
Multi-Residue Analysis of Thyreostats in Animal Muscle Tissues by Hydrophilic Interaction Liquid Chromatography Tandem Mass Spectrometry: A Thorough Chromatographic Study

 , , , and
, , , and 
Abstract
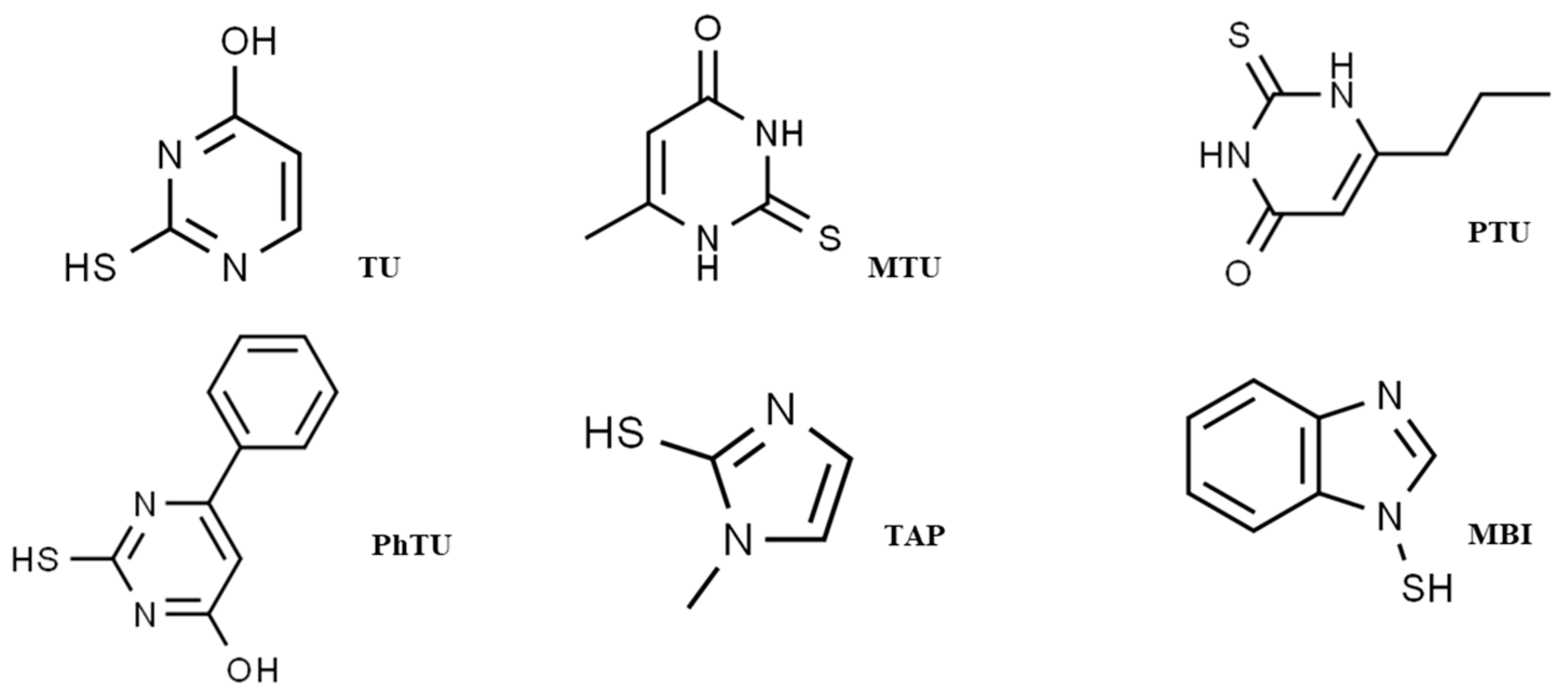
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Samples
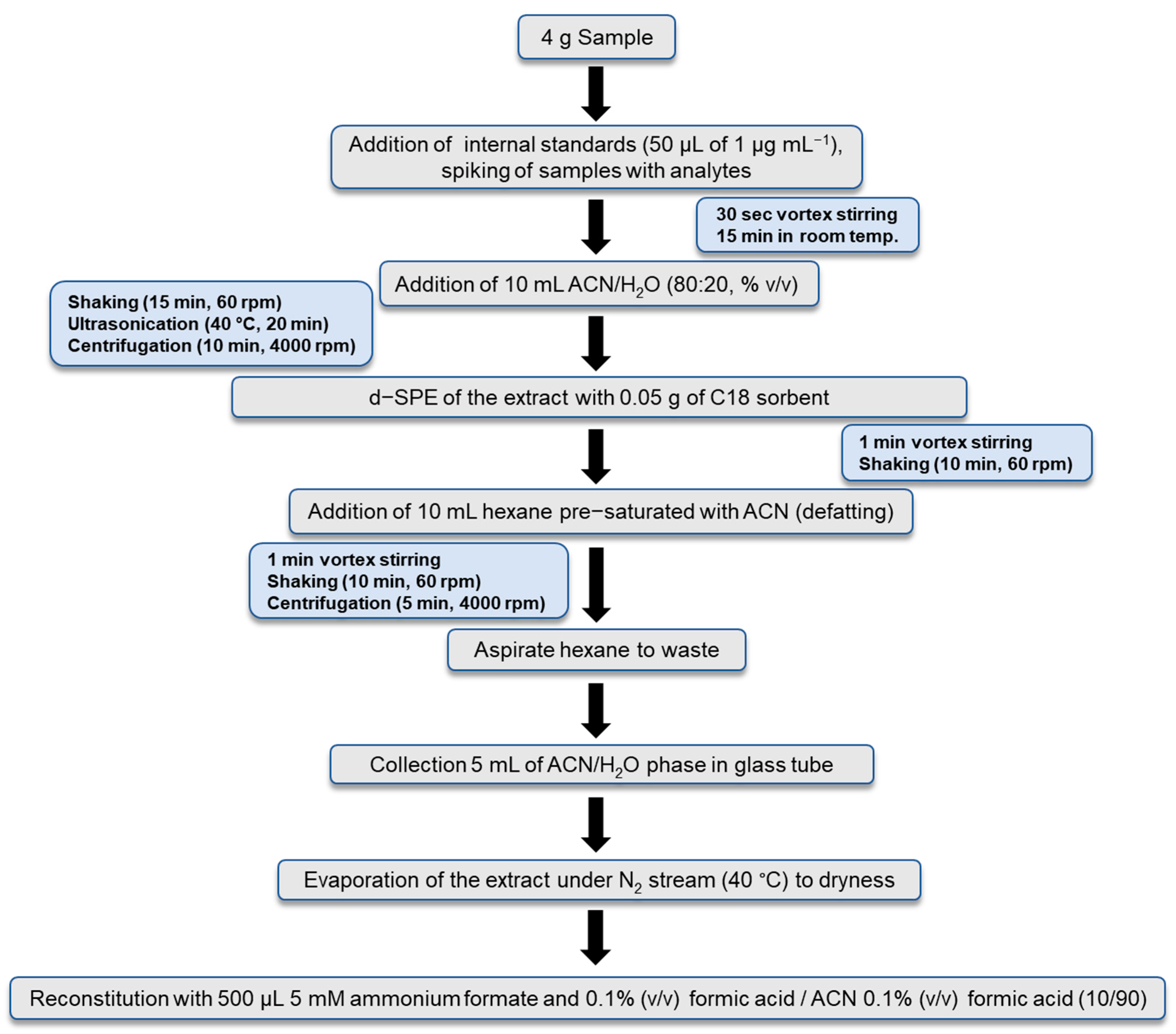
2.3. Sample Preparation Protocol
2.4. HILIC-ESI-MS/MS Measurements
2.4.1. HILIC-ESI-MS/MS Optimisation
2.4.2. Investigation of Retention Mechanism
2.4.3. HILIC-ESI-MS/MS Final Conditions
2.5. Validation
2.6. Application of the Method to Real Samples
3. Results and Discussion
3.1. HILIC-ESI-MS/MS Method Development
3.1.1. Comparison of Ionisation Polarities with Different Mobile Phases
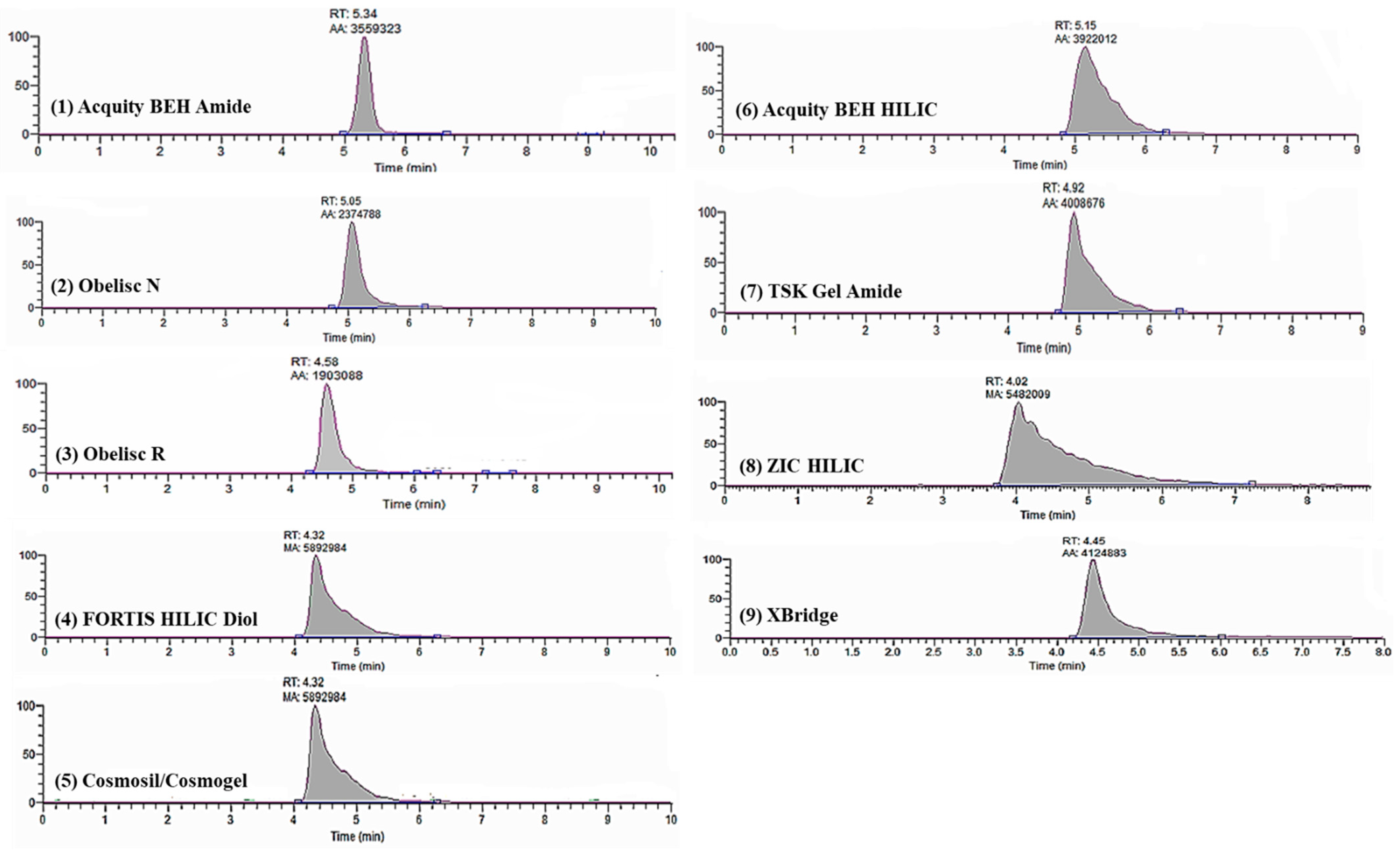
3.1.2. Hydrophilic Interaction Liquid Chromatography
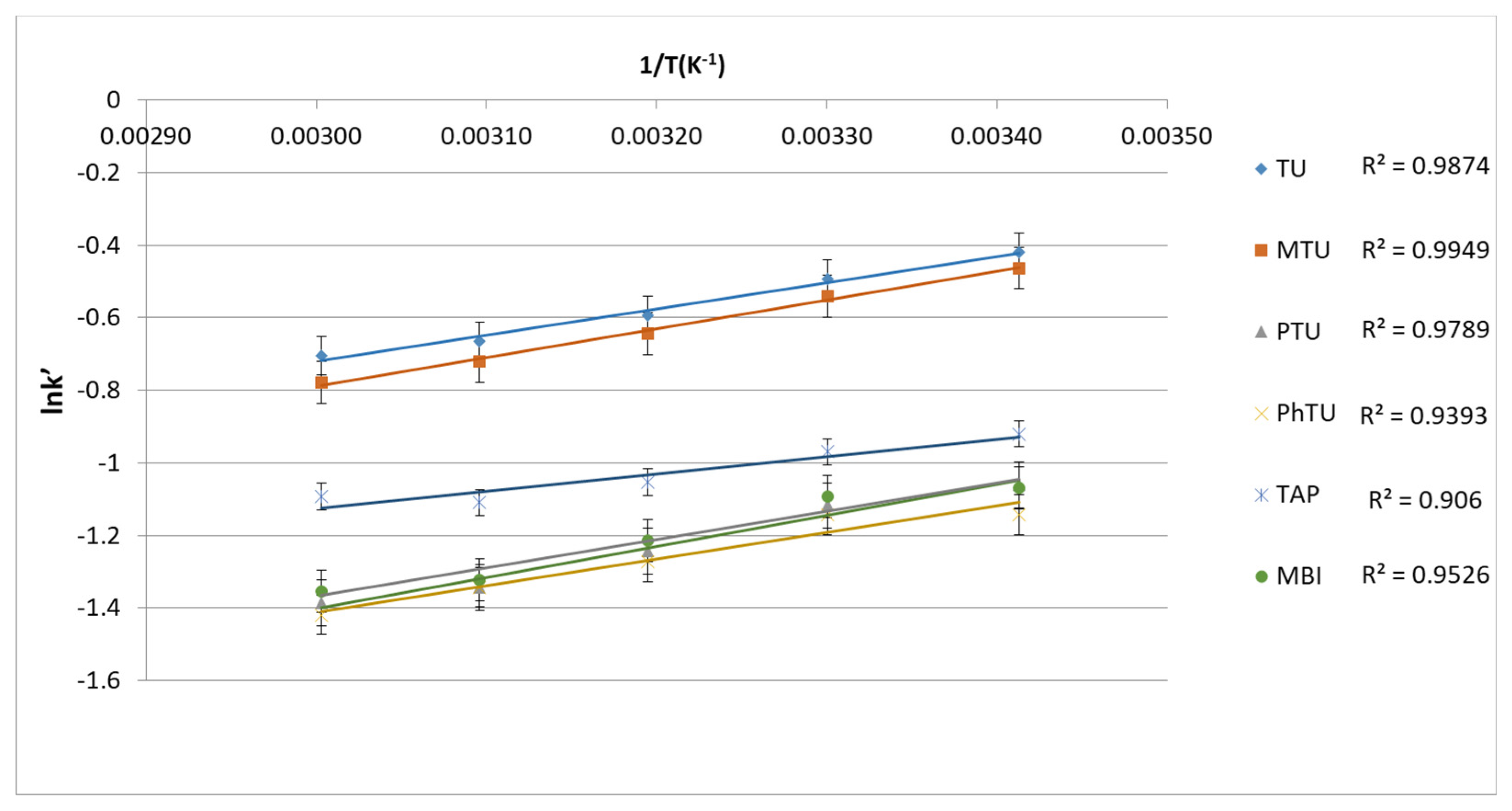
3.1.3. Retention Mechanism of TSs in BEH Amide
3.2. Method Validation
3.3. Application to Real Samples
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Clemente-Suárez, V.J.; Beltrán-Velasco, A.I.; Redondo-Flórez, L.; Martín-Rodríguez, A.; Tornero-Aguilera, J.F. Global Impacts of Western Diet and Its Effects on Metabolism and Health: A Narrative Review. Nutrients 2023, 15, 2749. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, K.S. In-house validation and factorial effect analysis of a liquid chromatography–tandem mass spectrometry method for the determination of thyreostats in bovine blood plasma. Anal. Bioanal. Chem. 2014, 406, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Kusmierek, K.; Bald, E. Determination of methimazole in urine by liquid chromatography. Talanta 2007, 71, 2121–2125. [Google Scholar] [CrossRef]
- Vanden Bussche, J.; Vanhaecke, L.; Deceuninck, Y.; Verheyden, K.; Wille, K.; Bekaert, K.; Le Bizec, B.; De Brabander, H.F. Development and validation of an ultra-high performance liquid chromatography tandem mass spectrometry method for quantifying thyreostats in urine without derivatisation. J. Chromatogr. A 2010, 1217, 4285–4293. [Google Scholar] [CrossRef]
- Blokland, M.H.; van Tricht, F.E.; Groot, M.J.; Van Ginkel, L.A.; Sterk, S.S. Discrimination between the exogenous and endogenous origin of thiouracil in farm animals, the final chapter? Food Addit. Contam. Part A 2021, 38, 2077–2090. [Google Scholar] [CrossRef]
- World Health Organization International Agency for Research on Cancer. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans—Some Thyrotropic Agents; IARC Press: Lyon, France, 2001; Volume 79. [Google Scholar]
- Council Directive 81/602/EEC of 31 July 1981 concerning the prohibition of certain substances having a hormonal action and of any substances having a thyrostatic action. Off. J. EC 1981, L222, 32–33.
- European Union Reference Laboratories. Eurl Guidance on Minimum Method Performance Requirements (Mmprs) for Specific Pharmacologically Active Substances in Specific Animal Matrices, Version 2.0 June 2022. Available online: https://eurl-residues.eu/wp-content/uploads/2020/12/EURL_MMPR_guidance-paper_final.pdf (accessed on 12 May 2024).
- Commission Implementing Regulation (EU). 2021/808 of 22 March 2021 on the performance of analytical methods for residues of pharmacologically active substances used in food-producing animals and on the interpretation of results as well as on the methods to be used for sampling and repealing Decisions 2002/657/EC and 98/179/EC. Off. J. EC 2021, L180, 84–109. [Google Scholar]
- COMMISSION DECISION 2002/657/EC of 12 August 2002 implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results. Off. J. EC 2002, L221, 8–36.
- Církva, A.; Šťastný, K. Method for the determination of thyreostats in milk samples using LC-MS/MS. Food Addit. Contam. Part A 2013, 30, 983–986. [Google Scholar] [CrossRef] [PubMed]
- Bertuzzi, T.; Rastelli, S.; Mulazzi, A.; Pietri, A. Trace Monitoring of Goitrin and Thiouracil in Milk and Cheese. Food Anal. Methods 2016, 9, 2952–2959. [Google Scholar] [CrossRef]
- Ji, Z.; Yu, Y.; Jin, Q.; Li, G.; Hu, N.; Zhou, W.; Suo, Y.; Sun, Z.; You, J. Determination of naturally occurring thyreostats in bovine milk by high performance liquid chromatography combined with fluorescence detection. Microchem. J. 2019, 145, 892–898. [Google Scholar] [CrossRef]
- Gentili, A.; De Pratti, V.; Caretti, F.; Pérez-Fernández, V.; Tomai, P.; Curini, R.; Mainero Rocca, L. Residue analysis of thyreostats in baby foods via matrix solid phase dispersion and liquid chromatography—Dual-polarity electrospray—Tandem mass spectrometry. Food Addit. Contam. Part A 2016, 33, 1793–1802. [Google Scholar] [CrossRef] [PubMed]
- Kiebooms, J.A.L.; Wauters, J.; Vanden Bussche, J.; Vanhaecke, L. Validation of a quantitative method using liquid chromatography coupled to multiple mass spectrometry for thiouracil in feedstuffs used in animal husbandry. Anal. Bioanal. Chem. 2015, 407, 4373–4384. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, B.; Zuchowska, I.M.; Zmudzki, J.; Jedziniak, P.; Korycinska, B.; Sielska, K.; Witek, S.; Klopot, A. Liquid chromatography tandem mass spectrometry with ion trap and triple quadrupole analyzers for determination of thyreostatic drugs in urine and muscle tissue. Anal. Chim. Acta. 2011, 700, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Purves, R.W.; Souster, K.; West, M.; Huda, A.M.; Fisher, C.M.E.; Belford, M.W.; Shurmer, B.O. Improved Thyreostatic Drug Detection in Animal Tissues Using Liquid Chromatography–High-Field Asymmetric Waveform Ion Mobility Spectrometry–Mass Spectrometry. J. Agric. Food Chem. 2022, 70, 4785–4791. [Google Scholar] [CrossRef] [PubMed]
- Wang, T. Polyethyleneimine-modified hybrid silica sorbent for hydrophilic solid-phase extraction of thyreostats in animal tissues. J. Chromatogr. A 2018, 1581–1582, 16–24. [Google Scholar] [CrossRef]
- Rocchi, S.; Caretti, F.; Mainero Rocca, L.; Pérez-Fernández, V.; Tomai, P.; Dal Bosco, C.; Curini, R.; Gentili, A. Subcritical water extraction of thyreostats from bovine muscle followed by liquid chromatography-tandem mass spectrometry. Food Addit. Contam. Part A 2018, 35, 1472–1483. [Google Scholar] [CrossRef]
- Casey, C.R.; Andersen, W.C.; Williams, N.T.; Nickel, T.J.; Ayres, P.R. Multiclass, Multiresidue Method for the Quantification and Confirmation of 112 Veterinary Drugs in Game Meat (Bison, Deer, Elk, and Rabbit) by Rapid Polarity Switching Liquid Chromatography-Tandem Mass Spectrometry. J. Agric. Food Chem. 2021, 69, 1175–1186. [Google Scholar] [CrossRef]
- Nuñez, A.; Lehotay, S.J.; Lightfield, A.R. Structural characterization of product ions of regulated veterinary drugs by electrospray ionization and quadrupole time-of-flight mass spectrometry. Part 3, Anthelmintics and thyreostats. Rapid Commun. Mass Spectrom. 2016, 30, 813–822. [Google Scholar] [CrossRef]
- Guarducci, M.A.; Fochetti, A.; Ciogli, A.; Mazzoccanti, G.A. Compendium of the Principal Stationary Phases Used in Hydrophilic Interaction Chromatography: Where Have We Arrived? Separations 2023, 10, 22. [Google Scholar] [CrossRef]
- Logotheti, M.; Theochari, K.; Kostakis, M.; Pasias, I.N.; Thomaidis, N.S. Development and validation of a HILIC-UV method for the determination of nucleotides in fish samples. Food Chem. 2018, 248, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Kokotou, M.G.; Thomaidis, N.S. Characterization of the Retention of Artificial Sweeteners by Hydrophilic Interaction Liquid Chromatography. Anal. Lett. 2018, 51, 49–72. [Google Scholar] [CrossRef]
- Geis-Asteggiante, L.; Lehotay, S.J.; Lightfield, A.R.; Dutko, T.; Ng, C.; Bluhm, L. Ruggedness testing and validation of a practical analytical method for >100 veterinary drug residues in bovine muscle by ultrahigh performance liquid chromatography–tandem mass spectrometry. J. Chromatogr. A 2012, 1258, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Dasenaki, M.E.; Thomaidis, N.S. Multi-residue methodology for the determination of 16 coccidiostats in animal tissues and eggs by hydrophilic interaction liquid chromatography – Tandem mass spectrometry. Food Chem. 2019, 275, 668–680. [Google Scholar] [CrossRef]
- Dasenaki, M.E.; Thomaidis, N.S. Multi-residue determination of 115 veterinary drugs and pharmaceutical residues in milk powder, butter, fish tissue and eggs using liquid chromatography–tandem mass spectrometry. Anal. Chim. Acta. 2015, 880, 103–121. [Google Scholar] [CrossRef]
- Dasenaki, M.E.; Michali, C.S.; Thomaidis, N.S. Analysis of 76 veterinary pharmaceuticals from 13 classes including aminoglycosides in bovine muscle by hydrophilic interaction liquid chromatography–tandem mass spectrometry. J. Chromatogr. A 2016, 1452, 67–80. [Google Scholar] [CrossRef]
- Lega, F.; Contiero, L.; Biancotto, G.; Angeletti, R. Determination of thyreostats in muscle and thyroid tissues by QuEChERS extraction and ultra-performance liquid chromatography tandem mass spectrometry. Food Addit. Contam. Part A 2013, 30, 949–957. [Google Scholar] [CrossRef] [PubMed]
- Maragou, N.C.; Thomaidis, N.S.; Koupparis, M.A. Optimization and comparison of ESI and APCI LC-MS/MS methods: A case study of Irgarol 1051, Diuron, and their degradation products in environmental samples. J. Am. Soc. Mass Spectrom. 2011, 22, 1826–1838. [Google Scholar] [CrossRef]
- McCalley, D.V. A study of column equilibration time in hydrophilic interaction chromatography. J. Chromatogr. A 2018, 1554, 61–70. [Google Scholar] [CrossRef]
- Shamshir, A.; Dinh, N.P.; Jonsson, T.; Sparrman, T.; Irgum, K. Probing the retention mechanism of small hydrophilic molecules in hydrophilic interaction chromatography using saturation transfer difference nuclear magnetic resonance spectroscopy. J. Chromatogr. A 2020, 1623, 461130. [Google Scholar] [CrossRef]
- Wang, F.; Yang, F.; Liu, J.; Bai, Q. Studies on the retention mechanism of solutes in hydrophilic interaction chromatography using stoichiometric displacement theory II. HILIC/RPLC dual-retention mechanism of solutes in hydrophilic interaction chromatography over the entire range of water concentration in mobile phase. Talanta 2023, 256, 124858. [Google Scholar] [CrossRef]
- Guo, Y.; Gaiki, S. Retention behavior of small polar compounds on polar stationary phases in hydrophilic interaction chromatography. J. Chromatogr. A 2005, 1074, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Baran, D. Hydrophilic Partitioning or Surface Adsorption? A Quantitative Assessment of Retention Mechanisms for Hydrophilic Interaction Chromatography (HILIC). Molecules 2023, 28, 6459. [Google Scholar] [CrossRef] [PubMed]
- Buszewski, B.; Noga, S. Hydrophilic interaction liquid chromatography (HILIC)—A powerful separation technique. Anal. Bioanal. Chem. 2012, 402, 231–247. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analytes | RT (min) | Tube Lens (V) | SRM1 (Q) SRM2 (C) | CE (eV) |
|---|---|---|---|---|
| 6-phenyl-2-thiouracil (PhTU) | 4.53 | 78 | 205.0 > 103.2 | 26 |
| 205.0 > 146.1 | 19 | |||
| 2-mercaptobenzimidazole (MBI) | 4.55 | 75 | 151.0 > 93.3 | 20 |
| 151.0 > 118.2 | 25 | |||
| 6-propyl-2-thiouracil (PTU) | 4.56 | 75 | 171.1 > 154.1 | 16 |
| 171.1 > 112.2 | 19 | |||
| 6-propyl-2-thiouracil-d5 (PTU-d5) (IS) | 4.56 | 79 | 176.0 > 117.2 | 19 |
| 176.0 > 159.1 | 18 | |||
| Methimazole (TAP) | 4.70 | 63 | 115.0 > 88.2 | 18 |
| 115.0 > 57.4 | 16 | |||
| Methimazole-d3 (TAP-d3) (IS) | 4.71 | 63 | 118.0 > 91.2 | 17 |
| 118.0 > 60.4 | 20 | |||
| 5,6-dimethyl-2-thiouracil (DMTU) (IS) | 5.12 | 74 | 157.0 > 140.1 | 16 |
| 157.0 > 98.2 | 18 | |||
| 6-methyl-2-thiouracil (MTU) | 5.47 | 68 | 143.1 > 126.1 | 16 |
| 143.1 > 84.3 | 17 | |||
| 2-thiouracil (TU) | 5.57 | 67 | 129.0 > 112.2 | 15 |
| 129.0 > 84.3 | 28 |
| Thyreostats | 5 ng g−1 | 10 ng g−1 | 15 ng g−1 | |||
|---|---|---|---|---|---|---|
| Average %Rec. | %RSD | Average %Rec. | %RSD | Average %Rec. | %RSD | |
| 2-thiouracil | 84.5 | 9.5 | 82.8 | 8.6 | 88.7 | 5.5 |
| 6-methyl-2-thiouracil | 89.2 | 10 | 93.6 | 14 | 91.7 | 8.5 |
| 6-propyl-2-thiouracil | 98.9 | 6.7 | 103 | 7.3 | 98.5 | 11 |
| 6-phenyl-2-thiouracil | 80.7 | 10.3 | 79.5 | 14 | 76.8 | 8.8 |
| Methimazole | 96.2 | 6.9 | 100 | 6 | 102 | 5.7 |
| 2-mercaptobenzimidazole | 80.2 | 12 | 84.7 | 13 | 76.6 | 9.7 |
| Thyreostats | LOD | LOQ | CCα | CCβ |
|---|---|---|---|---|
| 2-thiouracil | 4.1 | 9.6 | 3.7 | 4.8 |
| 6-methyl-2-thiouracil | 2.9 | 9.4 | 5.2 | 8.1 |
| 6-propyl-2-thiouracil | 3.3 | 7.1 | 8 | 9.3 |
| 6-phenyl-2-thiouracil | 2.8 | 8.3 | 7.5 | 9.0 |
| Methimazole | 2.9 | 8.7 | 6.1 | 8.4 |
| 2-mercaptobenzimidazole | 3.8 | 9.2 | 9.7 | 11 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kritikou, A.S.; Dasenaki, M.E.; Maragou, N.C.; Kostakis, M.G.; Thomaidis, N.S. Multi-Residue Analysis of Thyreostats in Animal Muscle Tissues by Hydrophilic Interaction Liquid Chromatography Tandem Mass Spectrometry: A Thorough Chromatographic Study. Separations 2024, 11, 269. https://doi.org/10.3390/separations11090269
Kritikou AS, Dasenaki ME, Maragou NC, Kostakis MG, Thomaidis NS. Multi-Residue Analysis of Thyreostats in Animal Muscle Tissues by Hydrophilic Interaction Liquid Chromatography Tandem Mass Spectrometry: A Thorough Chromatographic Study. Separations. 2024; 11(9):269. https://doi.org/10.3390/separations11090269
Chicago/Turabian StyleKritikou, Anastasia S., Marilena E. Dasenaki, Niki C. Maragou, Marios G. Kostakis, and Nikolaos S. Thomaidis. 2024. "Multi-Residue Analysis of Thyreostats in Animal Muscle Tissues by Hydrophilic Interaction Liquid Chromatography Tandem Mass Spectrometry: A Thorough Chromatographic Study" Separations 11, no. 9: 269. https://doi.org/10.3390/separations11090269
APA StyleKritikou, A. S., Dasenaki, M. E., Maragou, N. C., Kostakis, M. G., & Thomaidis, N. S. (2024). Multi-Residue Analysis of Thyreostats in Animal Muscle Tissues by Hydrophilic Interaction Liquid Chromatography Tandem Mass Spectrometry: A Thorough Chromatographic Study. Separations, 11(9), 269. https://doi.org/10.3390/separations11090269