Propeller-Shaped Aluminum Complexes with an Azaperylene Core in the Ligands

 , , ,
, , ,
Abstract
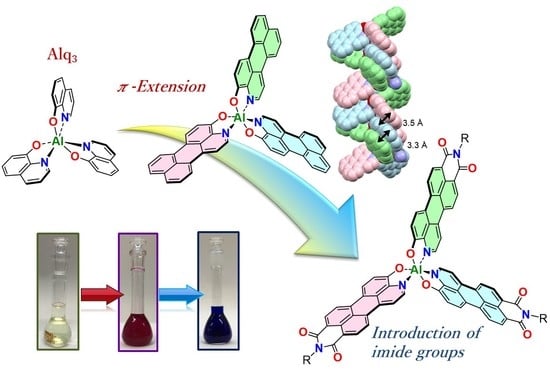
1. Introduction
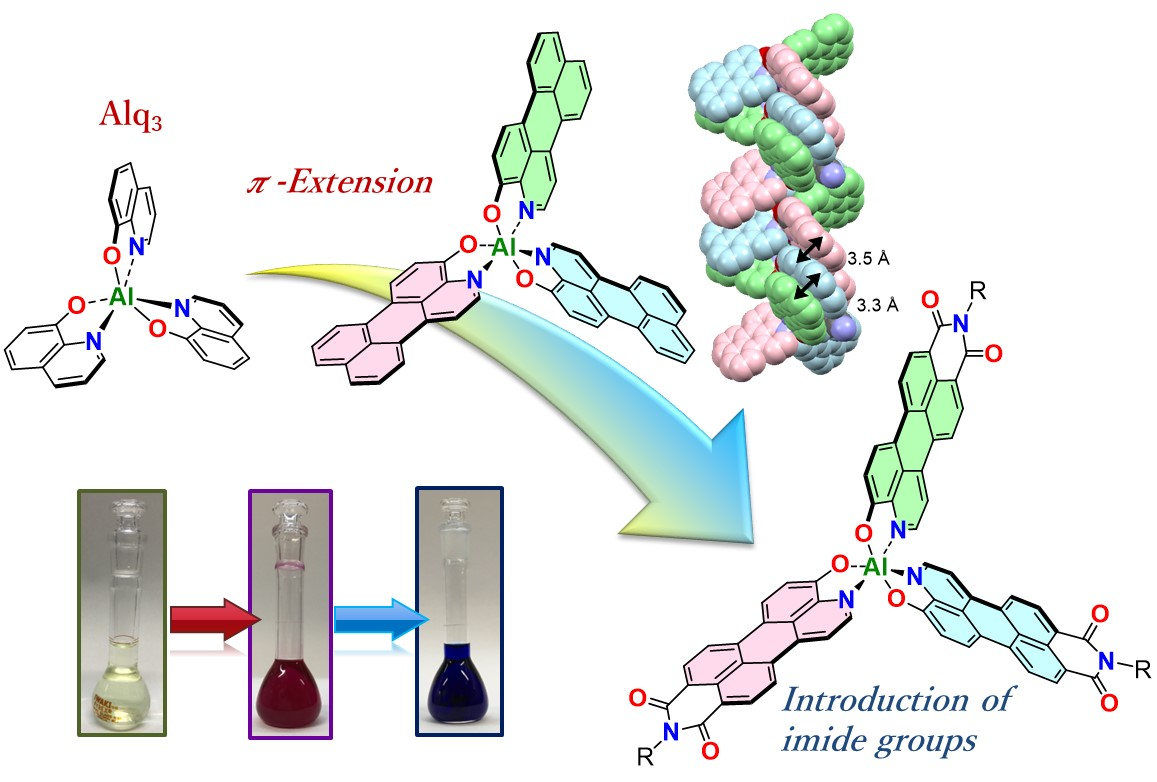
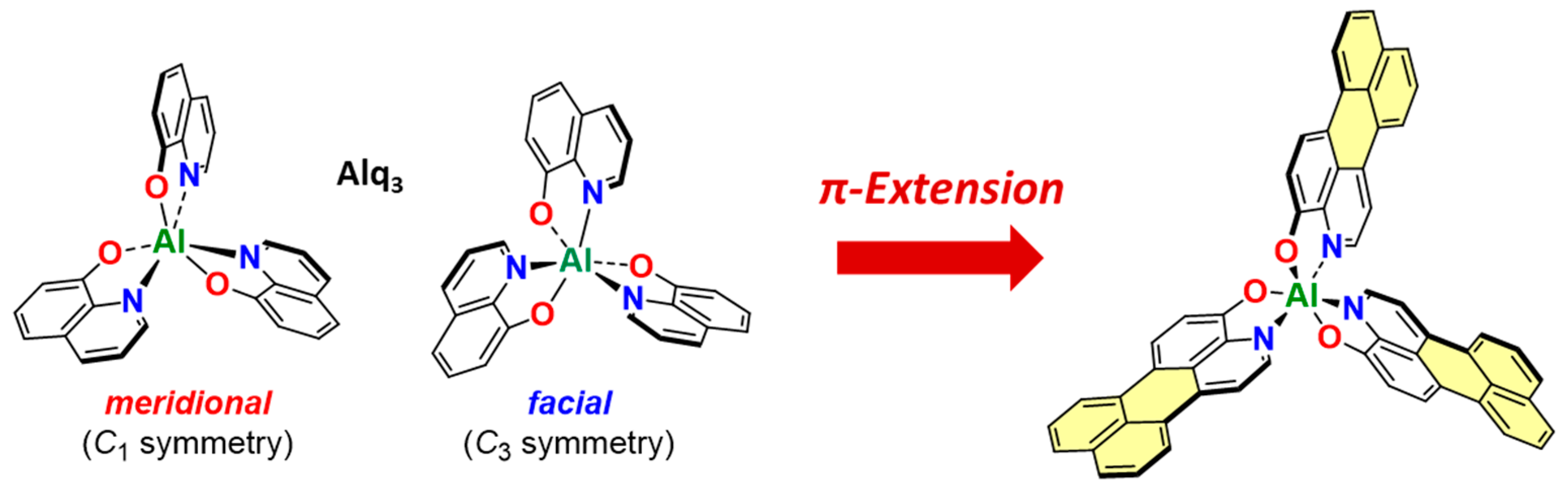
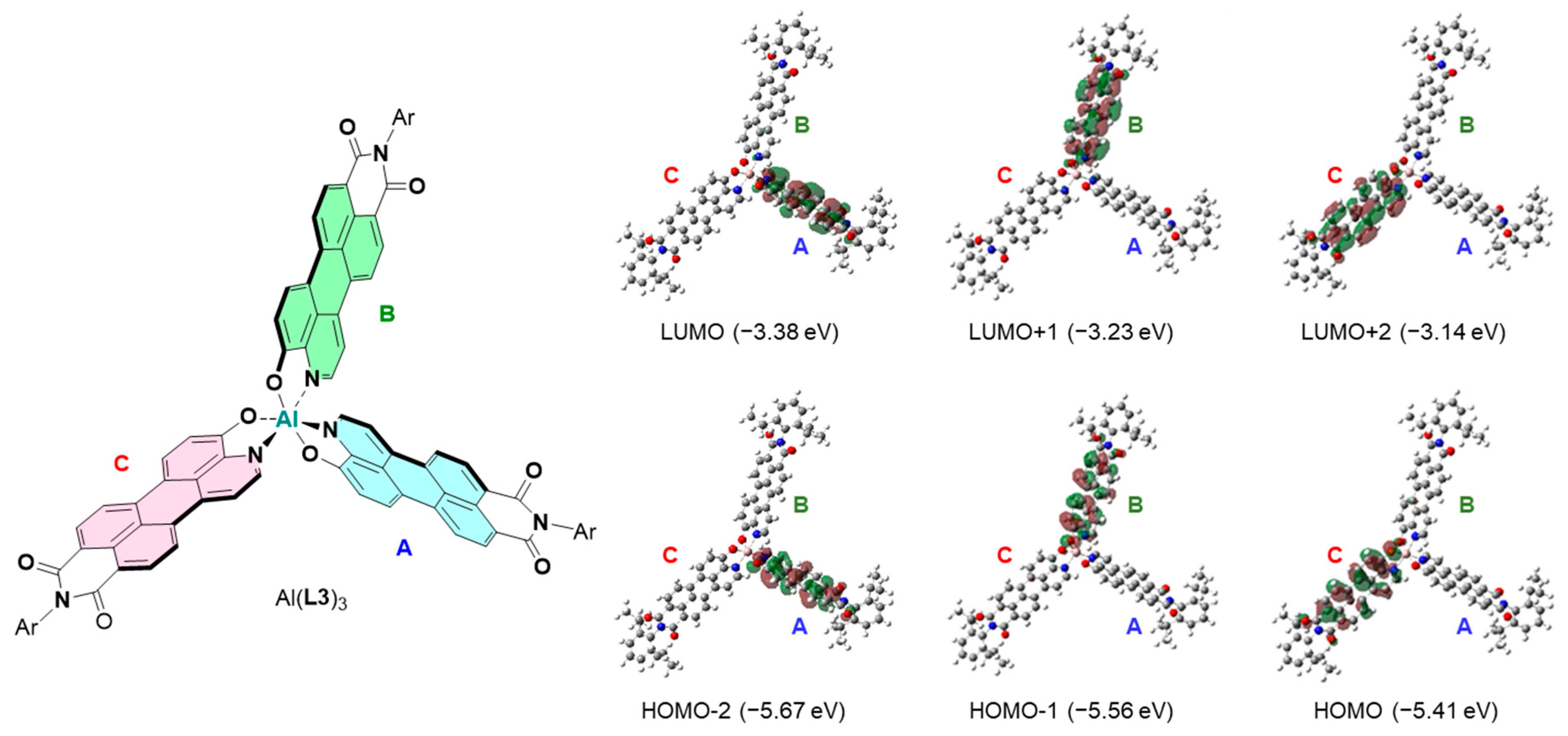
2. Results and Discussion
3. Materials and methods
3.1. Synthesis of Compound 1
3.2. Synthesis of Compounds 2 and 3
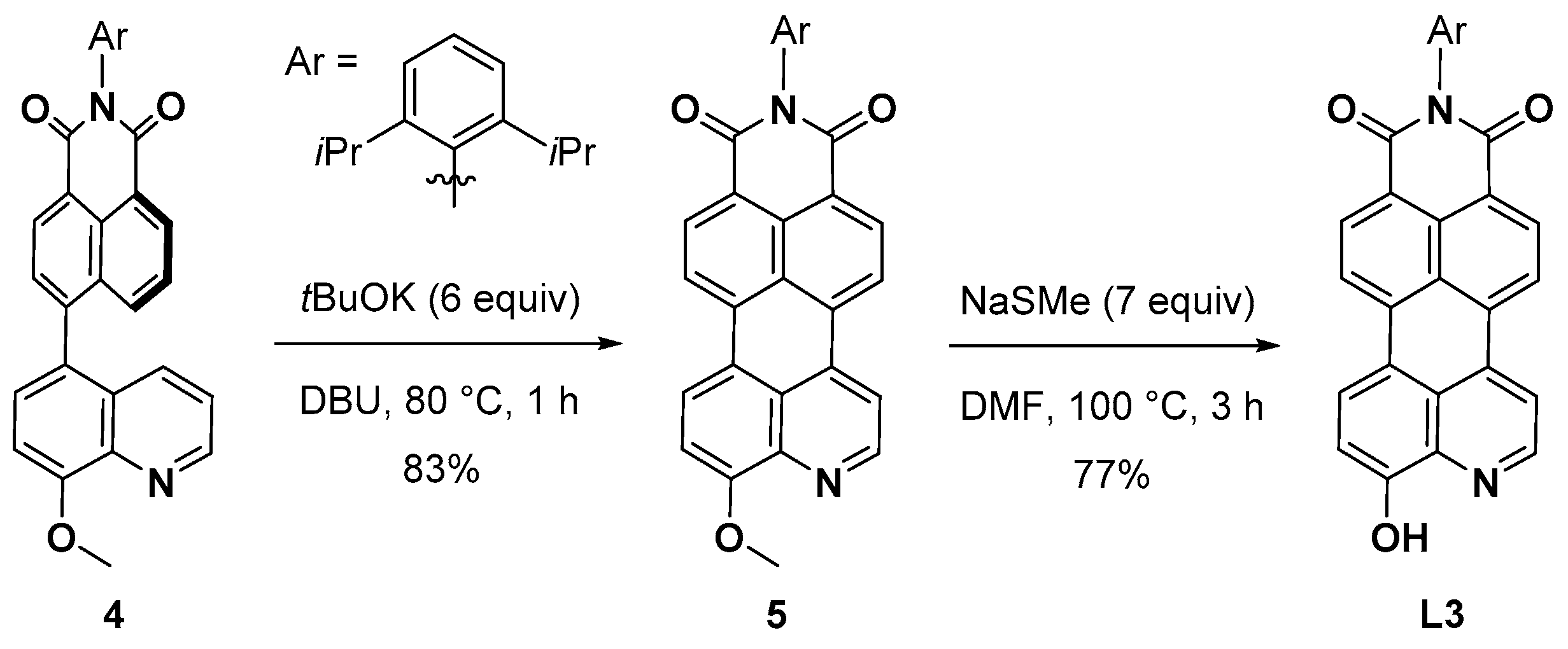
3.3. Synthesis of Compound 4
3.4. Synthesis of Compound 5
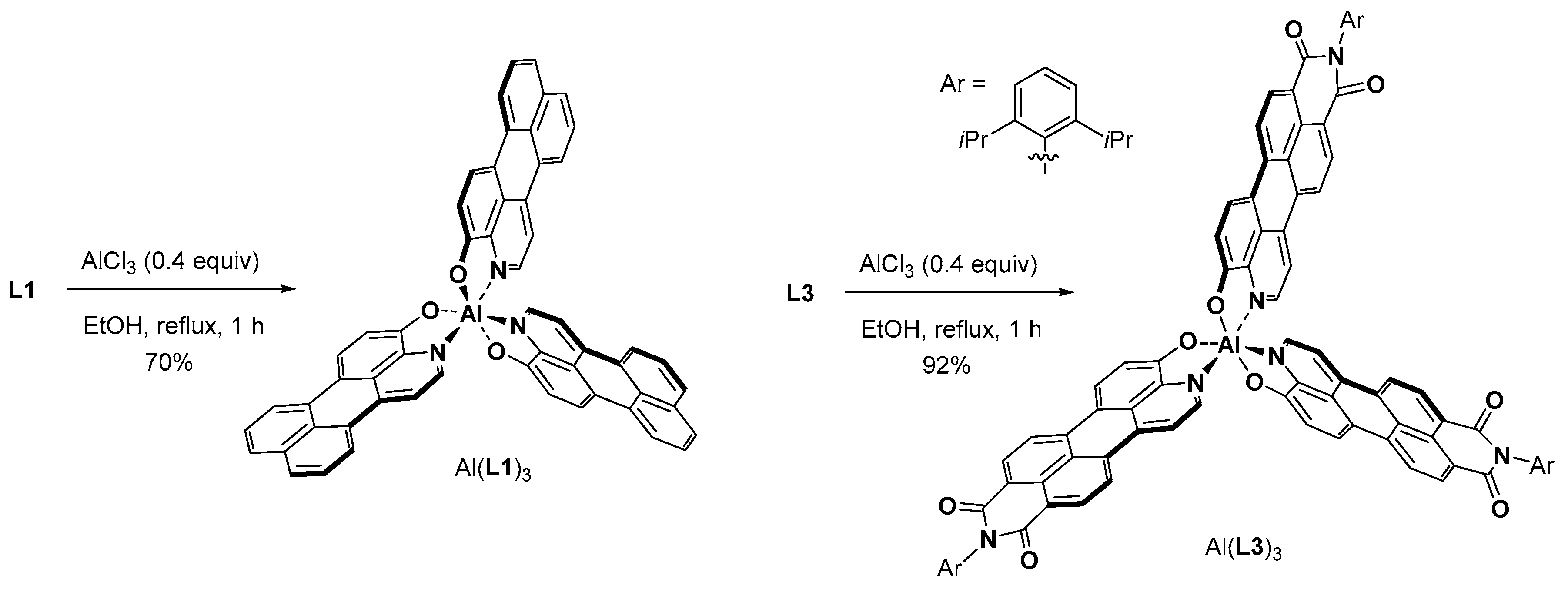
3.5. Synthesis of Compound L1
3.6. Synthesis of Compound L2
3.7. Synthesis of Compound L3
3.8. Synthesis of Compound Al(L1)3
3.9. Synthesis of Compound Al(L3)3
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhang, Z.; Burkholder, E.; Zubieta, J. Non-merohedrally twinned crystals of N,N′-bis(3-methylphenyl)-N,N′-diphenyl-1,1′-biphenyl-4,4′-diamine: An excellent triphenylamine-based hole transporter. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 2004, 60, 452–454. [Google Scholar] [CrossRef] [PubMed]
- Wakamiya, A.; Nishimura, H.; Fukushima, T.; Suzuki, F.; Saeki, A.; Seki, S.; Osaka, I.; Sasamori, T.; Murata, M.; Murata, Y.; et al. On-Top π-Stacking of Quasiplanar Molecules in Hole-Transporting Materials: Inducing Anisotropic Carrier Mobility in Amorphous Films. Angew. Chem. 2014, 126, 5910–5914. [Google Scholar] [CrossRef]
- Nishimura, H.; Ishida, N.; Shimazaki, A.; Wakamiya, A.; Saeki, A.; Scott, L.T.; Murata, Y. Hole-Transporting Materials with a Two-Dimensionally Expanded π-System around an Azulene Core for Efficient Perovskite Solar Cells. J. Am. Chem. Soc. 2015, 137, 15656–15659. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Kumar, B.; Oh, S.; Trinh, M.T.; Wu, Y.; Elbert, K.; Li, P.; Zhu, X.; Xiao, S.; Ng, F.; et al. Helical Ribbons for Molecular Electronics. J. Am. Chem. Soc. 2014, 136, 8122–8130. [Google Scholar] [CrossRef] [PubMed]
- Lv, L.; Roberts, J.; Xiao, C.; Jia, Z.; Jiang, W.; Zhang, G.; Risko, C.; Zhang, L. Triperyleno[3,3,3]propellane triimides: Achieving a new generation of quasi-D3h symmetric nanostructures in organic electronics. Chem. Sci. 2019, 10, 4951–4958. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Earmme, T.; Ren, G.; Saeki, A.; Yoshikawa, S.; Murari, N.M.; Subramaniyan, S.; Crane, M.J.; Seki, S.; Jenekhe, S.A. Beyond Fullerenes: Design of Nonfullerene Acceptors for Efficient Organic Photovoltaics. J. Am. Chem. Soc. 2014, 136, 14589–14597. [Google Scholar] [CrossRef] [PubMed]
- Menke, E.H.; Lami, V.; Vaynzof, Y.; Mastalerz, M. π-Extended rigid triptycene-trisaroylenimidazoles as electron acceptors. Chem. Commun. 2016, 52, 1048–1051. [Google Scholar] [CrossRef] [PubMed]
- Meng, D.; Fu, H.; Fan, B.; Zhang, J.; Li, Y.; Sun, Y.; Wang, Z. Rigid Nonfullerene Acceptors Based on Triptycene-Perylene Dye for Organic Solar Cells. Chem. Asian J. 2017, 12, 1286–1290. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.M.; Wu, Q.; Zhao, D.; Chen, W.; Yu, L. Covalently Bound Clusters of Alpha-Substituted PDI—Rival Electron Acceptors to Fullerene for Organic Solar Cells. J. Am. Chem. Soc. 2016, 138, 7248–7251. [Google Scholar]
- Gao, G.; Liang, N.; Geng, H.; Jiang, W.; Fu, H.; Feng, J.; Hou, J.; Feng, X.; Wang, Z. Spiro-Fused Perylene Diimide Arrays. J. Am. Chem. Soc. 2017, 139, 15914–15920. [Google Scholar] [CrossRef]
- Zhang, J.; Li, Y.; Huang, J.; Hu, H.; Zhang, G.; Ma, T.; Chow, P.C.Y.; Ade, H.; Pan, D.; Yan, H. Ring-Fusion of Perylene Diimide Acceptor Enabling Efficient Nonfullerene Organic Solar Cells with a Small Voltage Loss. J. Am. Chem. Soc. 2017, 139, 16092–16095. [Google Scholar] [CrossRef] [PubMed]
- Peurifoy, S.R.; Castro, E.; Liu, F.; Zhu, X.-Y.; Ng, F.; Jockusch, S.; Steigerwald, M.L.; Echegoyen, L.; Nuckolls, C.; Sisto, T.J. Three-Dimensional Graphene Nanostructures. J. Am. Chem. Soc. 2018, 140, 9341–9345. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.W.; Vanslyke, S.A. Organic Electroluminescent Diodes. Appl. Phys. Lett. 1987, 51, 913–915. [Google Scholar] [CrossRef]
- Desai, P.; Shakya, P.; Kreouzis, T.; Gillin, W.; Morley, N.; Gillin, W.P.; Morley, N.A.; Gibbs, M.R.J. Magnetoresistance and efficiency measurements of Alq3-based OLEDs. Phys. Rev. B 2007, 75, 094423. [Google Scholar] [CrossRef]
- Utz, M.; Nandagopal, M.; Mathai, M.; Papadimitrakopoulos, F. Characterization of isomers in aluminum tris(quinoline-8-olate) by one-dimensional [sup 27]Al nuclear magnetic resonance under magic-angle spinning. Appl. Phys. Lett. 2003, 83, 4023. [Google Scholar] [CrossRef]
- Utz, M.; Chen, C.; Morton, M.; Papadimitrakopoulos, F. Ligand Exchange Dynamics in Aluminum Tris-(Quinoline-8-olate): A Solution State NMR Study. J. Am. Chem. Soc. 2003, 125, 1371–1375. [Google Scholar] [CrossRef] [PubMed]
- Kaji, H.; Kusaka, Y.; Onoyama, G.; Horii, F. CP/MAS13C NMR Characterization of the Isomeric States and Intermolecular Packing in Tris(8-hydroxyquinoline) Aluminum(III) (Alq3). J. Am. Chem. Soc. 2006, 128, 4292–4297. [Google Scholar] [CrossRef] [PubMed]
- Pohl, R.; Anzenbacher, P. Emission Color Tuning in AlQ3 Complexes with Extended Conjugated Chromophores. Org. Lett. 2003, 5, 2769–2772. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Kiburu, I.; Shah, S.; Jäkle, F. Luminescence Tuning of Organoboron Quinolates through Substituent Variation at the 5-Position of the Quinolato Moiety. Org. Lett. 2006, 8, 5227–5230. [Google Scholar] [CrossRef]
- D’Souza, F.; Maligaspe, E.; Zandler, M.E.; Subbaiyan, N.K.; Ohkubo, K.; Fukuzumi, S. Metal Quinolinolate-Fullerene(s) Donor-Acceptor Complexes: Evidence for Organic LED Molecules Acting as Electron Donors in Photoinduced Electron-Transfer Reactions. J. Am. Chem. Soc. 2008, 130, 16959–16967. [Google Scholar] [CrossRef]
- Zlojutro, V.; Hudson, Z.M.; Sun, Y.; Wang, S. Triarylboron-functionalized 8-hydroxyquinolines and their aluminium(iii) complexes. Chem. Commun. 2011, 47, 3837. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, H.; Wan, L.; Bian, Y.; Jiang, J. 8-Hydroxyquinoline-Substituted Boron–Dipyrromethene Compounds: Synthesis, Structure, and OFF–ON–OFF Type of pH-Sensing Properties. J. Org. Chem. 2011, 76, 3774–3781. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Wang, S.; Zhang, L.; Liu, S.; Yuan, G. 8-Hydroxyquinolinate-Based Metal–Organic Frameworks: Synthesis, Tunable Luminescent Properties, and Highly Sensitive Detection of Small Molecules and Metal Ions. Inorg. Chem. 2019, 58, 2444–2453. [Google Scholar] [CrossRef] [PubMed]
- Campeau, L.-C.; Parisien, M.; Jean, A.; Fagnou, K. Catalytic Direct Arylation with Aryl Chlorides, Bromides, and Iodides: Intramolecular Studies Leading to New Intermolecular Reactions. J. Am. Chem. Soc. 2006, 128, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Lafrance, M.; Fagnou, K. Palladium-Catalyzed Benzene Arylation: Incorporation of Catalytic Pivalic Acid as a Proton Shuttle and a Key Element in Catalyst Design. J. Am. Chem. Soc. 2006, 128, 16496–16497. [Google Scholar] [CrossRef] [PubMed]
- Gorelsky, S.I.; Lapointe, D.; Fagnou, K. Analysis of the Concerted Metalation-Deprotonation Mechanism in Palladium-Catalyzed Direct Arylation across a Broad Range of Aromatic Substrates. J. Am. Chem. Soc. 2008, 130, 10848–10849. [Google Scholar] [CrossRef]
- Baghbanzadeh, M.; Pilger, C.; Kappe, C.O. Palladium-Catalyzed Direct Arylation of Heteroaromatic Compounds: Improved Conditions Utilizing Controlled Microwave Heating. J. Org. Chem. 2011, 76, 8138–8142. [Google Scholar] [CrossRef] [PubMed]
- Alberico, D.; Scott, M.E.; Lautens, M. Aryl−Aryl Bond Formation by Transition-Metal-Catalyzed Direct Arylation. Chem. Rev. 2007, 107, 174–238. [Google Scholar] [CrossRef]
- Neufeldt, S.R.; Sanford, M.S. Controlling Site Selectivity in Palladium-Catalyzed C–H Bond Functionalization. Acc. Chem. Res. 2012, 45, 936–946. [Google Scholar] [CrossRef]
- Tan, Y.; Hartwig, J.F. Assessment of the Intermediacy of Arylpalladium Carboxylate Complexes in the Direct Arylation of Benzene: Evidence for C–H Bond Cleavage by “Ligandless” Species. J. Am. Chem. Soc. 2011, 133, 3308–3311. [Google Scholar] [CrossRef]
- Seifert, S.; Schmidt, D.; Shoyama, K.; Würthner, F. Base-Selective Five- versus Six-Membered Ring Annulation in Palladium-Catalyzed C–C Coupling Cascade Reactions: New Access to Electron-Poor Polycyclic Aromatic Dicarboximides. Angew. Chem. Int. Ed. 2017, 56, 7595–7600. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision E.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Ueda, K.; Yanagisawa, S.; Yamaguchi, J.; Itami, K. A General Catalyst for the β-Selective C–H Bond Arylation of Thiophenes with Iodoarenes. Angew. Chem. Int. Ed. 2010, 49, 8946–8949. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.-Y.; Guo, Q.-X.; Fu, Y. Mechanistic Origin of Ligand-Controlled Regioselectivity in Pd-Catalyzed C-H Activation/Arylation of Thiophenes. Chem. A Eur. J. 2011, 17, 13866–13876. [Google Scholar] [CrossRef] [PubMed]
- Hashikawa, Y.; Murata, M.; Wakamiya, A.; Murata, Y. Palladium-Catalyzed Cyclization: Regioselectivity and Structure of Arene-Fused C60 Derivatives. J. Am. Chem. Soc. 2017, 139, 16350–16358. [Google Scholar] [CrossRef] [PubMed]
- Ohkawara, T.; Suzuki, K.; Nakano, K.; Mori, S.; Nozaki, K. Facile Estimation of Catalytic Activity and Selectivities in Copolymerization of Propylene Oxide with Carbon Dioxide Mediated by Metal Complexes with Planar Tetradentate Ligand. J. Am. Chem. Soc. 2014, 136, 10728–10735. [Google Scholar] [CrossRef] [PubMed]
- Andrew, T.L.; VanVeller, B.; Swager, T.M. The Synthesis of Azaperylene-9,10-dicarboximides. Synlett 2010, 2010, 3045–3048. [Google Scholar]
- Sakamoto, T.; Pac, C. A “Green” Route to Perylene Dyes: Direct Coupling Reactions of 1,8-Naphthalimide and Related Compounds under Mild Conditions Using a “New” Base Complex Reagent, t-BuOK/DBN. J. Org. Chem. 2001, 66, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Amati, M.; Lelj, F. Monomolecular isomerization processes of aluminum tris(8-hydroxyquinolinate) (Alq3): A DFT study of gas-phase reaction paths. Chem. Phys. Lett. 2002, 363, 451–457. [Google Scholar] [CrossRef]
- Katakura, R.; Koide, Y. Configuration-Specific Synthesis of the Facial and Meridional Isomers of Tris(8-hydroxyquinolinate)aluminum (Alq3). Inorg. Chem. 2006, 45, 5730–5732. [Google Scholar] [CrossRef]
- Brinkmann, M.; Gadret, G.; Muccini, M.; Taliani, C.; Masciocchi, N.; Sironi, A. Correlation between Molecular Packing and Optical Properties in Different Crystalline Polymorphs and Amorphous Thin Films of mer-Tris(8-hydroxyquinoline)aluminum(III). J. Am. Chem. Soc. 2000, 122, 5147–5157. [Google Scholar] [CrossRef]
- Hummelen, J.C.; Knight, B.W.; LePeq, F.; Wudl, F.; Yao, J.; Wilkins, C.L. Preparation and Characterization of Fulleroid and Methanofullerene Derivatives. J. Org. Chem. 1995, 60, 532–538. [Google Scholar] [CrossRef]
- Morinaka, Y.; Nobori, M.; Murata, M.; Wakamiya, A.; Sagawa, T.; Yoshikawa, S.; Murata, Y. ChemInform Abstract: Synthesis and Photovoltaic Properties of Acceptor Materials Based on the Dimerization of Fullerene C 60 for Use in Efficient Polymer Solar Cells. Chem. Commun. 2013, 49, 3670–3672. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Additive | Temp. (°C) | Yield (%) 1 | |
|---|---|---|---|---|
| 2 | 3 | |||
| 1 | – | 170 | 48 | 12 |
| 2 | PivOH | 170 | 47 | 21 |
| 3 | PivOH | 120 | 0 | 20 |
| λcalculated (nm) | Oscillator Strength | Contributing MOs (%) 1 | Character |
|---|---|---|---|
| 546 | 0.916 | HOMO → LUMO+2 (58) HOMO–2 → LUMO (19) HOMO → LUMO+1 (10) | π→π* π→π* ICT |
| 536 | 0.861 | HOMO–2 → LUMO (45) HOMO–1 → LUMO+1 (36) HOMO → LUMO+1 (5) | π→π* π→π* ICT |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsukao, M.; Hashikawa, Y.; Toyama, N.; Muraoka, M.; Murata, M.; Sasamori, T.; Wakamiya, A.; Murata, Y. Propeller-Shaped Aluminum Complexes with an Azaperylene Core in the Ligands. Inorganics 2019, 7, 109. https://doi.org/10.3390/inorganics7090109
Tsukao M, Hashikawa Y, Toyama N, Muraoka M, Murata M, Sasamori T, Wakamiya A, Murata Y. Propeller-Shaped Aluminum Complexes with an Azaperylene Core in the Ligands. Inorganics. 2019; 7(9):109. https://doi.org/10.3390/inorganics7090109
Chicago/Turabian StyleTsukao, Masahiro, Yoshifumi Hashikawa, Nana Toyama, Masahiro Muraoka, Michihisa Murata, Takahiro Sasamori, Atsushi Wakamiya, and Yasujiro Murata. 2019. "Propeller-Shaped Aluminum Complexes with an Azaperylene Core in the Ligands" Inorganics 7, no. 9: 109. https://doi.org/10.3390/inorganics7090109
APA StyleTsukao, M., Hashikawa, Y., Toyama, N., Muraoka, M., Murata, M., Sasamori, T., Wakamiya, A., & Murata, Y. (2019). Propeller-Shaped Aluminum Complexes with an Azaperylene Core in the Ligands. Inorganics, 7(9), 109. https://doi.org/10.3390/inorganics7090109