
Determination of Levamisole and Mebendazole and Its Two Metabolite Residues in Three Poultry Species by HPLC-MS/MS

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Preparation of Solution
2.3. HPLC-MS/MS Instruments and Conditions
2.4. Preparation of Sample
2.5. Method Validation
3. Results and Discussion
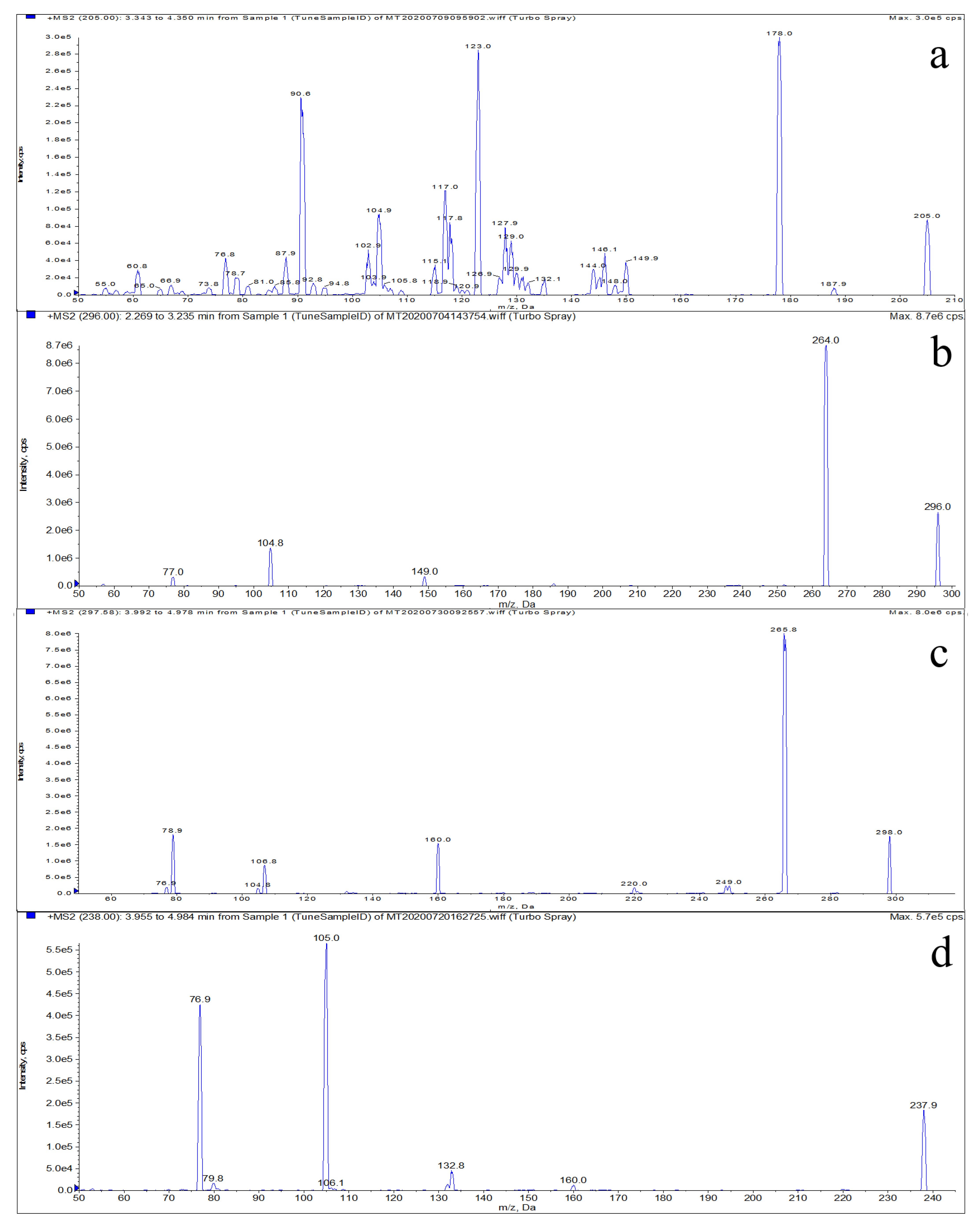
3.1. Optimization of HPLC-MS/MS Conditions
3.2. Optimization of Sample Preparation
3.3. Method Validation
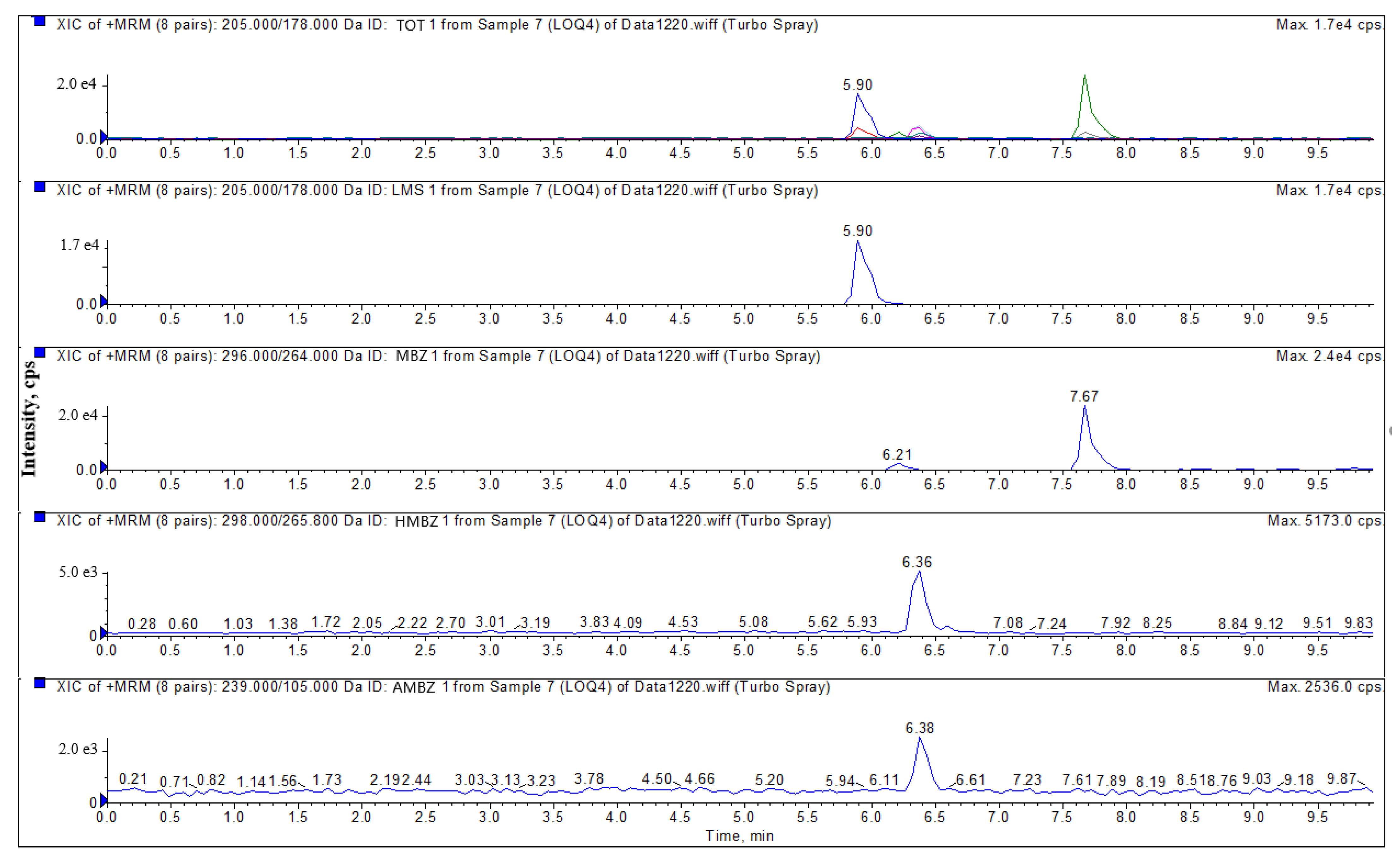
3.3.1. Specificity
3.3.2. Linearity
3.3.3. Matrix Effect
3.3.4. LODs and LOQs
3.3.5. CCα and CCβ
3.3.6. Recovery and Precision
3.4. Comparison with Other Methods
3.5. Real Sample Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Janssen, P. The levamisole story. Prog. Drug Res. 1976, 20, 347–383. [Google Scholar] [CrossRef] [PubMed]
- Cuesta, A.; Esteban, M.A.; Meseguer, J. Levamisole is a potent enhancer of gilthead seabream natural cytotoxic activity. Vet. Immunol. Immunolp. 2002, 89, 169–174. [Google Scholar] [CrossRef]
- Friedman, P.A.; Platzer, E.G. Interaction of anthelmintic benzimidazoles with AscarisSuum embryonic tubulin. BBA-Gen. Subjects. 1980, 630, 271–278. [Google Scholar] [CrossRef]
- Albonico, M.; Bickle, Q.; Ramsan, M.; Montresor, A.; Savioli, L.; Taylor, M. Efficacy of mebendazole and levamisole alone or in combination against intestinal nematode infections after repeated targeted mebendazole treatment in Zanzibar. B. World. Health. Organ. 2003, 81, 343–352. [Google Scholar] [CrossRef]
- Bennet, E.M.; Behm, C.; Bryant, C. Effects of mebendazole and levamisole on tetrathyridia of Mesocestoides corti in the mouse. Int. J. Parasitol. 1978, 8, 463–466. [Google Scholar] [CrossRef]
- Heath, D.D.; Christie, M.J.; Chevis, R. The lethal effect of mebendazole on secondary Echinococcus granulosus, cysticerci of Taenia pisiformis and tetrathyridia of Mesocestoides corti. Parasitology 1975, 70, 273–285. [Google Scholar] [CrossRef]
- European Commission. Commission Regulation (EU) No 37/2010 of 22 December 2009 on Pharmacologically Active Substances and Their Classification Regarding Maximum Residue Limits in Foodstuffs of Animal Origin. Off. J. Eur. Communities 2010, L15, 1–72. Available online: https://ec.europa.eu/health/sites/default/files/files/eudralex/vol5/reg_2010_37/reg_2010_37_en.pdf (accessed on 15 June 2020).
- FDA. CFR-Code of Federal Regulations Title 21. 2019. Available online: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfCFR/CFRSearch.cfm?CFRPart=556&showFR=1 (accessed on 15 June 2020).
- Korea Food & Drug Administration. Notice No. 2012-59 of the Korea Food & Drug Administration. 2012. Available online: https://www.mfds.go.kr/eng/brd/m_60/view.do?seq=67277 (accessed on 15 June 2020).
- Ejlertsen, B.; Mouridsen, H.T.; Jensen, M.B.; Andersson, M.; Kamby, C.; Knoop, A.S. Cyclophosphamide, methotrexate, and fluorouracil; oral cyclophosphamide; levamisole; or no adjuvant therapy for patients with high-risk, premenopausal breast cancer. Cancer 2010, 116, 2081–2089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, L.L.; Wu, X.L.; Liu, L.Q.; Kuang, H.; Xu, C.L. Gold nanoparticle-based paper sensor for simultaneous detection of 11 benzimidazoles by one monoclonal antibody. Small 2018, 14, 1701782. [Google Scholar] [CrossRef] [PubMed]
- Chappell, C.G.; Creaser, C.S. Modified on-column interface for coupled high-performance liquid chromatography-gas chromatography and its application to the determination of levamisole in milk. J. Chromatogr. A 1992, 626, 223–230. [Google Scholar] [CrossRef]
- Ragno, G.; Risoli, A.; Ioele, G.; Luca, M.D. Photo- and thermal-stability studies on benzimidazole anthelmintics by HPLC and GC-MS. Chem. Pharm. Bull. 2006, 54, 802–806. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Tao, Y.; Liu, Z.; Huang, L.; Wang, Y.; Pan, Y.; Peng, D.; Dai, M.; Yuan, Z. Development of a high-performance liquid chromatography method to monitor the residues of benzimidazoles in bovine milk. J. Chromatogr. B 2010, 878, 2928–2932. [Google Scholar] [CrossRef] [PubMed]
- Vichapong, J.; Santaladchaiyakit, Y.; Burakham, R.; Kanchanamayoon, W.; Srijaranai, S. Determination of benzimidazole anthelmintics using HPLC after vortex-assisted mixed anionic–cationic surfactant-enhanced emulsification microextraction with solidification of floating organic droplet procedure. J. Food Compos. Anal. 2015, 35, 30–37. [Google Scholar] [CrossRef]
- Tejada, C.; Olmo, M.D.; Lara, F.J.; García, A.M.; Lara, F.J. Green and simple analytical method to determine benzimidazoles in milk samples by using salting-out assisted liquid-liquid extraction and capillary liquid chromatography. J. Chromatogr. B 2018, 1091, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Kolanovic, B.S.; Bilandzic, N.; Kos, B.; Suskovic, J.; Cvetnic, L.; Varenina, I.; Luburic, D.B.; Varga, I.; Pavlicek, D.; Lugomer, M.D. Distribution and elimination of levamisole in eggs and tissues after oral administration to laying hens, determined by LC-MS/MS. Food. Addit. Contam. A 2019, 36, 729–739. [Google Scholar] [CrossRef]
- Wei, H.M.; Tao, Y.F.; Chen, D.M.; Xie, S.Y.; Pan, Y.H.; Liu, Z.L.; Huang, L.L.; Yuan, Z.H. Development and validation of a multi-residue screening method for veterinary drugs, their metabolites and pesticides in meat using liquid chromatography-tandem mass spectrometry. Food. Addit. Contam. A 2015, 32, 686–701. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Yu, Q.W.; He, H.B.; Lu, Q.; Feng, Y.Q. Nickel oxide nanoparticle-deposited silica composite solid-phase extraction for benzimidazole residue analysis in milk and eggs by liquid chromatography-mass spectrometry. J. Agric. Food Chem. 2016, 64, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Tao, Y.; Zhang, H.; Pan, Y.; Liu, Z.; Huang, L.; Wang, Y.; Peng, D.; Xu, W.; Dai, M. Development of a liquid chromatography–tandem mass spectrometry with pressurized liquid extraction method for the determination of benzimidazole residues in edible tissues. J. Chromatogr. B 2011, 879, 1659–1667. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Dong, J.; Yang, Y.B.; Liu, Y.T.; Yang, Q.H.; Ai, X.H. Development of a liquid chromatography-tandem mass spectrometry method with modified QuEChERS extraction for the quantification of mebendazole and its metabolites, albendazole and its metabolites, and levamisole in edible tissues of aquatic animals. Food Chem. 2018, 269, 442–449. [Google Scholar] [CrossRef]
- Cooper, K.M.; Whelan, M.; Kennedy, D.G.; Trigueros, G.; Danaher, M. Anthelmintic drug residues in beef: UPLC-MS/MS method validation, European retail beef survey, and associated exposure and risk assessments. Food. Addit. Contam. A 2012, 29, 746–760. [Google Scholar] [CrossRef] [PubMed]
- Yamada, R.; Kozono, M.; Ohmori, M.; Morimatsu, F.; Kitayama, M. Simultaneous determination of residual veterinary drugs in bovine, porcine, and chicken muscle using liquid chromatography coupled with electrospray ionization tandem mass spectrometry. Biosci. Biotech. Bioch. 2006, 70, 54–65. [Google Scholar] [CrossRef] [Green Version]
- Jedziniak, P.; Szprengier-Juszkiewicz, T.; Olejnik, M. Determination of benzimidazoles and levamisole residues in milk by liquid chromatography–mass spectrometry: Screening method development and validation. J. Chromatogr. A 2009, 1216, 8165–8172. [Google Scholar] [CrossRef]
- European Commission. Commission Decision of 12 August 2002 Implementing Council Directive 96/23/EC Concerning the Performance of Analytical Methods and the Interpretation of Results (2002/657/EC). Off. J. Eur. Communities 2002, L221, 8–36. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32002D0657&from=ES (accessed on 15 June 2020).
- Sun, L.J.; Hee, C.S.; Mi, L.C.; Chang, M.I.; Jin, J.H.; Hojae, B.; Jin, H.P. A liquid chromatography—Tandem mass spectrometry approach for the identification of mebendazole residue in pork, chicken, and horse. PLoS ONE 2017, 12, 0169597. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Li, X.; Liu, X.; Zhang, J.; Cao, Y.; Shi, Z.; Sun, H. Multi-class, multi-residue analysis of trace veterinary drugs in milk by rapid screening and quantification using ultra-performance liquid chromatography–quadrupole time-of-flight mass spectrometry. J. Dairy Sci. 2015, 98, 8433–8444. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, M.; Take, B.K.; Fujinuma, K.; Jimbo, K.; Miyazaki, T. Determination of levamisole in livestock products using high performance liquid chromatography. J. Food. Hyg. Soc. Jpn. 2002, 43, 6–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Z.G.; Luo, X.Q.; Guo, F.X.; Zhang, Z.R.; Peng, L. Determination of flunixin in swine plasma, urine and feces by UPLC-MS/MS and its application in the real samples. Curr. Pharm. Anal. 2019, 15, 51–60. [Google Scholar] [CrossRef]
- Abdelrahman, M.M. Solid-Phase Extraction and HPLC-DAD for determination of salbutamol in urine samples. Anal. Chem. Let. 2018, 8, 35–45. [Google Scholar] [CrossRef]
- SANTE. Guidance Document on Analytical Quality Control and Method Validation Procedures for Pesticides Residues Analysis in Food and Feed European Commission Document no SANTE/11945/2015; European Union: Brussels, Belgium, 2015. [Google Scholar]
- Yoshikawa, S.; Nagano, C.; Kanda, M.; Hayashi, H.; Matsushima, Y.; Nakajima, T.; Tsuruoka, Y.; Nagata, M.; Koike, H.; Sekimura, K.; et al. Simultaneous determination of multi-class veterinary drugs in chicken processed foods and muscle using solid-supported liquid extraction clean-up. J. Chromatogr. B 2017, 1507, 15–23. [Google Scholar] [CrossRef]
- Zhu, W.X.; Yang, J.Z.; Wang, Z.X.; Wang, C.J.; Liu, Y.F.; Zhang, L. Rapid determination of 88 veterinary drug residues in milk using automated TurborFlow online clean-up mode coupled to liquid chromatography-tandem mass spectrometry. Talanta 2016, 148, 401–411. [Google Scholar] [CrossRef]
- Martinez-Villalba, A.; Moyano, E.; Galceran, M.T. Ultra-high performance liquid chromatography-atmospheric pressure chemical ionization-tandem mass spectrometry for the analysis of benzimidazole compounds in milk samples. J. Chromatogr. A 2013, 1313, 119–131. [Google Scholar] [CrossRef]
- Kim, E.; Park, S.; Park, H.; Choi, J.; Yoon, H.J.; Kim, J.H. Determination of anthelmintic and antiprotozoal drug residues in fish using liquid chromatography-tandem mass spectrometry. Molecules 2021, 26, 2575. [Google Scholar] [CrossRef] [PubMed]
- Lehotay, S.J.; Lightfield, A.R. Extract-and-Inject Analysis of Veterinary Drug Residues in Catfish and Ready-to-Eat Meats by Ultrahigh-Performance Liquid Chromatography—Tandem Mass Spectrometry. J. Aoac. Int. 2020, 103, 584–606. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Compound | Molecular Weight | Retention Time (min) | Mass Transition (m/z) | Declustering Potential (V) | Collision Energy (eV) |
|---|---|---|---|---|---|
| LMS | 205 | 5.91 | 205 > 178.0 * | 110 | 29 |
| 205 > 123.0 | 38 | ||||
| MBZ | 296 | 7.68 | 296 > 264.0 * | 115 | 28 |
| 296 > 104.8 | 23 | ||||
| HMBZ | 298 | 6.37 | 298 > 265.8 * | 121 | 24 |
| 298 > 160.0 | 35 | ||||
| AMBZ | 238 | 6.38 | 238 > 105.0 * | 155 | 33 |
| 238 > 76.9 | 45 |
| Analyte | Ion Ratio of Standard Solutions | Maximum Permitted Tolerance According to Decision 2002/657/EC | Ion Ratio of Fortified Samples |
|---|---|---|---|
| LMS | 0.26 | 0.26 ± 25% (0.11–0.51) | 0.22–0.36 |
| MBZ | 0.20 | 0.20 ± 25% (0.10–0.45) | 0.13–0.25 |
| HMBZ | 0.98 | 0.98 ± 20% (0.78–1.18) | 0.78–1.11 |
| AMBZ | 0.62 | 0.62 ± 20% (0.42–0.82) | 0.50–0.74 |
| Matrix | Analyte | Regression Equation | Determination Coefficient (R2) | Linearity Range (μg/kg) |
|---|---|---|---|---|
| Chicken muscle | LMS | y = 891,231x − 13,830 | 0.9995 | 0.22–25 |
| MBZ | y = 298,982x + 168,358 | 0.9996 | 0.15–150 | |
| HMBZ | y = 89,829x − 4855 | 0.9995 | 0.60–150 | |
| AMBZ | y = 31,616x − 4274 | 0.9996 | 0.80–150 | |
| Duck muscle | LMS | y = 496,991x + 336,140 | 0.9995 | 0.20–25 |
| MBZ | y = 207,883x + 79,368 | 0.9994 | 0.12–150 | |
| HMBZ | y = 37,850x + 4580 | 0.9994 | 0.50–150 | |
| AMBZ | y = 22,019x + 1032 | 0.9995 | 0.62–150 | |
| Goose muscle | LMS | y = 858,472x + 38,029 | 0.9998 | 0.16–25 |
| MBZ | y = 313,426x + 148,749 | 0.9997 | 0.16–150 | |
| HMBZ | y = 83,271x − 12,277 | 0.9995 | 0.55–150 | |
| AMBZ | y = 30,999x − 1204 | 0.9997 | 0.70–150 |
| Analyte | Chicken Muscle | Duck Muscle | Goose Muscle |
|---|---|---|---|
| LMS | −7.2 | −5.2 | −13.6 |
| MBZ | 4.9 | 12.9 | 9.2 |
| HMBZ | 3.6 | −8.8 | −10.6 |
| AMBZ | 7.8 | 13.1 | 17.3 |
| Matrix | Analyte | LOD (µg/kg) | LOQ (µg/kg) | CCα (µg/kg) | CCβ (µg/kg) |
|---|---|---|---|---|---|
| Chicken muscle | LMS | 0.07 | 0.22 | 14.58 | 19.15 |
| MBZ | 0.06 | 0.15 | 63.33 | 66.66 | |
| HMBZ | 0.16 | 0.60 | 63.46 | 66.92 | |
| AMBZ | 0.25 | 0.80 | 63.80 | 63.60 | |
| Duck muscle | LMS | 0.06 | 0.20 | 13.25 | 16.49 |
| MBZ | 0.04 | 0.12 | 63.69 | 67.38 | |
| HMBZ | 0.15 | 0.50 | 63.15 | 66.30 | |
| AMBZ | 0.30 | 0.62 | 63.41 | 66.82 | |
| Goose muscle | LMS | 0.05 | 0.16 | 16.45 | 22.90 |
| MBZ | 0.05 | 0.16 | 64.30 | 68.60 | |
| HMBZ | 0.18 | 0.55 | 56.18 | 70.36 | |
| AMBZ | 0.23 | 0.70 | 67.35 | 74.70 |
| Matrix | Analyte | Addition Level (µg/kg) | Recovery (%) | RSD (%) | Intraday RSD (%) | Interday RSD (%) |
|---|---|---|---|---|---|---|
| Chicken muscle | LMS | 0.22 | 88.01 ± 2.29 | 2.29 | 3.51 | 3.83 |
| 5 | 91.44 ± 1.93 | 2.11 | 2.07 | 4.03 | ||
| 10 α | 93.30 ± 2.79 | 2.99 | 3.68 | 4.34 | ||
| 20 | 93.89 ± 3.22 | 3.43 | 3.30 | 4.24 | ||
| MBZ | 0.15 | 86.77 ± 2.24 | 2.58 | 3.51 | 3.98 | |
| 30 | 94.05 ± 1.96 | 2.09 | 2.07 | 2.62 | ||
| 60 α | 94.38 ± 2.03 | 2.15 | 3.68 | 3.99 | ||
| 120 | 94.49 ± 2.18 | 2.31 | 3.30 | 3.73 | ||
| HMBZ | 0.60 | 88.36 ± 3.65 | 4.13 | 3.21 | 4.60 | |
| 30 | 91.42 ± 2.01 | 2.20 | 4.56 | 4.69 | ||
| 60 α | 93.21 ± 2.11 | 2.26 | 2.39 | 2.96 | ||
| 120 | 91.53 ± 2.53 | 2.77 | 4.55 | 5.07 | ||
| AMBZ | 0.80 | 90.70 ± 1.90 | 2.09 | 2.60 | 3.06 | |
| 30 | 92.07 ± 1.95 | 2.11 | 2.49 | 2.95 | ||
| 60 α | 94.49 ± 2.32 | 2.45 | 3.92 | 4.25 | ||
| 120 | 96.47 ± 4.46 | 4.63 | 3.25 | 4.96 | ||
| Duck muscle | LMS | 0.20 | 90.85 ± 2.95 | 3.25 | 3.17 | 4.06 |
| 5 | 89.57 ± 2.05 | 2.29 | 3.56 | 3.89 | ||
| 10 α | 92.04 ± 1.98 | 2.16 | 2.52 | 2.99 | ||
| 20 | 95.95 ± 2.67 | 2.78 | 5.13 | 5.37 | ||
| MBZ | 0.12 | 88.68 ± 2.83 | 3.19 | 3.85 | 4.51 | |
| 30 | 91.08 ± 1.94 | 2.13 | 2.46 | 2.95 | ||
| 60 α | 95.14 ± 2.25 | 2.36 | 2.13 | 2.87 | ||
| 120 | 95.21 ± 1.97 | 2.07 | 3.52 | 3.77 | ||
| HMBZ | 0.50 | 91.33 ± 2.62 | 2.86 | 4.99 | 5.28 | |
| 30 | 95.63 ± 3.71 | 3.88 | 2.47 | 4.03 | ||
| 60 α | 92.26 ± 1.92 | 2.08 | 4.03 | 4.18 | ||
| 120 | 93.86 ± 2.06 | 2.20 | 2.52 | 3.03 | ||
| AMBZ | 0.62 | 89.00 ± 2.23 | 2.51 | 3.19 | 3.69 | |
| 30 | 89.95 ± 3.32 | 3.69 | 3.64 | 4.63 | ||
| 60 α | 95.58 ± 2.08 | 2.17 | 2.68 | 3.12 | ||
| 120 | 96.94 ± 2.93 | 3.02 | 3.04 | 3.85 | ||
| Goose muscle | LMS | 0.16 | 87.58 ± 2.21 | 2.52 | 4.36 | 4.62 |
| 5 | 90.54 ± 4.07 | 5.19 | 3.01 | 5.22 | ||
| 10 α | 95.03 ± 3.93 | 4.14 | 2.34 | 4.12 | ||
| 20 | 95.88 ± 2.81 | 2.93 | 3.73 | 4.29 | ||
| MBZ | 0.16 | 87.62 ± 1.87 | 2.13 | 4.67 | 4.75 | |
| 30 | 90.05 ± 2.31 | 2.57 | 2.52 | 3.23 | ||
| 60 α | 95.18 ± 2.62 | 2.75 | 3.55 | 4.12 | ||
| 120 | 93.68 ± 2.97 | 3.17 | 2.92 | 3.83 | ||
| HMBZ | 0.55 | 90.25 ± 2.12 | 2.35 | 3.34 | 3.73 | |
| 30 | 93.28 ± 4.03 | 4.32 | 4.33 | 5.52 | ||
| 60 α | 94.79 ± 3.16 | 3.33 | 3.23 | 4.16 | ||
| 120 | 92.97 ± 2.92 | 3.14 | 3.15 | 4.03 | ||
| AMBZ | 0.70 | 92.71 ± 3.31 | 3.57 | 3.73 | 4.66 | |
| 30 | 92.84 ± 2.13 | 2.29 | 1.75 | 2.54 | ||
| 60 α | 96.00 ± 4.48 | 4.67 | 2.08 | 4.39 | ||
| 120 | 95.49 ± 2.96 | 3.10 | 3.04 | 3.91 |
| Matrix | Analyte | Chromatographic Conditions | Sample Preparation Method | Recovery (%) | LODs (μg/kg) | LOQs (μg/kg) |
|---|---|---|---|---|---|---|
| Chicken muscle [32] | 37 veterinary drugs (including LMS, MBZ, HMBZ) | Inert Sustain Swift C18 column (250 mm × 4.6 mm, 5 μm) Mobile phase: 0.1% formic acid in water and 10 mM ammonium acetate in methanol HPLC-MS/MS | Extract with ethyl acetate solution and acetonitrile | 76–84 | 0.4 | - |
| Milk [14] | 21 veterinary drugs (including MBZ, HMBZ, AMBZ) | Waters XTerra C18 column (250 × 4.6 mm, 5 μm) Mobile phase: 0.02 M ammonium acetate and acetonitrile HPLC-UVD | Extract with acetonitrile; Purification of MCX solid phase extraction column | 78–109 | 3 | 10 |
| Beef, pork and poultry muscle [28] | LMS | ODS-80Ts column (4.6 mm × 150 mm, 5 μm) Mobile phase: 0.02 mol/L aqueous potassium phosphate solution and acetonitrile HPLC-DAD | Extract with ethyl acetate solution; Purification of SCX solid phase extraction column | 78.30–99.80 | 5 | - |
| Pork, chicken, horse muscle [26] | MBZ, HMBZ, AMBZ | Unison UK C18 column (100 mm × 2 mm, 3 μm) Mobile phase: 10 mM ammonium formate in water and methanol HPLC-MS/MS | Extract with ethyl acetate solution | 86.30–101.28 | 0.07 | 0.2 |
| Milk [33] | 88 veterinary drugs (including 13 benzimidazoles) | PAK C18 MG column (150 mm × 2.1 mm, 5 μm) Mobile phase: acetonitrile and 0.1% formic acid aqueous solution HPLC-MS/MS | Extract with disodium ethylenediaminetetraacetate and acetonitrile solution | 75.5–104.5 | 0.5 | 2.0 |
| Aquatic product [21] | LMS, MBZ, HMBZ, AMBZ | Poroshell 120 EC C18 column (250 mm × 4.6 mm, 5 μm) Mobile phase: 5 mM ammonium formate in methanol and acetonitrile LC-MS/MS | Extract with acetonitrile | 80–113.7 | 0.3 | 1 |
| Beef [22] | 37 veterinary drugs (including LMS and MBZ) | Hss T3 C18 column (100 mm × 2.1 mm, 1.8 μm) Mobile phase: acetonitrile:0.01% formic acid in water (10:90, v/v) and methanol:5 mM ammonium formate UHPLC-MS/MS | Extract with acetonitrile | 81–101 | 0.13–1.55 | 0.22–2.64 |
| Milk [34] | 19 veterinary drugs (including MBZ, HMBZ, AMBZ) | Ascentis Express C18 column (150 mm × 2.1 mm, 2.7 μm) Mobile phase: 0.1% formic acid and acetonitrile UHPLC-MS/MS | Extract with 10 mL of acetonitrile with 0.1% NH3 followed by 5 g of MgSO4:NaCl (4:1, w/w) | 65–100 | 1.0–10 | 1.6–18 |
| Fish [35] | 71 veterinary drugs (including LMS and MBZ) | X-SELECT HSS C18 (150 mm × 2.1 mm, 3.5 μm) Mobile phase: 0.1% formic acid in 2 mM ammonium formate in water and 0.1% formic acid in acetonitrile LC-MS/MS | Extract with 0.1% formic acid in MeCN/MeOH (95:5, v/v) clean up with PSA and C18 d-SPE sorbent | 60–119 | - | 0.02–4.8 |
| Animal-derived food [36] | 176 veterinary drugs (including LMS and MBZ) | Hss T3 C18 column (100 mm × 2.1 mm, 1.8 μm) Mobile phase: water with 0.1% formic acid and acetonitrile/MeOH (50:50, v/v) with 0.1% HCO2H UHPLC-MS/MS | Extract with acetonitrile/water (4:1, v/v) Clean up with EMR-Lipid cartridge | 70–120 | - | - |
| Chicken, duck, goose muscle (The study) | LMS, MBZ, HMBZ, AMBZ | XbridgeTM C18 column (4.6 mm × 150 mm, 5 μm) Mobile phase: 0.1% formic acid in water and acetonitrile HPLC-MS/MS | Extract with ethyl acetate; Purification of MCX solid phase extraction column | 86.77–96.94 | 0.04–0.30 | 0.12–0.80 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, P.; Zhang, P.; Guo, Y.; He, Z.; Dong, Y.; Tang, Y.; Guan, F.; Zhang, T.; Xie, K. Determination of Levamisole and Mebendazole and Its Two Metabolite Residues in Three Poultry Species by HPLC-MS/MS. Foods 2021, 10, 2841. https://doi.org/10.3390/foods10112841
Gao P, Zhang P, Guo Y, He Z, Dong Y, Tang Y, Guan F, Zhang T, Xie K. Determination of Levamisole and Mebendazole and Its Two Metabolite Residues in Three Poultry Species by HPLC-MS/MS. Foods. 2021; 10(11):2841. https://doi.org/10.3390/foods10112841
Chicago/Turabian StyleGao, Pengfei, Peiyang Zhang, Yawen Guo, Zhaoyuan He, Yuhao Dong, Yayun Tang, Fanxun Guan, Tao Zhang, and Kaizhou Xie. 2021. "Determination of Levamisole and Mebendazole and Its Two Metabolite Residues in Three Poultry Species by HPLC-MS/MS" Foods 10, no. 11: 2841. https://doi.org/10.3390/foods10112841