
Anti-Interference Detection of Vibrio parahaemolyticus from Aquatic Food Based on Target-Cyclized RCA with Dynamic Adapter Followed by LAMP

 , , and
, , and
Abstract
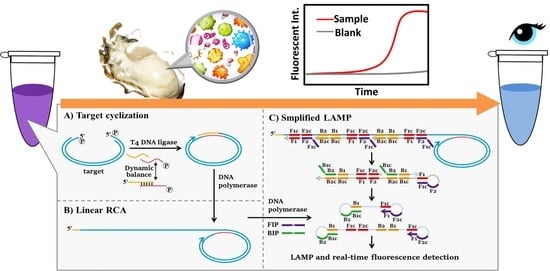
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Activation and Cultivation of V. parahaemolyticus
2.3. Preparation of Artificially Contaminated Samples
2.4. Genomic DNA Extraction from V. parahaemolyticus and Oyster
2.5. Purified Target Fragment Preparation by PCR
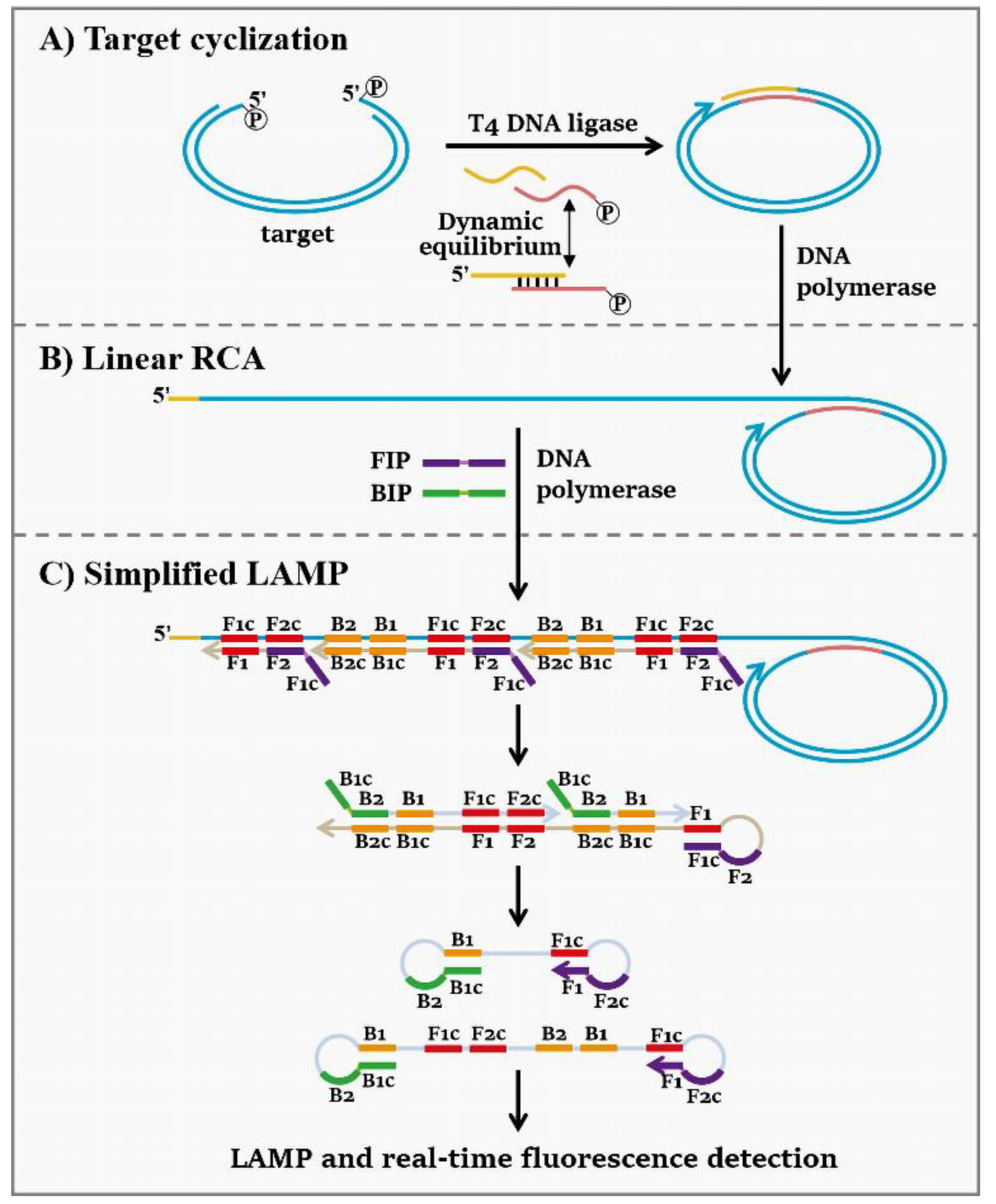
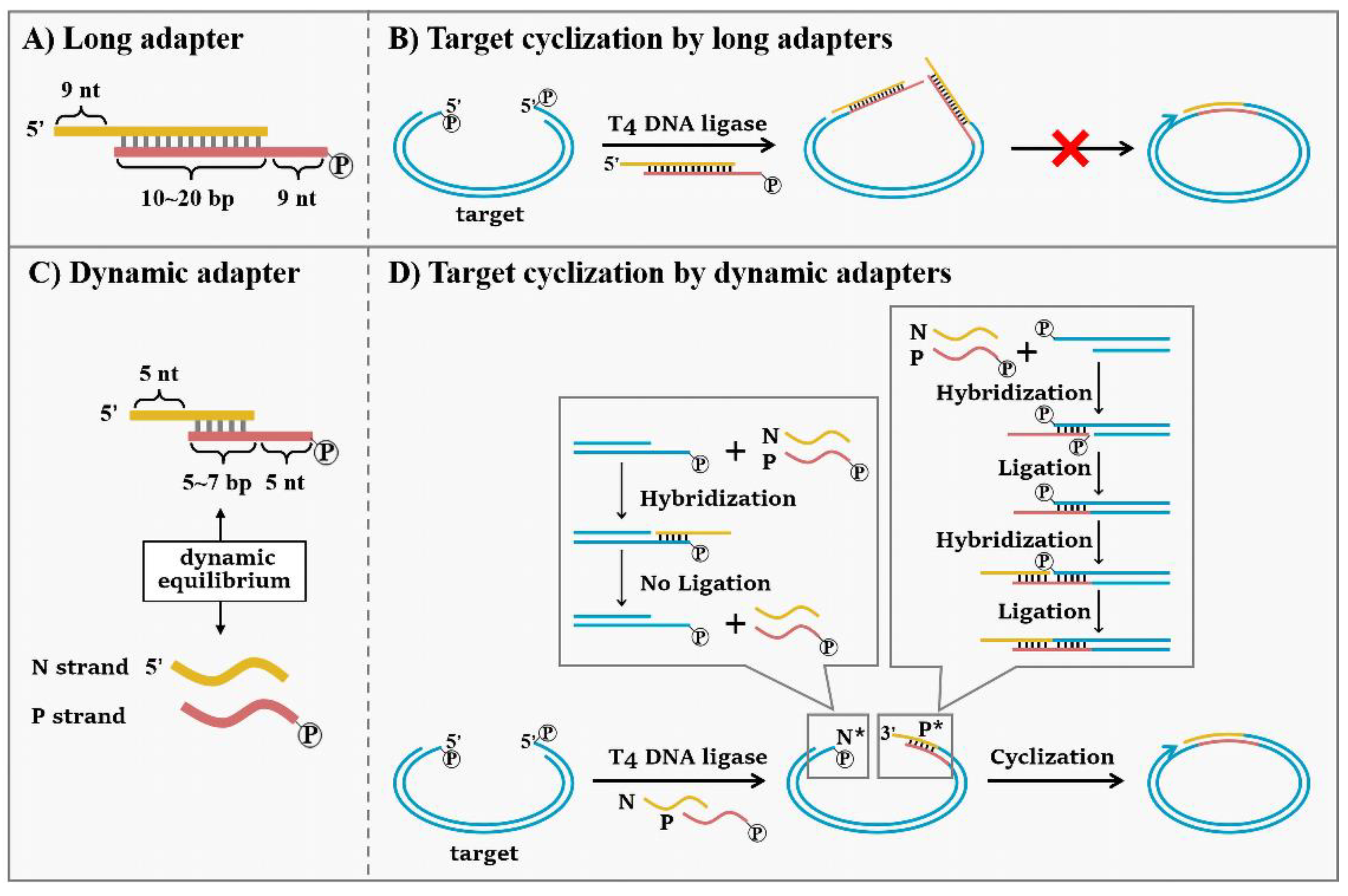
2.6. Target Cyclization by Dynamic Adapter
2.7. tRCA-Lamp Reaction
2.8. Tm Simulation by Mfold
3. Results
3.1. Strategy for the Combination of Target-Cyclized RCA and Simplified LAMP (tRCA-Lamp)
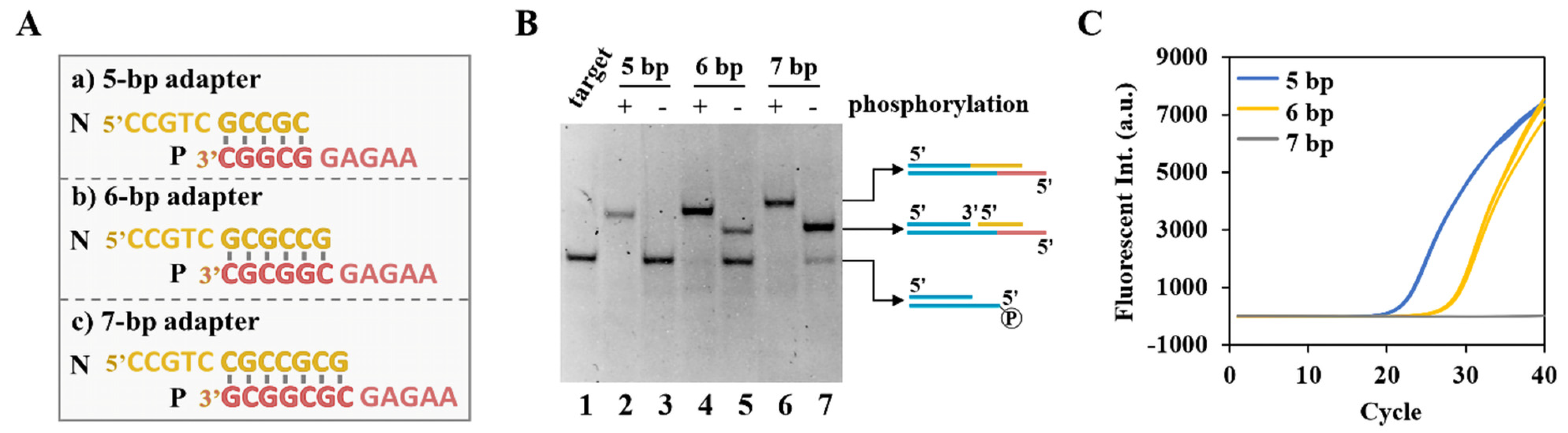
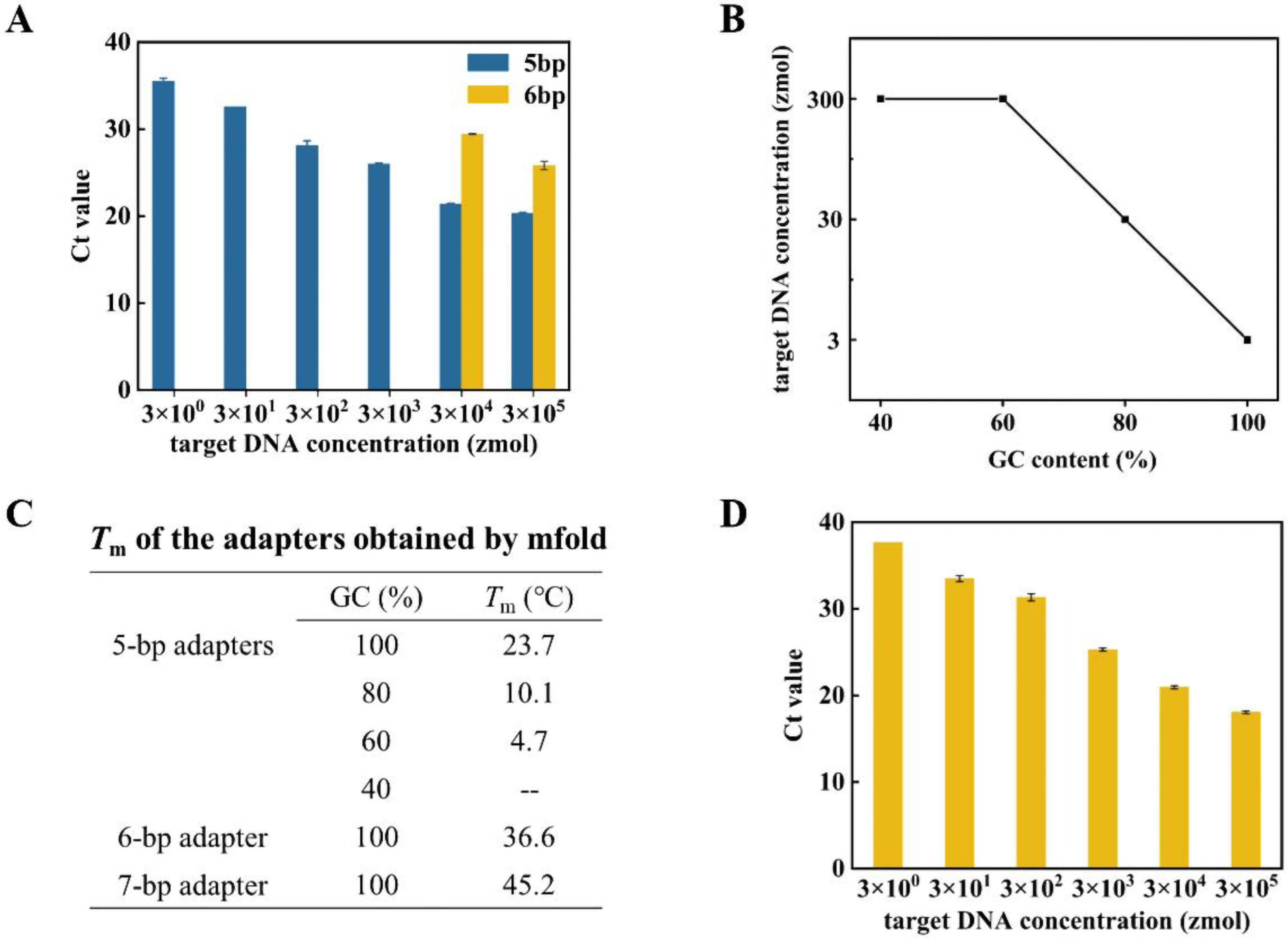
3.2. High Sensitivity Capture and Cyclization of Target with Dynamic Adapters
3.3. Anti-Interference Detection of V. parahaemolyticus by tRCA-Lamp
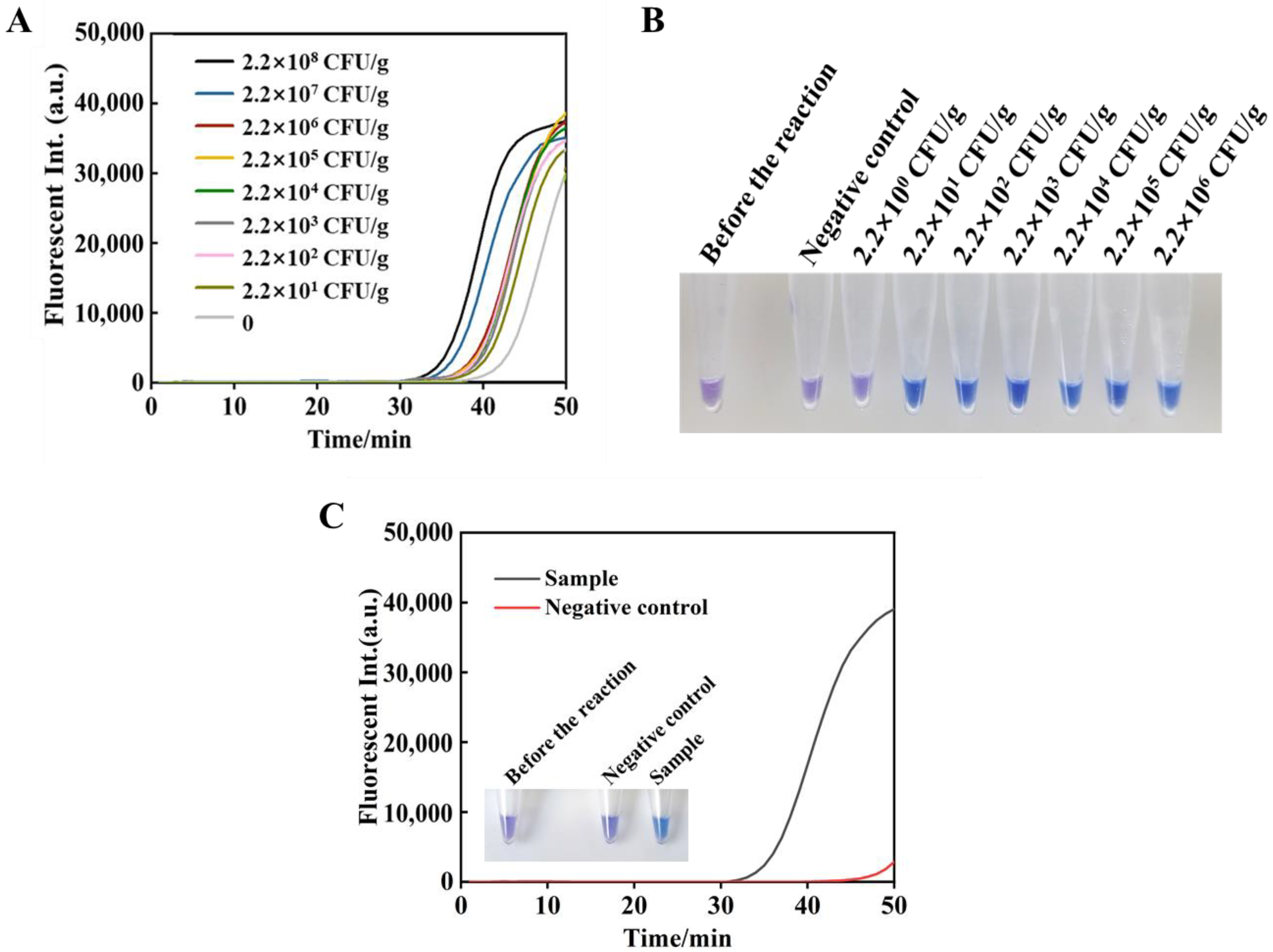
3.4. Sensitivity of the tRCA-Lamp
3.5. Fluorescence and Visual Detection of Artificially Contaminated Oyster Samples
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hartnell, R.E.; Stockley, L.; Keay, W.; Rosec, J.P.; Hervio-Heath, D.; Van den Berg, H.; Leoni, F.; Ottaviani, D.; Henigman, U.; Denayer, S.; et al. A Pan-European ring trial to validate an international standard for detection of Vibrio cholerae, Vibrio parahaemolyticus and Vibrio vulnificus in seafoods. Int. J. Food Microbiol. 2019, 288, 58–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Y.C.; Liu, C. Vibrio parahaemolyticus: A concern of seafood safety. Food Microbiol. 2007, 24, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Todd, E.C.D. Foodborne diseases: Overview of biological hazards and foodborne diseases. Encycl. Food Saf. 2014, 1, 221–242. [Google Scholar]
- Vidic, J.; Vizzini, P.; Manzano, M.; Kavanaugh, D.; Ramarao, N.; Zivkovic, M.; Radonic, V.; Knezevic, N.; Giouroudi, I.; Gadjanski, I. Point-of-Need DNA testing for detection of foodborne pathogenic bacteria. Sensors 2019, 19, 1100. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Yu, S.; Shang, J.; Chen, Y.; Wang, Q.; Liu, X.; Wang, F. Construction of an Exonuclease III-propelled integrated DNAzyme amplifier for highly efficient microRNA detection and intracellular imaging with ultralow background. Anal. Chem. 2020, 92, 15069–15078. [Google Scholar] [CrossRef]
- Jones, J.L.; Hara-Kudo, Y.; Krantz, J.A.; Benner, R.A.; Smith, A.B.; Dambaugh, T.R.; Bowers, J.C.; DePaola, A. Comparison of molecular detection methods for Vibrio parahaemolyticus and Vibrio vulnificus. Food Microbiol. 2012, 30, 105–111. [Google Scholar] [CrossRef]
- Tomoyasu, T. Development of the immunomagnetic enrichment method selective for Vibrio-parahaemolyticus serotype-k and its application to food poisoning study. Appl. Environ. Microbiol. 1992, 58, 2679–2682. [Google Scholar] [CrossRef] [Green Version]
- Duan, N.; Wu, S.; Yu, Y.; Ma, X.; Xia, Y.; Chen, X.; Huang, Y.; Wang, Z. A dual-color flow cytometry protocol for the simultaneous detection of Vibrio parahaemolyticus and Salmonella typhimurium using aptamer conjugated quantum dots as labels. Anal. Chim. Acta 2013, 804, 151–158. [Google Scholar] [CrossRef]
- Zeng, J.; Wei, H.; Zhang, L.; Liu, X.; Zhang, H.; Cheng, J.; Ma, D.; Zhang, X.; Fu, P.; Liu, L. Rapid detection of Vibrio parahaemolyticus in raw oysters using immunomagnetic separation combined with loop-mediated isothermal amplification. Int. J. Food Microbiol. 2014, 174, 123–128. [Google Scholar] [CrossRef]
- Federici, S.; Serrazanetti, D.I.; Guerzoni, M.E.; Campana, R.; Ciandrini, E.; Baffone, W.; Gianotti, A. Development of a rapid PCR protocol to detect Vibrio parahaemolyticus in clams. J. Food Sci. Technol. 2018, 55, 749–759. [Google Scholar] [CrossRef]
- Xu, D.; Ji, L.; Wu, X.; Yan, W.; Chen, L. Detection and differentiation of Vibrio parahaemolyticus by multiplexed real-time PCR. Can. J. Microbiol. 2018, 64, 809–815. [Google Scholar] [CrossRef] [PubMed]
- Song, S.X.; Wang, X.Y.; Xu, K.; Xia, G.M.; Yang, X.B. Visualized detection of Vibrio parahaemolyticus in food samples using dual-functional aptamers and cut-assisted rolling circle amplification. J. Agric. Food Chem. 2019, 67, 1244–1253. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Shi, L.; Su, J.Y.; Ye, Y.X.; Zhong, Q.P. Detection of Vibrio parahaemolyticus in food samples using in situ loop-mediated isothermal amplification method. Gene 2013, 515, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Gijavanekar, C.; Strych, U.; Fofanov, Y.; Fox, G.E.; Willson, R.C. Rare target enrichment for ultrasensitive PCR detection using cot-rehybridization and duplex-specific nuclease. Anal. Biochem. 2012, 421, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Pathirana, E.; McPherson, A.; Whittington, R.; Hick, P. The role of tissue type, sampling and nucleic acid purification methodology on the inferred composition of Pacific oyster (Crassostrea gigas) microbiome. J. Appl. Microbiol. 2019, 127, 429–444. [Google Scholar] [CrossRef]
- Zhao, X.H.; Wang, L.; Chu, J.; Li, Y.Y.; Li, Y.M.; Xu, Z.B.; Li, L.; Shirtliff, M.E.; He, X.W.; Liu, Y.; et al. Rapid detection of Vibrio parahaemolyticus strains and virulent factors by loop-mediated isothermal amplification assays. Food Sci. Biotechnol. 2010, 19, 1191–1197. [Google Scholar] [CrossRef]
- Wang, X.; Yu, X.; Wang, X.; Suzuki, M.; Asanuma, H.; Dong, P.; Wu, W.; Liang, X. Highly specific DNA detection from massive background nucleic acids based on rolling circle amplification of target dsDNA. RSC Adv. 2014, 4, 38293–38299. [Google Scholar] [CrossRef]
- Liu, J.; Xie, G.; Xiong, Q.; Liang, T.; Xu, H. Sensitive dual readout assays based on rolling circle amplification for fluorescent and colorimetric detection of Cronobacter spp. in powdered infant formula. Food Control 2021, 124, 107840. [Google Scholar] [CrossRef]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef]
- Wang, J.; Zhu, J.; Wang, C.; Zhou, G.; Yu, X.; Fan, H.; An, R.; Komiyama, M.; Liang, X. Thermus thermophilus DNA ligase connects two fragments having exceptionally short complementary termini at high temperatures. Biochemistry 2020, 59, 400–406. [Google Scholar] [CrossRef]
- Zhu, J.; Wang, J.; Cheng, K.; Chen, H.; An, R.; Zhang, Y.; Komiyama, M.; Liang, X. Effective characterization of DNA ligation kinetics by high-resolution melting analysis. Chembiochem 2020, 21, 785–788. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Liu, M.; Luo, X.; Pan, J. Draft genome sequence of Strain ATCC 17802(T), the type strain of Vibrio parahaemolyticus. Mar. Genom. 2015, 24, 203–205. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, R.; Fockler, C.; Dollinger, G.; Watson, R. Kinetic PCR analysis: Real-Time monitoring of DNA amplification reactions. Biotechnology 1993, 11, 1026–1030. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, S.; Almasi, M.A.; Fatehi, F.; Struik, P.C.; Moradi, A. Visual detection of potato leafroll virus by one-step reverse transcription loop-mediated isothermal amplification of DNA with hydroxynaphthol blue dye. J. Phytopathol. 2013, 161, 120–124. [Google Scholar] [CrossRef]
- An, R.; Li, Q.; Fan, Y.; Li, J.; Pan, X.; Komiyama, M.; Liang, X. Highly efficient preparation of single-stranded DNA rings by T4 ligase at abnormally low Mg(II) concentration. Nucleic Acids Res. 2017, 45, e139. [Google Scholar] [CrossRef] [Green Version]
- Shuman, S. DNA ligases: Progress and prospects. J. Biol. Chem. 2009, 284, 17365–17369. [Google Scholar] [CrossRef] [Green Version]
- Mahmoudian, L.; Kaji, N.; Tokeshi, M.; Nilsson, M.; Baba, Y. Rolling circle amplification and circle-to-circle amplification of a specific gene integrated with electrophoretic analysis on a single chip. Anal. Chem. 2008, 80, 2483–2490. [Google Scholar] [CrossRef]
- Jiang, Q.; Wei, L.; Gai, C.; Yu, W.; He, C.; Chen, M.; Zhang, Z.; Song, H.; Wang, X.; Wang, X. An improved extraction method reveals varied DNA content in different parts of the shells of Pacific oysters. Aquat. Living Resour. 2019, 32, 5. [Google Scholar] [CrossRef]
- Zhang, G.; Fang, X.; Guo, X.; Li, L.; Luo, R.; Xu, F.; Yang, P.; Zhang, L.; Wang, X.; Qi, H.; et al. The oyster genome reveals stress adaptation and complexity of shell formation. Nature 2012, 490, 49–54. [Google Scholar] [CrossRef] [Green Version]
- Tian, W.; Li, P.; He, W.; Liu, C.; Li, Z. Rolling circle extension-actuated loop-mediated isothermal amplification (RCA-LAMP) for ultrasensitive detection of microRNAs. Biosens. Bioelectron. 2019, 128, 17–22. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, B.; Sun, W.; Ran, L.; Wang, C.; Wang, J.; An, R.; Liang, X. Anti-Interference Detection of Vibrio parahaemolyticus from Aquatic Food Based on Target-Cyclized RCA with Dynamic Adapter Followed by LAMP. Foods 2022, 11, 352. https://doi.org/10.3390/foods11030352
Zhang B, Sun W, Ran L, Wang C, Wang J, An R, Liang X. Anti-Interference Detection of Vibrio parahaemolyticus from Aquatic Food Based on Target-Cyclized RCA with Dynamic Adapter Followed by LAMP. Foods. 2022; 11(3):352. https://doi.org/10.3390/foods11030352
Chicago/Turabian StyleZhang, Boying, Wenhua Sun, Lingling Ran, Chenru Wang, Jing Wang, Ran An, and Xingguo Liang. 2022. "Anti-Interference Detection of Vibrio parahaemolyticus from Aquatic Food Based on Target-Cyclized RCA with Dynamic Adapter Followed by LAMP" Foods 11, no. 3: 352. https://doi.org/10.3390/foods11030352