, B.; Audinot-Bouchez, V.; Boutin, J.A.; Dacquet, C.; Ktorza, A.; Viaud-Massuard, M.C. 4,4-Dimethyl-1,2,3,4-tetrahydroquinoline-based PPARα/γ agonists. Part I: Synthesis and pharmacological evaluation. Bioorg. Med. Chem. Lett. 2008, 18, 1617–1622. [Google Scholar] [CrossRef]
- Cho, M.C.; Lee, D.H.; Kim, E.J.; Lee, J.Y.; Kang, J.W.; Song, J.H.; Chong, Y.; Kim, Y.; Hong, J.T.; Yoon, D.Y. Novel PPARγ partial agonists with weak activity and no cytotoxicity; Identified by a simple PPARγ ligand screening system. Mol. Cell. Biochem. 2011, 358, 75–83. [Google Scholar] [CrossRef]
- Alemán-González-Duhart, D.; Tamay-Cach, F.; Correa-Basurto, J.; Padilla-Martínez, I.I.; Álvarez-Almazán, S.; Mendieta-Wejebe, J.E. In silico design, chemical synthesis and toxicological evaluation of 1,3-thiazolidine-2,4-dione derivatives as PPARγ agonists. Regul. Toxicol. Pharmacol. 2017, 86, 25–32. [Google Scholar] [CrossRef]
- Sethi, A.; Joshi, K.; Sasikala, K.; Alvala, M. Molecular Docking in Modern Drug Discovery: Principles and Recent Applications. In Drug Discovery and Development-New Advances; IntechOpen: London, UK, 2016; Volume i, p. 13. [Google Scholar]
- Molinspiration Cheminformatics Calculation of Molecular Properties and Bioactivity Score. Available online: https://www.molinspiration.com/cgi-bin/properties (accessed on 19 July 2020).
- Sander, T. Molecular Properties Prediction-Osiris Property Explorer. Available online: https://www.organic-chemistry.org/prog/peo/ (accessed on 19 July 2020).
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y. AdmetSAR: A comprehensive source and free tool for assessment of chemical ADMET properties. J. Chem. Inf. Model. 2012, 52, 3099–3105. [Google Scholar] [CrossRef] [PubMed]
- Nolte, R.T.; Wisely, G.B.; Westin, S.; Cobb, J.E.; Lambert, M.H.; Kurokawa, R.; Rosenfeld, M.G.; Willson, T.M.; Glass, C.K.; Milburn, M.V. Ligand binding and co-activator assembly of the peroxisome proliferator- activated receptor-γ. Nature 1998, 395, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Gumbart, J.C.; Luo, D. HBonds Plugin, Version 1.2. Available online: http://www.ks.uiuc.edu/Research/vmd/plugins/hbonds/ (accessed on 20 August 2020).
- Bakhtyari, N.G.; Raitano, G.; Benfenati, E.; Martin, T.; Young, D. Comparison of in silico models for prediction of mutagenicity. J. Environ. Sci. Heal. Part C Environ. Carcinog. Ecotoxicol. Rev. 2013, 31, 45–66. [Google Scholar] [CrossRef] [PubMed]
- Gfeller, D.; Grosdidier, A.; Wirth, M.; Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: A web server for target prediction of bioactive small molecules. Nucleic Acids Res. 2014, 42, W32–W38. [Google Scholar] [CrossRef] [PubMed]
- Stümpel, E.; Hartmann, H. Acute actions of insulin-like growth factor II on glucose metabolism in adult rats. Diabetology 1992, 35, 932–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derosa, G.; Cicero, A.F.G.; Gaddi, A.; Ragonesi, P.D.; Fogari, E.; Bertone, G.; Ciccarelli, L.; Piccinni, M.N. Metabolic effects of pioglitazone and rosiglitazone in patients with diabetes and metabolic syndrome treated with glimepiride: A twelve-month, multicenter, double-blind, randomized, controlled, parallel-group trial. Clin. Ther. 2004, 26, 744–754. [Google Scholar] [CrossRef]
- Elmazar, M.M.; El-abhar, H.S.; Schaalan, M.F.; Farag, N.A. Phytol/Phytanic Acid and Insulin Resistance: Potential Role of Phytanic Acid Proven by Docking Simulation and Modulation of Biochemical Alterations. PLoS ONE 2013, 8, e45638. [Google Scholar] [CrossRef] [Green Version]
- OECD. Acute Oral Toxicity-Up-and-Down-Procedure (UDP). In OECD Guidelines for the Testing of Chemicals; OECD: Paris, France, 2008; pp. 1–27. [Google Scholar]
- Al-Faris, N.A.; Al-sawadi, A.D.; Alokail, M.S. Effect of samh seeds supplementation (Mesembryanthemum forsskalei Hochst) on liver enzymes and lipid profiles of streptozotocin (STZ)-induced diabetic Wistar rats. Saudi J. Biol. Sci. 2010, 17, 23–28. [Google Scholar] [CrossRef] [Green Version]
- Zafar, M.; Naeem-ul-Hassan Naqvi, S.; Ahmed, M.; Kaimkhani, Z.A. Altered Kidney Morphology and Enzymes in Streptozotocin Induced Diabetic Rats. Int. J. Morphol. 2009, 27, 719–725. [Google Scholar] [CrossRef] [Green Version]
- Szkudelski, T.; Zywert, A.; Szkudelska, K. Metabolic disturbances and defects in insulin secretion in rats with streptozotocin-nicotinamide-induced diabetes. Physiol. Res. 2013, 62, 663–670. [Google Scholar] [CrossRef]
- Elrashidy, R.A.; Asker, M.E.; Mohamed, H.E. Beneficial effects of pioglitazone against cardiovascular injury are enhanced by combination with aliskiren in a rat model of diabetic nephropathy. J. Pharm. Pharmacol. 2012, 64, 862–871. [Google Scholar] [CrossRef]
- Ko, G.J.; Kang, Y.S.; Han, S.Y.; Lee, M.H.; Song, H.K.; Han, K.H.; Kim, H.K.; Han, J.Y.; Cha, D.R. Pioglitazone attenuates diabetic nephropathy through an anti-inflammatory mechanism in type 2 diabetic rats. Nephrol. Dial. Transplant. 2008, 23, 2750–2760. [Google Scholar] [CrossRef] [Green Version]
- Cabrera-Pérez, L.C.; Padilla-Martínez, I.I.; Cruz, A.; Mendieta-Wejebe, J.E.; Tamay-Cach, F.; Rosales-Hernández, M.C. Evaluation of a new benzothiazole derivative with antioxidant activity in the initial phase of acetaminophen toxicity. Arab. J. Chem. 2019, 12, 3871–3882. [Google Scholar] [CrossRef]
- Owens, C.W.; Belcher, R.V. A Colorimetric Micro-Method for the Determination of Glutathione. Biochem. J. 1965, 94, 705–711. [Google Scholar] [CrossRef]
- Da Costa Leite, L.F.C.; Veras Mourão, R.H.; de Lima, M.D.C.A.; Galdino, S.L.; Hernandes, M.Z.; de Assis Rocha Neves, F.; Vidal, S.; Barbe, J.; da Rocha Pitta, I. Synthesis, biological evaluation and molecular modeling studies of arylidene-thiazolidinediones with potential hypoglycemic and hypolipidemic activities. Eur. J. Med. Chem. 2007, 42, 1263–1271. [Google Scholar] [CrossRef]
- Alemán-González-Duhart, D.; Tamay-Cach, F.; Álvarez-Almazán, S.; Mendieta-Wejebe, J.E. Current Advances in the Biochemical and Physiological Aspects of the Treatment of Type 2 Diabetes Mellitus with Thiazolidinediones. PPAR Res. 2016, 2016, 7614270. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Petrie, I.D.; Levy, R.H.; Ragueneau-Majlessi, I. Mechanisms and clinical significance of pharmacokinetic-based drug-drug interactions with drugs approved by the U.S. Food and drug administration in 2017 s. Drug Metab. Dispos. 2019, 47, 135–144. [Google Scholar] [CrossRef] [Green Version]
- International Diabetes Federation Atlas de la Diabetes de la FID. Available online: https://www.diabetesatlas.org/upload/resources/material/20200302_133352_2406-IDF-ATLAS-SPAN-BOOK.pdf (accessed on 12 June 2021).
- Stead, A.G.; Hasselblad, V.; Creason, J.P.; Claxton, L. Modeling the Ames test. Mutat. Res. Mutagen. Relat. Subj. 1981, 85, 13–27. [Google Scholar] [CrossRef]
- Butler, A.; Helliwell, M.V.; Zhang, Y.; Hancox, J.C.; Dempsey, C.E. An update on the structure of HERG. Front. Pharmacol. 2020, 10, 1572. [Google Scholar] [CrossRef] [PubMed]
- Rashid, M. Design, synthesis and ADMET prediction of bis-benzimidazole as anticancer agent. Bioorg. Chem. 2020, 96, 103576. [Google Scholar] [CrossRef]
- Wigren, J.; Surapureddi, S.; Olsson, A.G.; Glass, C.K.; Hammarström, S.; Söderström, M. Differential recruitment of the coactivator proteins CREB-binding protein and steroid receptor coactivator-1 to peroxisome proliferator-activated receptor gamma/9-cis-retinoic acid receptor heterodimers by ligands present in oxidized low-density lipoprote. J. Endocrinol. 2003, 177, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.C.C.; Tsai, S.P.; Jhao, J.Y.; Jiang, W.K.; Tsao, C.K.; Chang, L.Y. Liver Fat, Hepatic Enzymes, Alkaline Phosphatase and the Risk of Incident Type 2 Diabetes: A Prospective Study of 132,377 Adults. Sci. Rep. 2017, 7, 4649. [Google Scholar] [CrossRef]
- Nazreen, S.; Alam, M.S.; Hamid, H.; Yar, M.S.; Dhulap, A.; Alam, P.; Pasha, M.A.Q.; Bano, S.; Alam, M.M.; Haider, S.; et al. Thiazolidine-2,4-diones derivatives as PPAR-γ agonists: Synthesis, molecular docking, in vitro and in vivo antidiabetic activity with hepatotoxicity risk evaluation and effect on PPAR-γ gene expression. Bioorg. Med. Chem. Lett. 2014, 24, 3034–3042. [Google Scholar] [CrossRef]
- Maxwell, D.B.; Fisher, E.A.; Ross-Clunis, H.A.; Estep, H.L. Serum Alkaline Phosphatase in Diabetes Mellitus. J. Am. Coll. Nutr. 1986, 5, 55–59. [Google Scholar] [CrossRef]
- Yuan, L.; Kaplowitz, N. Glutathione in liver diseases and hepatotoxicity. Mol. Aspects Med. 2008, 30, 29–41. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Aspects Med. 2009, 30, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Liu, C.; Zhang, H.; Sun, Z.; Wang, R. Myricitrin exhibits anti-atherosclerotic and anti-hyperlipidemic effects in diet-induced hypercholesterolemic rats. AMB Express 2019, 9, 204. [Google Scholar] [CrossRef]
- Kojima-Yuas, A.; Deguchi, Y.; Konishi, Y.; Matsui-Yuasa, I. Effect of 1,5-Anhydro-D-fructose on the inhibition of adipogenesis in 3T3-L1 adipocytes. Nat. Prod. Commun. 2012, 7, 1501–1506. [Google Scholar] [CrossRef] [Green Version]
- Shittu, M.; Ayo, J.O.; Ambali, S.F.; Fatihu, M.Y.; Onyeanusi, B.I.; Kawu, M.U. Chronic chlorpyrifos-induced oxidative changes in the testes and pituitary gland of Wistar rats: Ameliorative effects of vitamin C. Pestic. Biochem. Physiol. 2012, 102, 79–85. [Google Scholar] [CrossRef]
- Hansson, B.; Rippe, C.; Kotowska, D.; Wasserstrom, S.; Säll, J.; Göransson, O.; Swärd, K.; Stenkula, K.G. Rosiglitazone drives cavin-2/SDPR expression in adipocytes in a CEBP α -dependent manner. PLoS ONE 2017, 12, e173412. [Google Scholar] [CrossRef]
- Jamali, B.; Bjørnsdottir, I.; Nordfang, O.; Hansen, S.H. Investigation of racemisation of the enantiomers of glitazone drug compounds at different pH using chiral HPLC and chiral CE. J. Pharm. Biomed. Anal. 2008, 46, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Sheu, S.H.; Kaya, T.; Waxman, D.J.; Vajda, S. Exploring the binding site structure of the PPARγ ligand-binding domain by computational solvent mapping. Biochemistry 2005, 44, 1193–1209. [Google Scholar] [CrossRef]
- Bauters, D.; Cobbaut, M.; Geys, L.; Van Lint, J.; Hemmeryckx, B.; Lijnen, H.R. Loss of ADAMTS5 enhances brown adipose tissue mass and promotes browning of white adipose tissue via CREB signaling. Mol. Metab. 2017, 6, 715–724. [Google Scholar] [CrossRef]
- Ukkola, O.; Rankinen, T.; Lakka, T.; Leon, A.S.; Skinner, J.S.; Wilmore, J.H.; Rao, D.C.; Kesäniemi, Y.A.; Bouchard, C. Protein tyrosine phosphatase 1B variant associated with fat distribution and insulin metabolism. Obes. Res. 2005, 13, 829–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneditz, G.; Elias, J.E.; Pagano, E.; Zaeem Cader, M.; Saveljeva, S.; Long, K.; Mukhopadhyay, S.; Arasteh, M.; Lawley, T.D.; Dougan, G.; et al. GPR35 promotes glycolysis, proliferation, and oncogenic signaling by engaging with the sodium potassium pump. Sci. Signal. 2019, 12, eaau9048. [Google Scholar] [CrossRef] [PubMed]
- Sivenius, K.; Niskanen, L.; Voutilainen-Kaunisto, R.; Laakso, M.; Uusitupa, M. Aldose reductase gene polymorphisms and susceptibility to microvascular complications in Type 2 diabetes. Diabet. Med. 2004, 21, 1325–1333. [Google Scholar] [CrossRef]
- Watarai, A.; Nakashima, E.; Hamada, Y.; Watanabe, G.; Naruse, K.; Miwa, K.; Kobayashi, Y.; Kamiya, H.; Nakae, M.; Hamajima, N.; et al. Aldose reductase gene is associated with diabetic macroangiopathy in Japanese Type 2 diabetic patients. Diabet. Med. 2006, 23, 894–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crosas, B.; Hyndman, D.J.; Gallego, O.; Martras, S.; Parés, X.; Flynn, T.G.; Farrés, J. Human aldose reductase and human small intestine aldose reductase are efficient retinal reductases: Consequences for retinoid metabolism. Biochem. J. 2003, 373, 973–979. [Google Scholar] [CrossRef] [Green Version]
- Cheyssac, C.; Lecoeur, C.; Dechaume, A.; Bibi, A.; Charpentier, G.; Balkau, B.; Marre, M.; Froguel, P.; Gibson, F.; Vaxillaire, M. Analysis of common PTPN1 gene variants in type 2 diabetes, obesity and associated phenotypes in the French population. BMC Med. Genet. 2006, 7, 44. [Google Scholar] [CrossRef] [Green Version]
- Vomastek, T.; Iwanicki, M.P.; Burack, W.R.; Tiwari, D.; Kumar, D.; Parsons, J.T.; Weber, M.J.; Nandicoori, V.K. Extracellular Signal-Regulated Kinase 2 (ERK2) Phosphorylation Sites and Docking Domain on the Nuclear Pore Complex Protein Tpr Cooperatively Regulate ERK2-Tpr Interaction. Mol. Cell. Biol. 2008, 28, 6954–6966. [Google Scholar] [CrossRef] [Green Version]
- Roemer, H.C.; Weistenhofer, W.; Lohlein, D.; Geller, F.; Blomeke, B.; Golka, K. N-acetyltransferase 1 in colon and rectal cancer cases from an industrialized area. J. Toxicol. Environ. Health. A 2008, 71, 902–905. [Google Scholar] [CrossRef]
- Uppal, S.; Diggle, C.P.; Carr, I.M.; Fishwick, C.W.G.; Ahmed, M.; Ibrahim, G.H.; Helliwell, P.S.; Latos-Bieleńska, A.; Phillips, S.E.V.; Markham, A.F.; et al. Mutations in 15-hydroxyprostaglandin dehydrogenase cause primary hypertrophic osteoarthropathy. Nat. Genet. 2008, 40, 789–793. [Google Scholar] [CrossRef]
- Qian, Y.; Zhou, X.; Zhang, Y.; Gao, P.; Zhang, W.; Zhu, D. Study on insulin resistance and genetic polymorphisms in essential hypertension patients of two different kinds of TCM constitution. Chin. J. Integr. Med. 2007, 13, 175–179. [Google Scholar] [CrossRef]
- Slaaby, R.; Schäffer, L.; Lautrup-Larsen, I.; Andersen, A.S.; Shaw, A.C.; Mathiasen, I.S.; Brandt, J. Hybrid receptors formed by insulin receptor (IR) and insulin-like growth factor I receptor (IGF-IR) have low insulin and high IGF-1 affinity irrespective of the IR splice variant. J. Biol. Chem. 2006, 281, 25869–25874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saraiva, M.J.; Almeida, M.D.R.; Sherman, W.; Gawinowicz, M.; Costa, P.; Costa, P.P.; Goodman, D.S. A new transthyretin mutation associated with amyloid cardiomyopathy. Am. J. Hum. Genet. 1992, 50, 1027–1030. [Google Scholar]
- Morishita, D.; Katayama, R.; Sekimizu, K.; Tsuruo, T.; Fujita, N. Pim kinases promote cell cycle progression by phosphorylating and down-regulating p27Kip1 at the transcriptional and posttranscriptional levels. Cancer Res. 2008, 68, 5076–5085. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Value | Punctuation | Reference |
|---|---|---|---|
| LogP | 2–4 | 2 | [13,15] |
| 0–2 and 4–5 | 1 | ||
| <0 and >5 | 0 | ||
| TPSA [Å] | <140 | 1 | [15] |
| >140 | 0 | ||
| MW [g/mol] | <400 | 1 | |
| >400 | 0 | ||
| Binding mode similar to pioglitazone | Yes | 1 | [7,8] |
| No | 0 | ||
| Sum of the total number of H-bonds | <12 | 1 | [15] |
| >12 | 0 | ||
| Number of rotatable bonds | 8–10 | 2 | |
| 1–7 | 1 | ||
| Mutagenesis | Absence | 1 | [14] |
| Tumorigenesis | |||
| Irritability | Presence | 0 | |
| Reproductive effects | |||
| LogS | −4 to −2 | 2 | [14,15] |
| −6 to −4 and −2 to 0 | 1 | ||
| <−6 | 0 |
| Compound | ΔG (kcal/mol) | H-Bond Length (Å) | ||||
|---|---|---|---|---|---|---|
| Ser289 | His323 | Tyr473 | His449 | Gln286 | ||
| Pioglitazone (the current study) | −10.164 | 2.35 | 1.76 | 2.67 | 2.15 | 2.13 |
| Pioglitazone (reported) | −9.92 3,* | 1.20 1 | 0.70 1 | 1.80 1 | 2.76 1 | 2.18 3,* |
| Rosiglitazone (the current study) | −10.106 | 1.78 | 2.01 | 2.30 | 2.07 | 1.87 |
| Rosiglitazone (BRL-49653) (reported) | −10.22 2 | 1.93 2 | 1.86 2 | 1.81 1 | 1.39 1 | 1.74 3,# |
| Troglitazone (the current study) | −13.098 | 2.13 | 2.46 | 2.03 | 1.88 | 2.04 |
| Troglitazone # (reported) | −12.61 3 | 1.78 3 | 1.83 3 | 2.22 3 | 2.12 3 | 1.89 3 |
| Compound | (-)ΔG (Kcal/mol) | KI (nM) |
|---|---|---|
| Pioglitazone | 10.164 | 36.09 |
| Rosiglitazone | 10.106 | 41.5 |
| Troglitazone | 13.098 | 0.243 |
| Family 1 | 10.04 to 12.26 | 1.0 to 43.9 |
| Family 2 | 9.82 to 12.49 | 0.6 to 63.8 |
| Family 3 | 5.07 to 12.41 | 0.8 to 193.5 |
| Family 4 | 10.12 to 12.59 | 0.6 to 38.2 |
| Family 5 | 8.65 to 12.38 | 0.8 to 459.2 |
| Family 6 | 9.45 to 12.94 | 0.3 to 118.8 |
| Family 7 | 10.73 to 13.14 | 0.2 to 13.7 |
| Family 8 | 10.92 to 13.44 | 0.1 to 9.9 |
| Compound | LogP | TPSA (Å) | MW (g/mol) | nON | nOHNH | n-rotb | Volume (Å) | Violation of Lipinski’s Five Rules |
|---|---|---|---|---|---|---|---|---|
| Pioglitazone | 3.1 | 68.3 | 356.4 | 5 | 1 | 7 | 318.5 | 0 |
| Rosiglitazone | 2.3 | 71.5 | 357.4 | 6 | 1 | 7 | 314.5 | 0 |
| Troglitazone | 4.7 | 84.9 | 441.5 | 6 | 2 | 5 | 395.0 | 0 |
| Families 1–8 | 0.9–4.9 | 68.3–112.2 | 346–493 | 5–7 | 1–3 | 3–9 | 263–426 | 0 |
| Compound | M | T | I | Re | LogP | logS | MW | TPSA (Å) | Dl | Ds |
|---|---|---|---|---|---|---|---|---|---|---|
| PIO | 3.08 | −3.84 | 356 | 93.59 | 5.02 | 0.76 | ||||
| ROSI | 2.16 | −3.67 | 357 | 96.83 | 9.14 | 0.8 | ||||
| TRO | 4.36 | −5.31 | 441 | 110.1 | 4.02 | 0.49 | ||||
| Family 1 | 0.8 to 3.1 | −7.0 to −4.4 | 348 to 452 | 152.1 | −5.3 to 4.1 | 0.3 to 0.7 | ||||
| Family 2 | 0.5 to 2.9 | −6.8 to −4.4 | 347 to 451 | 154.9 to 155.3 | −3.3 to 3.1 | 0.1 to 0.4 | ||||
| Family 3 | 0.7 to 3.0 | −6.7 to −4.4 | 361 to 465 | 146.1 | −1.2 to 5.0 | 0.1 to 0.3 | ||||
| Family 4 | 1.2 to 3.5 | −7.0 to −4.7 | 375 to 479 | 146.1 | −4.9 to 1.2 | 0.1 to 0.2 | ||||
| Family 5 | 1.6 to 4.0 | −6.8 to −4.6 | 346 to 450 | 142.9 | −4.5 to 3.3 | 0.2 to 0.7 | ||||
| Family 6 | 2.1 to 4.4 | −7.1 to −4.7 | 360 to 464 | 142.9 | −2.0 to 4.8 | 0.2 to 0.7 | ||||
| Family 7 | 2.5 to 4.9 | −7.4 to −5.0 | 374 to 478 | 142.0 to 142.9 | −1.4 to 4.9 | 0.2 v 0.6 | ||||
| Family 8 | 2.7 to 5.0 | −7.5 to −5.2 | 388 to 492 | 142.9 | −4.3 to 3.6 | 0.2 to 0.6 |
| Concept | Result | Value |
|---|---|---|
| Absorption | ||
| Blood-brain barrier absorption | BBB+ | 90.49% |
| Human intestinal absorption | HIA+ | 98.65% |
| Caco-2 permeability | Caco2- | 58.90% |
| P-glycoprotein substrate | Non-substrate | 64.43% |
| P-glycoprotein inhibitor | Non-inhibitor | 74.50% |
| Non-inhibitor | 95.59% | |
| Renal organic cation transporter | Non-inhibitor | 85.55% |
| Metabolism | ||
| CYP450 2C9 substrate | Non-substrate | 84.33% |
| CYP450 2D6 substrate | Non-substrate | 80.86% |
| CYP450 3A4 substrate | Non-substrate | 63.45% |
| CYP450 1A2 inhibitor | Inhibitor | 58.04% |
| CYP450 2C9 inhibitor | Inhibitor | 62.32% |
| CYP450 2D6 inhibitor | Non-inhibitor | 67.90% |
| CYP450 2C19 inhibitor | Inhibitor | 55.12% |
| CYP450 3A4 inhibitor | Inhibitor | 58.98% |
| CYP inhibitory promiscuity | High CYP inhibitory promiscuity | 87.62% |
| Toxicity | ||
| Human ether-a-go-go-related gene inhibition | Weak inhibitor | 97.80% |
| Non-inhibitor | 89.45% | |
| Ames toxicity | No AMES toxicity | 74.27% |
| Carcinogen | Non-carcinogen | 85.65% |
| Fish toxicity | high FHMT | 99.08% |
| Tetrahymena pyriformis toxicity | high TPT | 99.39% |
| Honey bee toxicity | high HBT | 54.44% |
| Biodegradation | Not readily biodegraded | 85.42% |
| Acute oral toxicity | III | 56.36% |
| Carcinogenicity (three classes) | Not required | 46.12% |
| Absorption | ||
| Aqueous solubility | LogS | −3.703 |
| Caco-2 permeability | LogPapp, cm/s | 1.0474 |
| Toxicity | ||
| Rat acute toxicity | LD50, mol/kg | 2.3386 |
| Fish toxicity | pLC50, mg/L | 0.7877 |
| Tetrahymena pyriformis toxicity | pIGC50, ug/L | 0.8072 |
| Toxicity OECD/GHS | hERG Blocker | Probability of CYP450 Isoform Inhibition (%) | LD50 for Rats (mg/kg) | LD50 for Mice (mg/kg) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LD50 (mg/kg) | Category | % | 3A4 | 2D6 | 2C9 | 2C19 | 1A2 | ip | PO | ip | PO | IV | SC |
| 50< ≤2000 | III | 49 | 5 | 0 | 27 | 2 | 12 | 97 | 110 | 350 | 790 | 59 | 200 |
| Dose (mg/kg) | Weight Gain for 2-Weeks (g) | ALT (U/L) | AST (U/L) | Toxic Effects |
|---|---|---|---|---|
| 2000 | 31.5 | 14.3 | 50.8 | None |
| 550 | 25.0 | 21.4 | 55.1 | |
| 175 | 30.5 | 34.4 | 64.0 |
| Total Area | Citrate Buffer Vehicle | Type 2 Diabetes without Treatment | Type 2 Diabetes Treated with 1G | Type 2 Diabetes Treated with PIO |
|---|---|---|---|---|
| GTC week 5 | 34,141 | 135,504 | 124,415 | 118,846 |
| GTC week 6 | 29,598 | 137,939 | 126,309 | 116,448 |
| GTC week 7 | 29,931 | 138,256 | 120,404 | 121,853 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Álvarez-Almazán, S.; Navarrete-Vázquez, G.; Padilla-Martínez, I.I.; Correa-Basurto, J.; Alemán-González-Duhart, D.; Tamay-Cach, F.; Mendieta-Wejebe, J.E. A New Symmetrical Thiazolidinedione Derivative: In Silico Design, Synthesis, and In Vivo Evaluation on a Streptozotocin-Induced Rat Model of Diabetes. Processes 2021, 9, 1294. https://doi.org/10.3390/pr9081294
Álvarez-Almazán S, Navarrete-Vázquez G, Padilla-Martínez II, Correa-Basurto J, Alemán-González-Duhart D, Tamay-Cach F, Mendieta-Wejebe JE. A New Symmetrical Thiazolidinedione Derivative: In Silico Design, Synthesis, and In Vivo Evaluation on a Streptozotocin-Induced Rat Model of Diabetes. Processes. 2021; 9(8):1294. https://doi.org/10.3390/pr9081294
Chicago/Turabian StyleÁlvarez-Almazán, Samuel, Gabriel Navarrete-Vázquez, Itzia Irene Padilla-Martínez, José Correa-Basurto, Diana Alemán-González-Duhart, Feliciano Tamay-Cach, and Jessica Elena Mendieta-Wejebe. 2021. "A New Symmetrical Thiazolidinedione Derivative: In Silico Design, Synthesis, and In Vivo Evaluation on a Streptozotocin-Induced Rat Model of Diabetes" Processes 9, no. 8: 1294. https://doi.org/10.3390/pr9081294