1. Introduction
Areca catechu L. is a tropical fruit widely grown in China, India, and Southeast Asia [
1]. Historically, it has been used as a chewing substance, and it possesses a certain addictive nature. Moreover, the areca nut has been used in traditional Chinese medicine in China and Ayurvedic medicine in India, since the areca nut can remove parasites from the body and disperse effusions in the abdominal cavity [
2,
3]. On the other hand, the International Agency for Research on Cancer classified areca nut as a Class I carcinogen in 2003 because it contains a large number of alkaloids, such as arecoline, arecaidine, and guvacine, and these arecolines are carcinogenic [
4,
5]. The safety of areca nuts has become a hotly debated topic [
6]. Therefore, the new direction of areca nut product applications is to separate the alkaloids from the polyphenols and further focus on the research and development of the polyphenols in the areca nut.
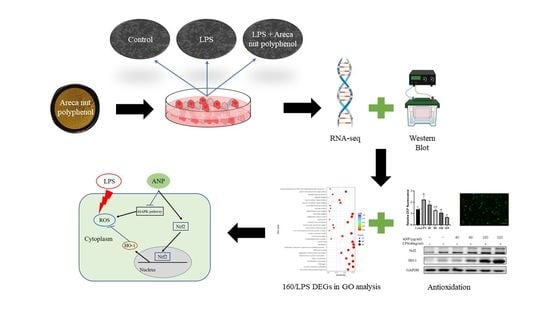
The antioxidant effects of plant polyphenols have received a lot of interest in recent years, since free radical reactive oxygen species (ROS) are often produced when the body is exposed to different biochemical conditions or pathological states. Therefore, the ability to remove ROS is an important part of antioxidants. Several studies have shown that areca leaf and areca nut extracts have antioxidant and radical scavenging activities [
7,
8]. However, the components of the areca nut contributing those antioxidant effects are unclear. Areca nut contains two kinds of major bioactive compounds, which are polyphenols and alkaloids. Alkaloids have been reported to be carcinogenic. RAW264.7 is a macrophage-like cell line, which is often used as a cellular model to study antioxidant and anti-inflammatory activities at the cellular level. Especially, the stimulation of lipopolysaccharides (LPS) can lead to the production of ROS and inflammation in RAW264.7 cells [
9,
10]. Therefore, in this study, we used RAW264.7 cells to investigate how areca nut polyphenols play a role against LPS-induced ROS and inflammation at the cellular level.
The balance between the generation and elimination of cellular ROS is critically regulated by cellular antioxidant enzyme genes containing the antioxidant/electrophile-responsive element (ARE/EpRE) in their promoters. Several molecules, such as nuclear factor erythroid 2-related factor 2 (Nrf2), transcription factor Jun (c-Jun), activating transcription factor 2 (ATF2), and activating transcription factor 4 (ATF4), are possible modulators of ARE/EpRE [
11]. Of these, Nrf2 is a powerful activator of ARE-mediated gene expression that regulates the expression of several antioxidant enzyme genes, including heme oxygenase 1 (HO-1) [
12]. Nrf2 could be activated by both Keap1-dependent and Keap1-independent pathways. The phosphorylation of protein kinases C, MAPKs, and PI3K is closely related to Nrf2 activity in a Keap1-independent way [
13,
14,
15,
16,
17,
18].
High-throughput RNA-sequencing technology has been shown to be an effective tool for studying the bioactive effects and mechanisms of natural compounds at the genome-wide gene expression level [
19,
20,
21]. Thus, we ultimately used this technology to evaluate the molecular mechanisms of antioxidant activity of APN in LPS-stimulated RAW264 cells.
2. Materials and Methods
2.1. Materials
LPS were purchased from Sigma (St. Louis, MO, USA). Dulbecco’s modified eagle media (DMEM) and fetal bovine serum (FBS) were purchased from Biological Industries (Kibbutz Beit Haemek, Israel). MTT cell proliferation and cytotoxicity assay kits were purchased from Solarbio (Beijing, China). Reactive oxygen species assay kits were purchased from Beyotime (Shanghai, China). Anti-Nrf2 antibody (Nrf2), anti-Heme Oxygenase 1 antibody (HO-1), and anti-GAPDH antibody (GAPDH) were purchased from Abcam (Cambridge, UK); anti-MEK1 antibody (MEK1), anti-p-MEK1 antibody (p-MEK1), anti-ERK antibody (ERK), anti-p-ERK antibody (p-ERK), anti-p-MKK3 antibody (p-MKK3), anti-p38 antibody (p38), anti-p-p38 antibody (p-p38), anti-MKK4 antibody (MKK4), anti-p-MKK4 antibody (p-MKK4), anti-JNK antibody (JNK), anti-p-JNK antibody (p-JNK), anti-α-tubulin antibody (α-tubulin), and anti-rabbit antibody were purchased from CST (Boston, MA, USA).
2.2. Preparation of Areca Nut Polyphenols
The raw materials of the areca nut, which was named by James A. Duke (Agricultural Research Service, USDA), were obtained in January 2018 from Wanning city, Hainan, China. Areca nut polyphenols (ANP) were extracted as described previously. In brief, fresh areca nut was freeze-dried and ground into powder. The crude polyphenol from areca nut powder was extracted with 50% (
v/
v) ethanol at 68 °C for 48 min and then freeze-dried. The crude polyphenols were purified with XAD-7 macroporous resin by washing them with 50% ethanol. The total polyphenol contents were estimated at 80% by the Folin-Ciocalteu method [
22]. In previous studies, the two kinds of polyphenol compositions of areca nuts were catechins (2060.44 ± 18.24 μg/mL) and proanthocyanidin B1 (2510.18 ± 62.40 μg/mL) [
23].
2.3. Antioxidant Activity Assays in LPS-Stimulated RAW264.7 Macrophages
2.3.1. Cell Culture
RAW264.7 macrophages were obtained from the American Type Culture Collection (VA, USA) and cultured in DMEM containing 10% FBS and 1% antibiotic at 37 °C in 5% CO2 humidified air.
2.3.2. Cell Viability Assay
Cell viability was assessed using the MTT cell proliferation and cytotoxicity assay kit [
24]. Briefly, RAW 264.7 cells (2 × 10
4 cells/well) were incubated into each well of a 96-well plate. After 24 h of culture, they were treated with different concentrations (40, 80, 160, 320, and 640 μg/mL) of ANP at different times (6, 9, 12, and 15 h). Then the culture medium was removed, and 90 μL of the new medium and 10 μL of MTT solution were added to continue the culture for 4 h. After the culture medium was removed, 150 μL culture medium was added and shaken at low speed for 10 min on a shaker with light protection and detected at 490 nm on a microplate reader (Thermo Scientific Multiskan, version 1.00.79, Vantaa, Finland). The absorbance of the ANP-treated group was divided by the absorbance of the control group to determine the percentage of cell viability.
2.3.3. Cellular ROS Detection by 2′,7′-Dichlorodihydrofluorescein Diacetate and Fluorescence Inverted Microscope Observation
The intracellular formation of ROS was detected using the fluorescent probe 2’,7’-dichlorodihydrofluorescein diacetate (DCFH-DA). This compound easily passes the cellular membrane into cells and is hydrolyzed by intracellular esterase to yield 2’,7’-dichlorodihydrofluorescein (DCFH). ROS produced by the cells oxidizes DCFH to the highly fluorescent compound 2’,7’-dichlorofluorescein (DCF). Thus, the fluorescence intensity is proportional to the amount of ROS produced by the cells. RAW264.7 cells were plated in 96-well plates pre-incubation for 24 h; the cells were then stimulated by LPS for 30 min, then treated with or without ANP for 12 h. After the prepared DCFH-DA was added to the 96-well plates at 10 μM and incubated for 30 min at 37 °C. Then the cells were washed three times with serum-free media. The fluorescence intensity was measured at an excitation wavelength of 488 nm and an emission wavelength of 530 nm using a fluorescence counter (FL 6500, Perkin-Elmer, Waltham, MA, USA) and observed simultaneously by fluorescence inverted microscopy [
25].
2.3.4. Western Blotting
The cells (1.2 × 106 cells/well) were incubated into each well of a 6-well plate. The cultured cells were placed on ice and the culture medium was removed. After cells were washed with 1 mL of pre-cooled phosphate-buffered saline (PBS) 3 times, the cell lysis solution of 200 μL was added and cells were lysed on ice for 20 min. The cells on the 6-well plate were scraped by the cell scraper and transfered to a 1.5-mL Eppendorf (EP) tube. After the EP tube was centrifuged (3500, KUBOTA, Naniwa, Osaka, Japan) at 14,000 rpm at 4 °C for 10 min, the supernatant was placed into the new EP tube. The protein content of the lysate was determined using a protein assay kit (A046-1-1, Nanjing Jiancheng Bioengineering Institute, Nanjing, Jiangsu, China) following the manufacturer’s protocols. The protein loading buffer was added into the EP tube and denatured in a thermostatic metal bath at 100 °C for 7 min. After cooling to room temperature, the EP tube was stored at −20 °C until used. Proteins (20 μg/lane) were separeted on 10% sodium dodecyl sulfate (SDS) polyacrylamide gels, transferred to polyvinylidene difluoride (PVDF) membranes, and blocked with 5% skim milk powder in phosphate buffered saline with Tween-20 (PBST). The membranes were treated with a primary monoclonal antibody overnight at 4 °C before being incubated for 1 h at room temperature with a secondary antibody that had been diluted in skim milk by 2000-fold. Blots were further treated with an enhanced chemiluminescence kit (1705040, Bio-Rad, Hercules, CA, USA) and detected on an ImageQuant LAS 4000 mini (Las 4000, GE, Boston, MA, USA). A quantitative density analysis was performed using ImageJ software.
2.4. Transcriptome Analysis
Cells were pre-cultured on 6-well plates for 24 h. LPS (40 ng/mL) was added for 30 min, followed by 160 µg/mL and 320 µg/mL of ANP, and incubated for more than 12 h. Four groups were set up: control group (Con), LPS group (LPS), LPS + 160 µg/mL group (160/LPS), and LPS + 320 µg/mL group (320/LPS). After incubation, the culture solution was removed and washed with PBS. After the removal of PBS, RIPA lysis solution was added for 20 min. After lysis, cells were scraped off with a cell scraper. Then, the cells were collected in 1.5-mL enzyme-free centrifuge tubes. Collected cells were frozen at −80 °C. RNA extraction and detection, library preparation for transcriptome sequencing, clustering, and sequencing followed the procedures of Novogene (Beijing, China). Differential gene analysis was performed using DESeq2, for significantly different expressions, a corrected
p-value of 0.05 and an absolute foldchange of 2 were established [
26]. Gene Ontology (GO) enrichment analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis of differentially expressed genes were implemented by the clusterProfiler R package [
27,
28].
2.5. Data Analysis
All data were analyzed using one-way ANOVA SPSS (23, IBM, Armonk, NY, USA) and the differences among treatment groups were evaluated using least significant difference (LSD) and Duncan’s multiple range tests, with a significance threshold of
p < 0.05 or
p < 0.01 [
29].
4. Discussion
Areca nuts are a popular tropical fruit in Asian areas, and they are rich in polyphenols. However, there are limited investigations on the benefit to health because areca nut contains some carcinogenic alkaloids, such as arecoline, arecaidine, and guvacine. The areca nut alkaloids have been reported to have reproductive toxicity and ROS production [
33,
34]. On the other hand, the extracts of areca nut were reported to have antioxidant potential [
35,
36], and antibacterial activity [
37,
38].
In our previous study, two kinds of polyphenols, catechin and proanthocyanidin B1 were found [
23]. To further investigate the antioxidant activity of ANP, we used a macrophage-like cell line, RAW264.7, which is a cellular model to study antioxidant and anti-inflammatory activities at the cellular level. As a result, ANP had a significant inhibitory effect on LPS-stimulated ROS (
Figure 1 and
Figure 2). Moreover, ANP significantly increased Nrf2 and HO-1 levels (
Figure 3). Moreover, continuous stimulation of LPS causes inflammation of the cells, and MAPK pathway is associated with the production of inflammation [
39,
40,
41]. Our data revealed that ANP further downregulated the activation of the MAPK signaling pathway (
Figure 8). Thus, ANP inhibited not only LPS-stimulated oxidative stress, but also the LPS-stimulated inflammation.
In order to further investigate how APN affects gene expression across the entire genome in RAW264 cells; we used RNA sequencing and bioinformatics. Our data revealed that ANP had a substantial influence on mitochondria, kinase activity, and transferase activity according to GO analysis (
Figure 5). Therefore, we speculate that ANP may alleviate oxidative stress by regulating mitochondrial activity and protease activity. KEGG analysis revealed that ANP affected some pathways that are related with diseases, NAFLD, virus infections, and bacterial infections. Most of them are related to the MAPK pathway (
Table 3). ANP inhibited the expressions of MAPK-related genes (
Table 4). These data also supported the finding that ANP prevented LPS-stimulated oxidative damage via the MAPK signaling pathways. A similar pathway was also reported: cryptochlorogenic acid activated the Nrf2/HO-1 signaling pathway via the ROS-dependent MAPK pathway, and reduced the LPS-stimulated inflammation [
42].
In the PPI analysis according to the DEGs data, we found that the cellular senescence pathway and the MAPK pathway interacted with each other (
Figure 6). In the 160/LPS, Mapk3, which is also known as Erk1 and activates the activity of many MAPK enzymes, had the highest connectivity degree and was associated with most of the genes. Erk1/2 is reported to play a key role in cellular senescence [
43]. In the 320/LPS, Mapk1, which is also known as Erk2, had the highest connectivity degree, can activate the activity of many protein kinases, and is involved in a variety of cellular processes (
Figure 7). A previous study showed that Erk1/2 and p38 play a key role in cellular senescence due to smoking [
44].











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}