
Extraction of Nucleotides from Dietary Supplements by Newly Synthesized Adsorbents


Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Instrumentation
2.3. Chromatographic Conditions
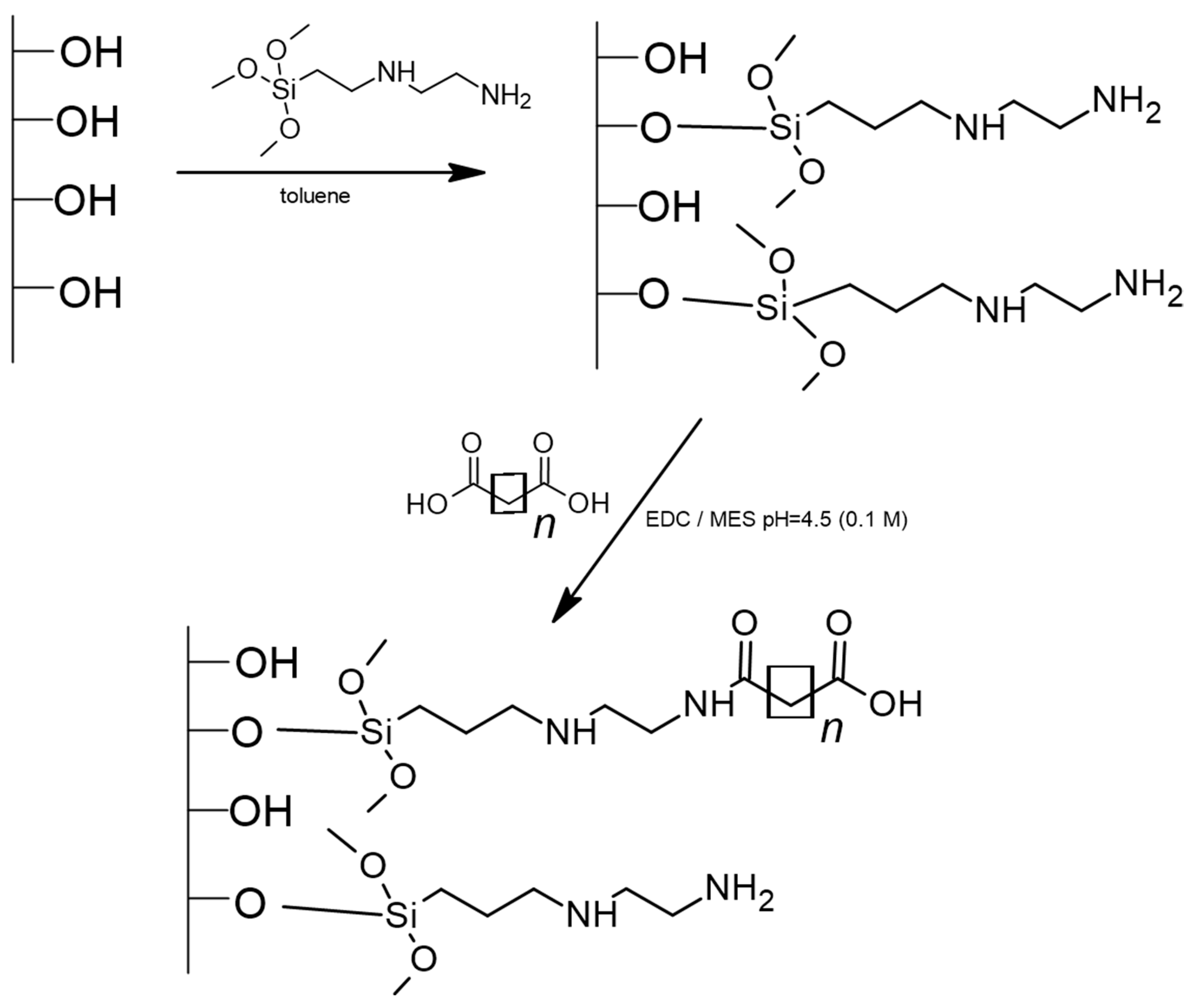
2.4. Synthesis of New Adsorbents
2.5. Nucleoside Adsorption and Desorption Tests for New Adsorbents
2.6. Adsorption Capacity
2.7. Nucleotides Extraction from Two Dietary Supplements
2.8. Recovery of Nucleotides from Dietary Supplements and Their Quantification
- A1—peak area in the extract of dietary supplement enriched after the extraction;
- A3—peak area in the dietary supplement extract;
- A2—peak area in the dietary supplement extract enriched before the extraction.
3. Results and Discussion
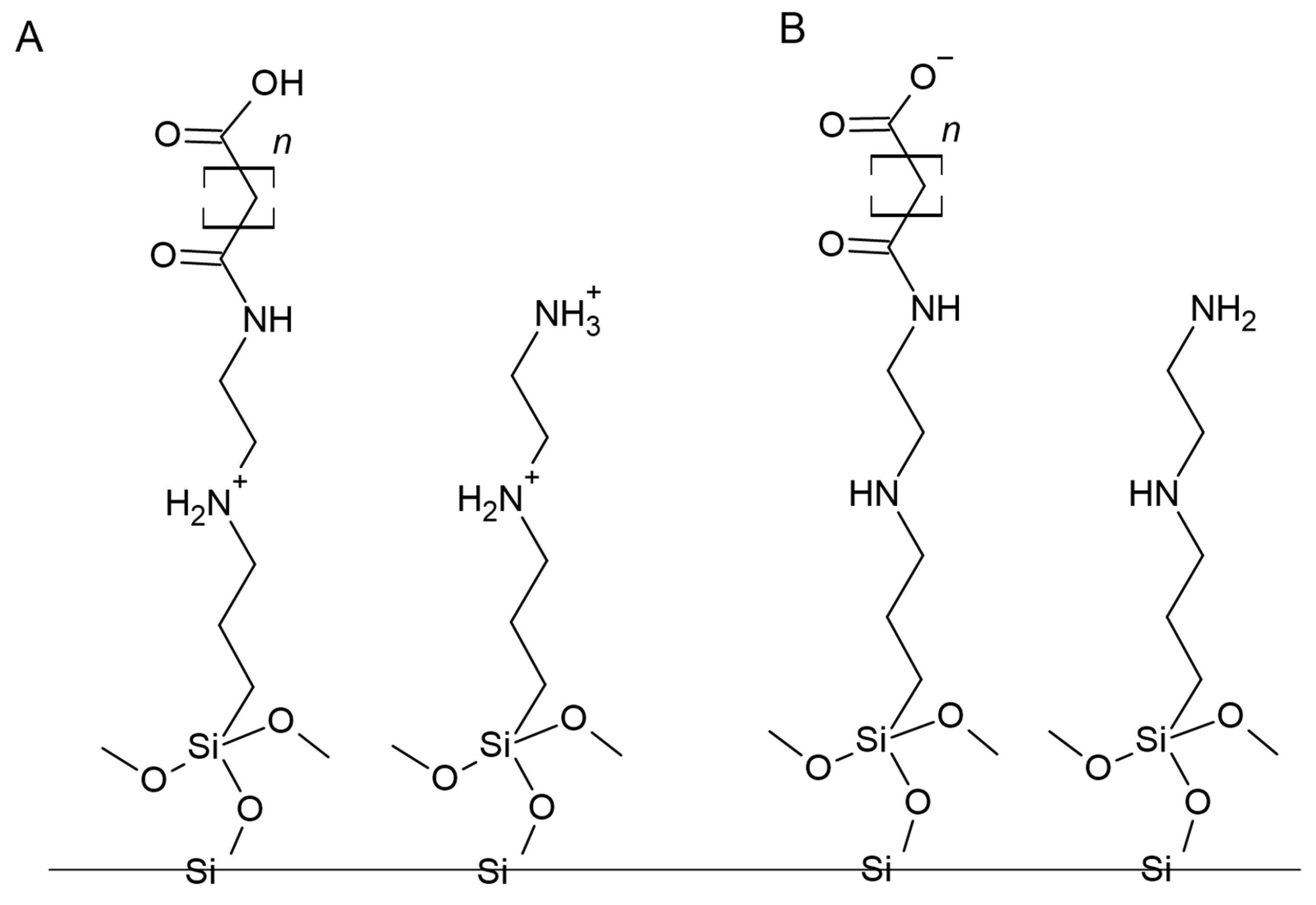
3.1. Characterization of Adsorbents
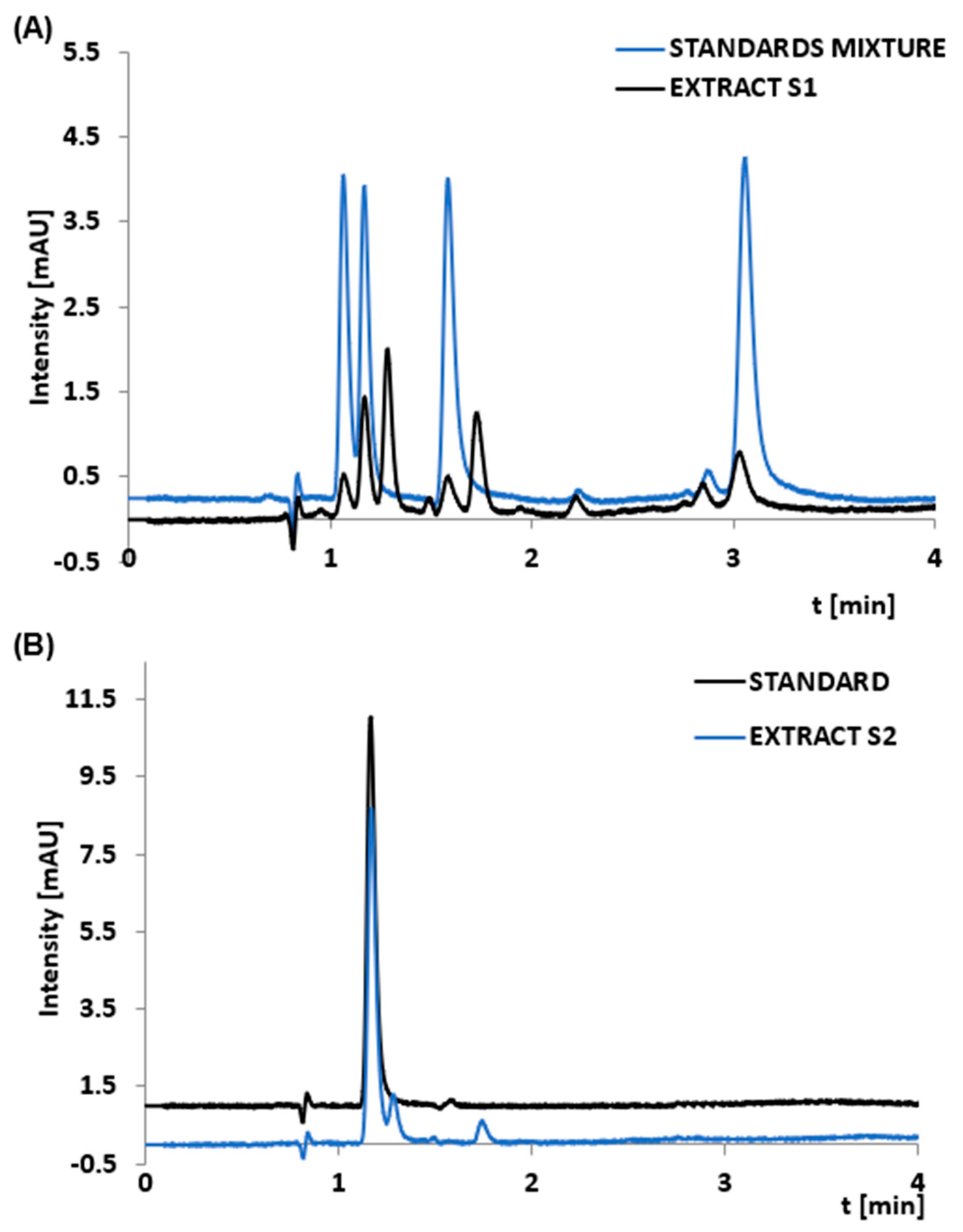
3.2. Nucleotides Analysis
3.3. Development of a dSPE Method for Nucleotides Using New Adsorbents
3.3.1. Adsorbent Structure and Probable Impact on Nucleotide Extraction
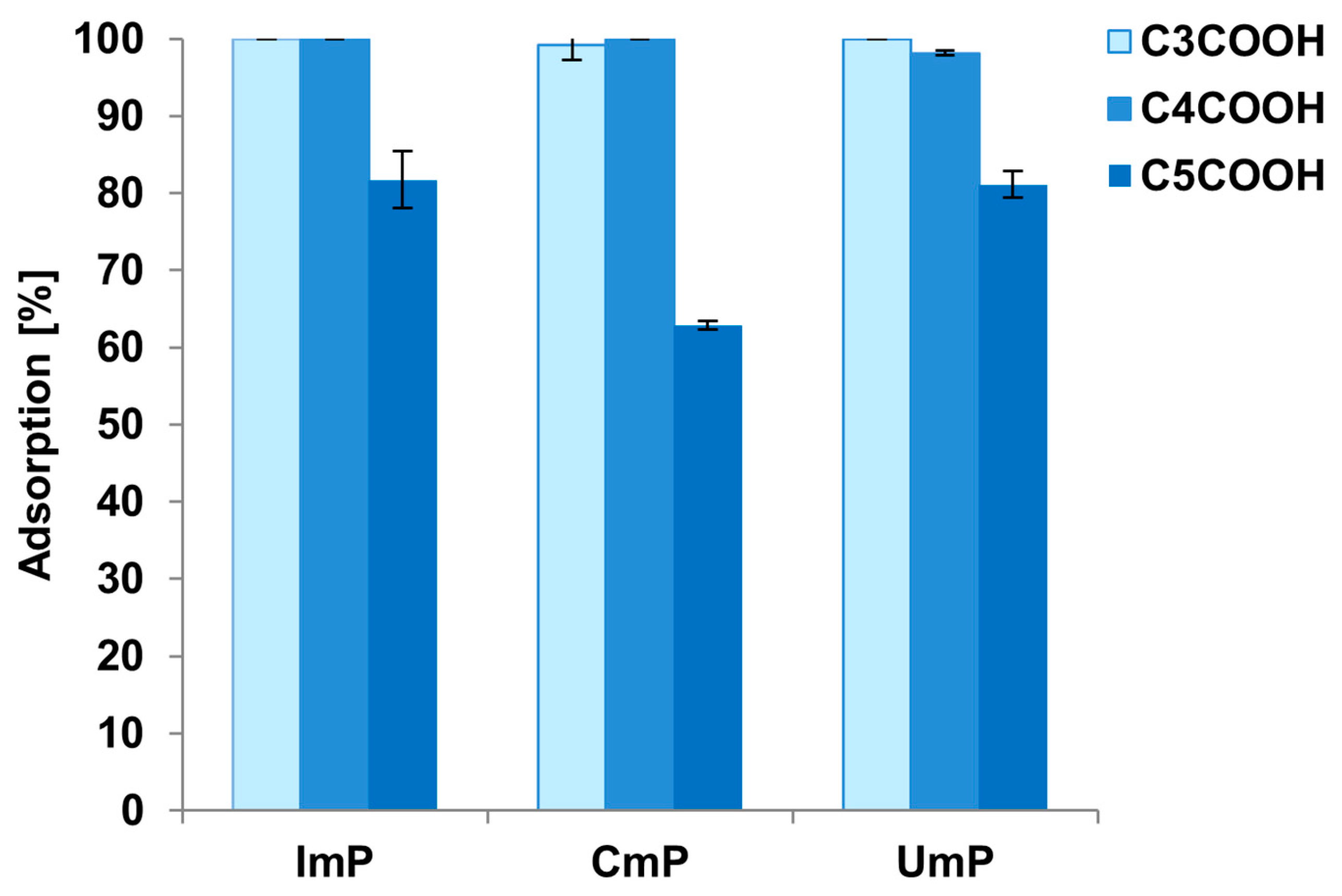
3.3.2. Selection of Solvent Additive for Adsorption
3.3.3. Selection of Solvent for Desorption
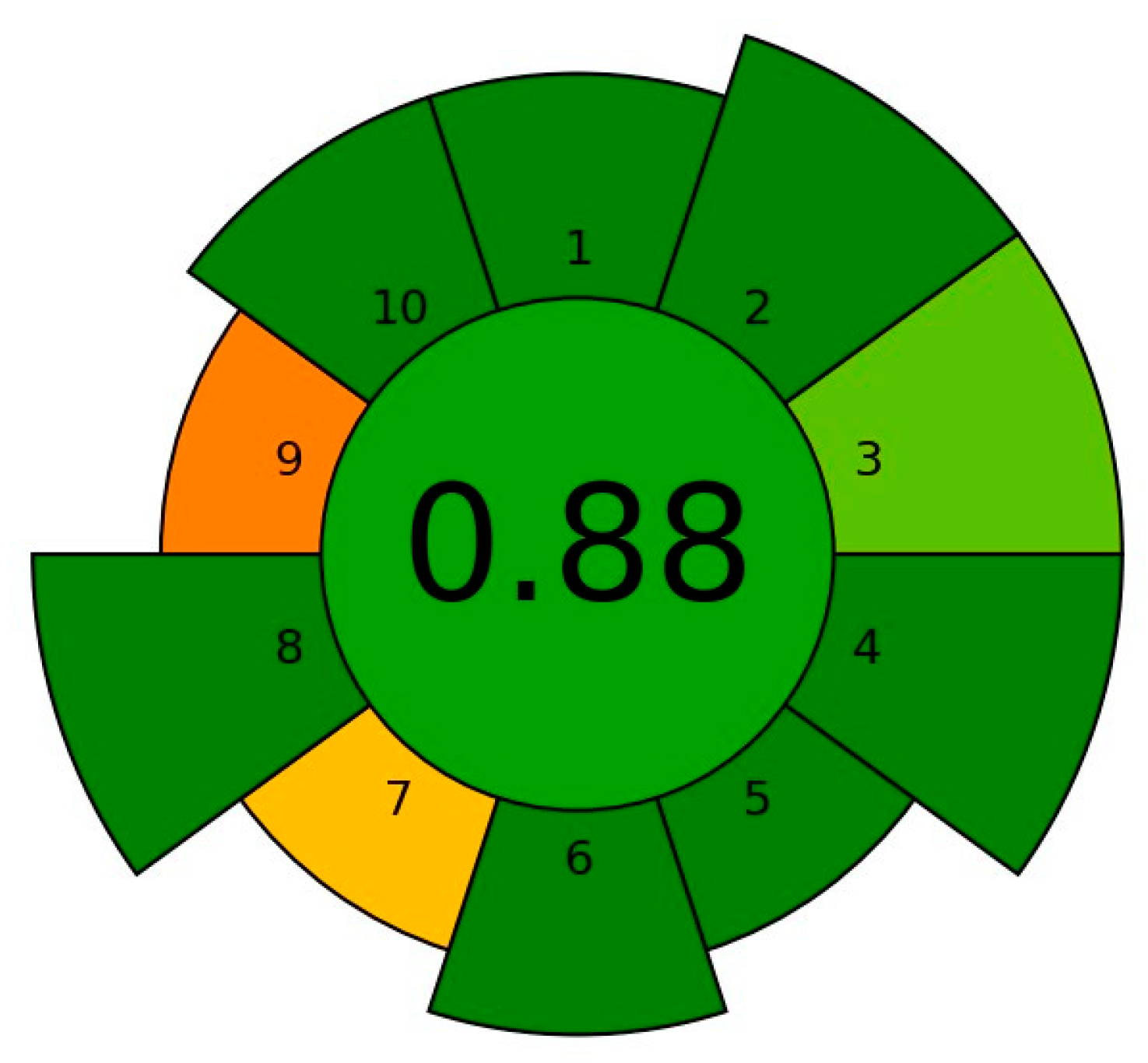
3.3.4. The Final Optimized dSPE Method: Its Advantages, Limitations and Greenness
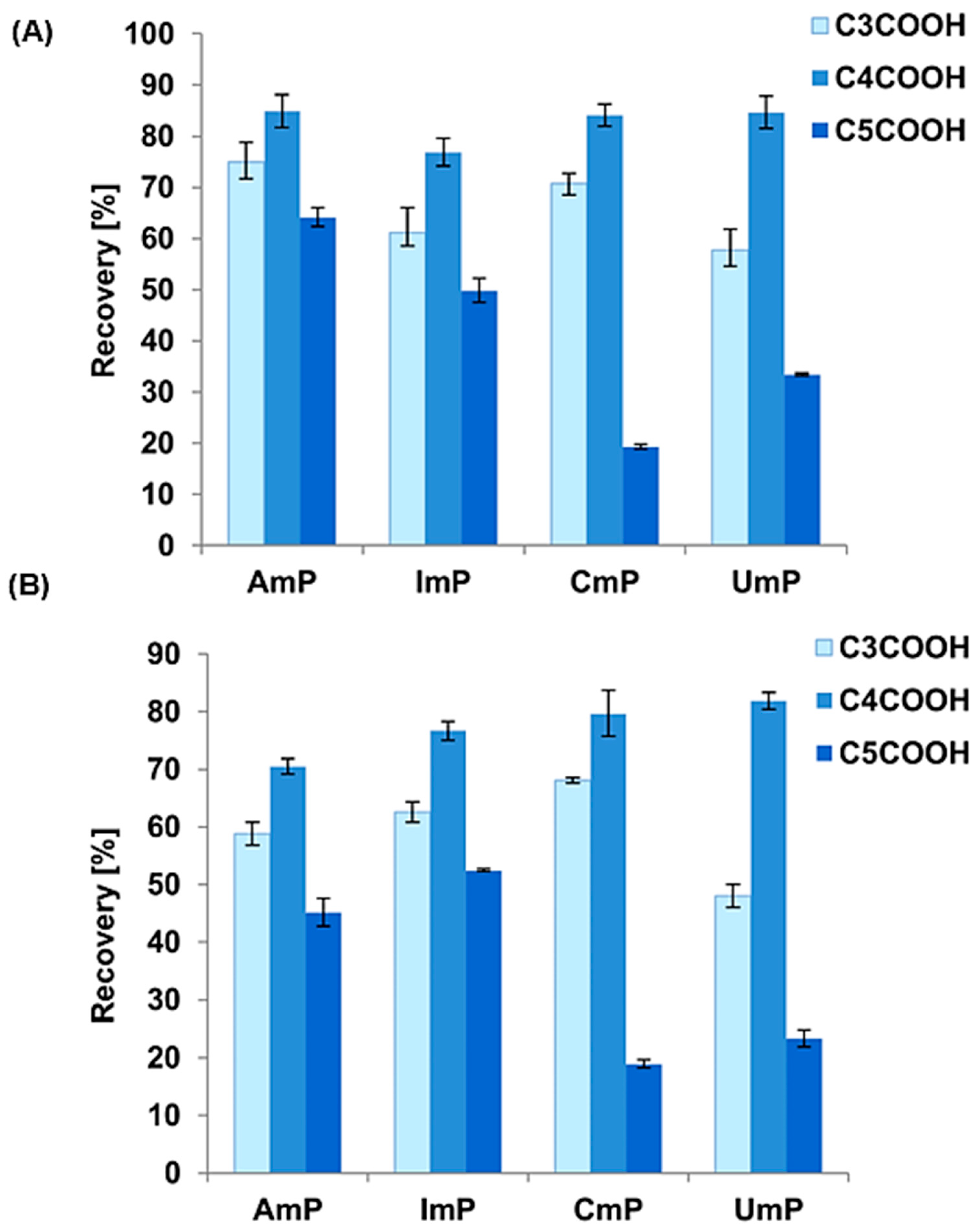
3.4. Application of the Developed Extraction Method to Dietary Supplement Samples
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Ding, T.; Song, G.; Liu, X.; Xu, M.; Li, Y. Nucleotides as Optimal Candidates for Essential Nutrients in Living Organisms: A Review. J. Funct. Foods 2021, 82, 104498. [Google Scholar] [CrossRef]
- Lane, A.N.; Fan, T.W.M. Regulation of Mammalian Nucleotide Metabolism and Biosynthesis. Nucleic Acids Res. 2015, 43, 2466–2485. [Google Scholar] [CrossRef]
- Carver, J.D.; Sosa, R.; Saste, M.; Kuchan, M. Dietary Nucleotides and Intestinal Blood Flow Velocity in Term Infants. J. Pediatr. Gastroenterol. Nutr. 2004, 39, 38–42. [Google Scholar] [CrossRef]
- Carver, J.D.; Saste, M.; Sosa, R.; Zaritt, J.; Kuchan, M.; Barness, L.A. The Effects of Dietary Nucleotides on Intestinal Blood Flow in Preterm Infants. Pediatr. Res. 2002, 52, 425–429. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Serrano, F.; Álvarez, P.; Caba, O.; Picón, M.; Marchal, J.A.; Perán, M.; Prados, J.; Melguizo, C.; Rama, A.R.; Boulaiz, H.; et al. Promotion of Human Adipose-Derived Stem Cell Proliferation Mediated by Exogenous Nucleosides. Cell Biol. Int. 2010, 34, 917–924. [Google Scholar] [CrossRef]
- Jang, K.B.; Kim, S.W. Supplemental Effects of Dietary Nucleotides on Intestinal Health and Growth Performance of Newly Weaned Pigs. J. Anim. Sci. 2019, 97, 4875–4882. [Google Scholar] [CrossRef] [PubMed]
- Singhal, A.; Kennedy, K.; Lanigan, J.; Clough, H.; Jenkins, W.; Elias-Jones, A.; Stephenson, T.; Dudek, P.; Lucas, A. Dietary Nucleotides and Early Growth in Formula-Fed Infants: A Randomized Controlled Trial. Pediatrics 2010, 126, e946–e953. [Google Scholar] [CrossRef] [PubMed]
- Dancey, C.P.; Attree, E.A.; Brown, K.F. Nucleotide Supplementation: A Randomised Double-Blind Placebo Controlled Trial of IntestAidIB in People with Irritable Bowel Syndrome [ISRCTN67764449]. Nutr. J. 2006, 5, 16. [Google Scholar] [CrossRef]
- Cordell, R.L.; Hill, S.J.; Ortori, C.A.; Barrett, D.A. Quantitative Profiling of Nucleotides and Related Phosphate-Containing Metabolites in Cultured Mammalian Cells by Liquid Chromatography Tandem Electrospray Mass Spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2008, 871, 115–124. [Google Scholar] [CrossRef]
- Soga, T.; Ishikawa, T.; Igarashi, S.; Sugawara, K.; Kakazu, Y.; Tomita, M. Analysis of Nucleotides by Pressure-Assisted Capillary Electrophoresis-Mass Spectrometry Using Silanol Mask Technique. J. Chromatogr. A 2007, 1159, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Studzińska, S.; Zalesińska, E. Development of an Ultra High Performance Liquid Chromatography Method for the Separation and Determination of Nucleotides and Nucleosides in Extracts from Infant Milk Formulas and Human Milk Samples. J. Food Compos. Anal. 2023, 115, 104949. [Google Scholar] [CrossRef]
- Cohen, S.; Jordheim, L.P.; Megherbi, M.; Dumontet, C.; Guitton, J. Liquid Chromatographic Methods for the Determination of Endogenous Nucleotides and Nucleotide Analogs Used in Cancer Therapy: A Review. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2010, 878, 1912–1928. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, H.; Wang, J.; Pan, C. Determination of Trace Level of CAMP in Locusta Migratoria Manilensis Meyen by HPLC with Fluorescence Derivation. Int. J. Mol. Sci. 2006, 7, 266–273. [Google Scholar] [CrossRef]
- Zhang, K. From Purification of Large Amounts of Phospho-Compounds (Nucleotides) to Enrichment of Phospho-Peptides Using Anion-Exchanging Resin. Anal. Biochem. 2006, 357, 225–231. [Google Scholar] [CrossRef]
- Yao, T.; Liao, Y.; Li, S.; Qiao, L.; Du, K. Bisphosphonated-Immobilized Porous Cellulose Monolith with Tentacle Grafting by Atom Transfer Radical Polymerization for Selective Adsorption of Lysozyme. J. Chromatogr. A 2021, 1651, 462337. [Google Scholar] [CrossRef]
- Yao, T.; Song, J.; Gan, Y.; Qiao, L.; Du, K. Preparation of Cellulose-Based Chromatographic Medium for Biological Separation: A Review. J. Chromatogr. A 2022, 1677, 463297. [Google Scholar] [CrossRef] [PubMed]
- Yao, T.; Song, J.; Hong, Y.; Gan, Y.; Ren, X.; Du, K. Application of Cellulose to Chromatographic Media: Cellulose Dissolution, and Media Fabrication and Derivatization. J. Chromatogr. A 2023, 1705, 464202. [Google Scholar] [CrossRef] [PubMed]
- Czarnecka, J.; Cieślak, M.; Michał, K. Application of Solid Phase Extraction and High-Performance Liquid Chromatography to Qualitative and Quantitative Analysis of Nucleotides and Nucleosides in Human Cerebrospinal Fluid. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2005, 822, 85–90. [Google Scholar] [CrossRef]
- Contreras-Sanz, A.; Scott-Ward, T.S.; Gill, H.S.; Jacoby, J.C.; Birch, R.E.; Malone-Lee, J.; Taylor, K.M.G.; Peppiatt-Wildman, C.M.; Wildman, S.S.P. Simultaneous Quantification of 12 Different Nucleotides and Nucleosides Released from Renal Epithelium and in Human Urine Samples Using Ion-Pair Reversed-Phase HPLC. Purinergic Signal. 2012, 8, 741–751. [Google Scholar] [CrossRef]
- Machon, C.; Jordheim, L.P.; Puy, J.Y.; Lefebvre, I.; Dumontet, C.; Guitton, J. Fully Validated Assay for the Quantification of Endogenous Nucleoside Mono- and Triphosphates Using Online Extraction Coupled with Liquid Chromatography-Tandem Mass Spectrometry. Anal. Bioanal. Chem. 2014, 406, 2925–2941. [Google Scholar] [CrossRef] [PubMed]
- Jendresen, C.B.; Kilstrup, M.; Martinussen, J. A Simplified Method for Rapid Quantification of Intracellular Nucleoside Triphosphates by One-Dimensional Thin-Layer Chromatography. Anal. Biochem. 2011, 409, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Magdenoska, O.; Martinussen, J.; Thykaer, J.; Nielsen, K.F. Dispersive Solid Phase Extraction Combined with Ion-Pair Ultra High-Performance Liquid Chromatography Tandem Mass Spectrometry for Quantification of Nucleotides in Lactococcus Lactis. Anal. Biochem. 2013, 440, 166–177. [Google Scholar] [CrossRef]
- Available online: https://mostwiedzy.pl/AGREEprep (accessed on 5 May 2023).
- Bocian, S.; Studzińska, S.; Buszewski, B. Functionalized Anion Exchange Stationary Phase for Separation of Anionic Compounds. Talanta 2014, 127, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Jaćkowska, M.; Bocian, S.; Buszewski, B. Dendrimer Modified Silica Gel for Anion Exchange Chromatographt: Synthesis, Characterization, and Application. Analyst 2012, 137, 4610–4617. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, G.E.; Galan, L. de Role of the Chain Length of Chemically Bonded Phases and the Retention Mechanism in Reversed-Phase Liquid Chromatography. J. Chromatogr. A 1980, 196, 21–37. [Google Scholar] [CrossRef]
- Balci, M. 13C chemical shifts of organic compounds. In Basic 1H- 13C-NMR Spectroscopy; Elsevier Science: Amsterdam, The Netherlands, 2005; pp. 293–324. [Google Scholar] [CrossRef]
- Rabenstein, D.L.; Sayer, T.L. Carbon-13 Chemical Shift Parameters for Amines, Carboxylic Acids, and Amino Acids. J. Magn. Reson. 1976, 24, 27–39. [Google Scholar] [CrossRef]
- Wojnowski, W.; Tobiszewski, M.; Pena-Pereira, F.; Psillakis, E. AGREEprep—Analytical Greenness Metric for Sample Preparation. TrAC Trends Anal. Chem. 2022, 149, 116553. [Google Scholar] [CrossRef]
- Pena-Pereira, F.; Wojnowski, W.; Tobiszewski, M. AGREE—Analytical GREEnness Metric Approach and Software. Anal. Chem. 2020, 92, 10076–10082. [Google Scholar] [CrossRef]
- Chen, X.; Wu, Y.; Huang, L.; Yang, L.; Hong, R.; Yao, H.; Li, S. Magnetic Dispersive Solid-Phase Micro-Extraction Combined with High-Performance Liquid Chromatography for Determining Nucleotides in Anoectochilus Roxburghii (Wall.) Lindl. J. Pharm. Biomed. Anal. 2019, 174, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.D.; Zhang, H.; Zhang, Q.; Qian, Z.M.; Li, W.J.; Li, C.H.; Yang, F.Q.; Chen, H. Preparation of Titanium Ion Functionalized Polydopamine Coated Ferroferric Oxide Core-Shell Magnetic Particles for Selective Extraction of Nucleotides from Cordyceps and Lentinus Edodes. J. Chromatogr. A 2019, 1591, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Cavalluzzi, M.M.; Lamonaca, A.; Rotondo, N.P.; Miniero, D.V.; Muraglia, M.; Gabriele, P.; Corbo, F.; De Palma, A.; Budriesi, R.; De Angelis, E.; et al. Microwave-Assisted Extraction of Bioactive Compounds from Lentil Wastes: Antioxidant Activity Evaluation and Metabolomic Characterization. Molecules 2022, 27, 7471. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Zhang, J.; Song, X.; Chen, X.; Li, D. Simultaneous Determination of 5′-Monophosphate Nucleotides in Infant Formulas by HPLC-MS. J. Chromatogr. Sci. 2011, 49, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Gill, B.D.; Indyk, H.E.; Kumar, M.C.; Sievwright, N.K.; Manley-Harris, M. A Liquid Chromatographic Method for Routine Analysis of 5’-Mononucleotides in Pediatric Formulas. J. AOAC Int. 2010, 93, 966–973. [Google Scholar] [CrossRef] [PubMed]
- Pastor-Belda, M.; Fernández-Caballero, I.; Campillo, N.; Arroyo-Manzanares, N.; Hernández-Córdoba, M.; Viñas, P. Hydrophilic interaction liquid chromatography to quadrupole-time-of-flight mass spectrometry for determination of nuclear and cytoplasmatic contents of nucleotides, nucleosides and their nucleobases in food yeasts. Talanta Open 2021, 4, 100064. [Google Scholar] [CrossRef]
- Büyüktuncel, S.E. Determination of ATP and its metabolites in dietary energy supplements by capillary electrophoresis. Braz. J. Pharm. Sci. 2022, 58, e201045. [Google Scholar] [CrossRef]
- Neubauer, S.; Rugova, A.; Chu, D.B. Mass spectrometry-based analysis of nucleotides, nucleosides, and nucleobases-application to feed supplements. Anal. Bioanal. Chem. 2012, 404, 799–808. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Adsorbent | ||||
|---|---|---|---|---|
| Di-Amine | C3COOH | C4COOH | C5COOH | |
| Carbon content [%] | 6.267 | 7.300 | 8.184 | 9.164 |
| Hydrogen content [%] | 2.709 | 2.018 | 2.506 | 2.103 |
| Nitrogen content [%] | 2.547 | 2.196 | 2.112 | 2.115 |
| Coverage density [µmol/m2] | 2.56 | 0.59 | 0.83 | 1.02 |
| Solvent | Adsorbent | Nucleotide | Percentage of Nucleotide Adsorbed at the Surface [%] |
|---|---|---|---|
| H2O pH = 4.5 (adjusted with acetic acid) | C3COOH | GmP | 97.4 ± 0.4 |
| AmP | 81.7 ± 2.9 | ||
| C4COOH | GmP | 96.4 ± 0.4 | |
| AmP | 99.8 ± 1.6 | ||
| C5COOH | GmP | 90.1 ± 1.8 | |
| AmP | 81.1 ± 4.8 | ||
| 10 mM CH3COONH4 pH = 4.5 (adjusted with acetic acid) | C3COOH | GmP | 85.5 ± 5.6 |
| AmP | 92.6 ± 0.6 | ||
| C4COOH | GmP | 90.5 ± 0.2 | |
| AmP | 82.5 ± 0.4 | ||
| C5COOH | GmP | 92.1 ± 4.8 | |
| AmP | 95.5 ± 5.6 | ||
| 10 mM CH3COONH4 pH = 5.5 (adjusted with acetic acid) | C3COOH | GmP | 80.8 ± 0.2 |
| AmP | 77.1 ± 1.0 | ||
| C4COOH | GmP | 88.8 ± 0.4 | |
| AmP | 86.9 ± 0.6 | ||
| C5COOH | GmP | 80.0 ± 4.8 | |
| AmP | 75.5 ± 5.6 |
| Solvent | Adsorbent | Percentage of Desorbed Nucleotide [%] |
|---|---|---|
| 10 mM CH3COONH4 pH = 9 | C3COOH | 67.5 ± 0.6 |
| C4COOH | 79.8 ± 1.9 | |
| C5COOH | 67.9 ± 3.3 | |
| 10 mM CH3COONH4 pH = 9.5 | C3COOH | 67.3 ± 2.8 |
| C4COOH | 72.8 ± 1.7 | |
| C5COOH | 67.1 ± 2.9 | |
| 10 mM CH3COONH4 pH = 10 | C3COOH | 75.3 ± 12.3 |
| C4COOH | 85.0 ± 9.9 | |
| C5COOH | 73.0 ± 6.4 | |
| 10 mM CH3COONH4 pH = 10.5 | C3COOH | 70.3 ± 4.7 |
| C4COOH | 73.2 ± 5.3 | |
| C5COOH | 85.5 ± 6.5 | |
| H2O pH = 10 | C3COOH | 19.8 ± 1.7 |
| C4COOH | 65.4 ± 1.7 | |
| C5COOH | 73.5 ± 0.6 | |
| H2O pH = 10.5 | C3COOH | 4.0 ± 0.2 |
| C4COOH | 15.2 ± 0.5 | |
| C5COOH | 44.2 ± 1.3 |
| Method | Adsorbent | Solvents | Recovery [%] | Time | Matrix | Reference |
|---|---|---|---|---|---|---|
| DMSPE | magnetic ferroferric oxide nanoparticles, Fe3O4@GO | 0.05 M NaOH pH 3.5 | 88–109 | ~20 min | plant | [31] |
| DMSPE | titanium-ion-functionalized ferroferric oxide magnetic particles | 0.02 M Na3PO4·12H2O | 76–95 | ~20 min | plant | [32] |
| dSPE | activated charcoal | ethanol, water, 1 M HCl, 2% NH3, and 50% acetonitrile in water | — | ~1 h | bacteria | [22] |
| SPE | Strata X | 0.025 M ethanolamine pH 8.0, water, methanol | 86–98 | ~1 h | cerebrospinal fluid | [18] |
| MAE | — | MeOH:H2O 80:20 (v/v) | — | ~1 h | lentil waste | [33] |
| MAE | not used | water, formic acid | 95–104 | ~30 min | infant formulas | [34] |
| SPE | strong cation exchanger | 0.5 M KH2PO4 pH 3.0; 0.3 M KBr | 81–102 | ~30 min | infant formulas | [35] |
| SPE | anion-exchanging | 0.1–0.3 M NH4H2PO4 (pH 3.0), NH4OH (pH 10.0) | — | ~1 h | phospho-peptides | [14] |
| SPE | polymeric reversed-phase Strata-X | 25 mM ethanolamine (pH 5); 30% methanol in 25 mM ethanolamine (pH 5) | 98–104 | renal cell line and urine | [19] | |
| dSPE | weak anion exchanger | water (pH 4.5), 0.01 M CH3COONH4 (pH 9.0) | 78–87 | ~40 min | dietary supplements | our method |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Studzińska, S.; Bocian, S.; Stypczyńska, P.; Wolan, A. Extraction of Nucleotides from Dietary Supplements by Newly Synthesized Adsorbents. Foods 2023, 12, 3675. https://doi.org/10.3390/foods12193675
Studzińska S, Bocian S, Stypczyńska P, Wolan A. Extraction of Nucleotides from Dietary Supplements by Newly Synthesized Adsorbents. Foods. 2023; 12(19):3675. https://doi.org/10.3390/foods12193675
Chicago/Turabian StyleStudzińska, Sylwia, Szymon Bocian, Paulina Stypczyńska, and Andrzej Wolan. 2023. "Extraction of Nucleotides from Dietary Supplements by Newly Synthesized Adsorbents" Foods 12, no. 19: 3675. https://doi.org/10.3390/foods12193675
APA StyleStudzińska, S., Bocian, S., Stypczyńska, P., & Wolan, A. (2023). Extraction of Nucleotides from Dietary Supplements by Newly Synthesized Adsorbents. Foods, 12(19), 3675. https://doi.org/10.3390/foods12193675