1. Introduction
Cronobacter spp. is a facultatively anaerobic, Gram-negative foodborne pathogen [
1]. The World Food and Agriculture Organization (FAO) and the World Health Organization (WHO) have classified
Cronobacter spp. as one of the pathogenic bacteria in category A of infant formula (PIF). Newborns and infants are high-risk groups for
Cronobacter infection, and PIF and its processing environment are the main pollution channels of
Cronobacter spp. [
2,
3,
4].
Cronobacter spp. includes seven species and three subspecies [
5,
6,
7]. Among them,
C. sakazakii and
C. malonaticus are the main pathogenic bacteria in the clinic. This bacterium is related to meningitis and necrotizing enterocolitis and infects infants by contaminating powdered infant formula (PIF) [
8,
9], with mortality rates ranging from 40% to 80% [
10,
11]. Even if cured, there is a possibility of severe neurological sequelae. Multilocus sequence typing (MLST) is a molecular typing method based on DNA sequence. MLST method for
Cronobacter spp. has become one of the most effective and common methods [
12,
13]. Currently, the
Cronobacter MLST database (
http://www.pubmlst.org/cronobacter accessed on 22 March 2022) has been established, which contains more than 3000
Cronobacter strains from different sources and is divided into more than 800 sequence types (STs). MLST technology has been gradually expanded in the typing research of
Cronobacter spp., because of its simple operation, high identification, and a high degree of sharing. At present, the study found that the STs of
Cronobacter have a certain relationship with its serotype [
14,
15]. In addition, Fei et al. used MLST technology to classify 70
Cronobacter strains isolated from PIF in China into 19 STs and found that the dominant STs of
Cronobacter spp. from PIF in China were ST4, ST1, and ST64 [
16]. It can be seen that the MLST method has been widely used in the identification and typing of
Cronobacter spp. and has been well accepted.
Currently, the most effective treatment for diseases caused by
Cronobacter spp. is antibiotic therapy [
17,
18]. Many studies have been carried out to evaluate the drug resistance of
Cronobacter spp. [
19,
20,
21]. Research reports in recent years have confirmed that
Cronobacter spp. has strong drug resistance and is increasing year by year [
22,
23,
24]. After research, penicillin, first- and second-generation cephalosporins and other commonly used antibiotics in hospitals have lost their inhibitory effect on
Cronobacter spp., and new
Cronobacter isolates with multi-drug resistance spectrum have been gradually discovered [
25,
26]. Although the multidrug-resistant operon mar was found in the genome of
Cronobacter spp., the drug resistance level of this genus is generally lower than that of other foodborne pathogens, and the drug resistance mechanism is not yet clear [
27,
28]. Cao et al. studied the reaction mechanism of
C. sakazakii CICC 21,544 under the combined stress of citral and carvacrol by transcriptome sequencing and found 25 significantly differentially expressed genes, which were mainly involved in the metabolism of various small molecules, ribosome function, and transmembrane transport systems [
29]. Xu et al. used transcriptome sequencing to explore the regulatory mechanism of
pmrA mutation on the biofilm formation of
C. sakazakii BAA-894 [
30]. The transcriptome research method can be used to determine the known tolerance mechanism. We can also find a new regulatory mechanism related to the tolerance of the bacteria from these differentially expressed genes and reveal the gene function and its possible metabolic pathway. Thereby, contributing to the prevention and control of pathogenic microorganisms in food and its processing environment [
31,
32,
33,
34].
Thirty-five Cronobacter strains isolated from PIF and its processing environment were used as the research objects in this investigation. The strains were identified and molecularly typed by 16S rRNA sequencing and MLST technology. On this basis, strains with different STs were selected for antibiotic resistance evaluation and analyzed. The drug resistance-related genes and metabolic pathways of Cronobacter spp. under antibiotic conditions were mined with the transcriptome analysis, thereby, revealing the resistance mechanism.
2. Materials and Methods
2.1. Bacterial Strains Information
Thirty-five
Cronobacter strains isolated from PIF and processing environments in different areas of China were selected in this investigation. The detailed information of strains was listed in
Table S1. Meanwhile, eight type strains (
C. sakazakii ATCC 29544T,
C. sakazakii ATCC BAA-894T,
C. malonaticus LMG 23826T,
C. turicensis DSM 18703T,
C. muytjensii ATCC 51329T,
C.universalis NCTC 9529T,
C. dublinensis DSM 18705T,
C. condimenti LMG 26250T) and two reference strains (
C. sakazakii ATCC 12868,
C. sakazakii ATCC 29004) were also chosen for comparison.
2.2. Reagents and Instruments
A 2× Taq PCR MasterMix and DNA Marker were purchased from TIANGEN Biotech Co., Ltd. (Beijing, China). Primers were synthesized by Sangon Biotech Co., Ltd. (Shanghai, China). EZNATM bacterial total RNA extraction kit was obtained from Omega Bio-Tek Inc. (Norcross, GA, USA). PrimeScriptTM reverse transcription kit was obtained from TaKaRa BIO Co., Ltd. (Dalian, China). Absorbance was measured by SpectraMax i3x multifunctional microplate reader (Molecular Devices Co., Ltd., Sunnyvale, CA, USA). The concentration and purity of the DNA was measured by NanoDrop NC2000 spectrophotometer (Thermo Fisher Scientific Co., Ltd., Shanghai, China).
2.3. Strains Culture and DNA Extraction
All strains were cultured at 37 °C, 150 r/min overnight, and then purified on tryptic soy agar (TSA) plates at 37 °C for 14–16 h. A single colony was selected in a tryptic soy broth (TSB) liquid medium and cultured for 8 h at 37 °C 150 r/min to obtain strains at the end of the logarithmic growth phase. In this experiment, TIANGEN bacterial genomic DNA extraction kit was used for rapid extraction of strain DNA and stored at −20 °C for further tests.
2.4. 16S rRNA-Based PCR Identification and MLST Technology
The extracted DNA was amplified by PCR using bacterial 16S rRNA universal primers 27-F and 1492-R. The PCR assay was carried out in a 50 μL mixture system including 16 μL of 2× Taq PCR MasterMix, 27 μL of sterile distilled water, 1 μL of each primer with a concentration of 10 μM, and 5 μL of template DNA. The reaction conditions consisted of an initial predenaturation at 95 °C for 5 min, followed by 30 cycles of 94 °C for 30 s, an annealing at 55 °C for 30 s, an elongation at 72 °C for 30 s, and a final extension at 72 °C for 5 min. The amplified products were sequenced to complete the molecular identification of the strains.
At the same time, 7 pairs of housekeeping gene primers of
Cronobacter spp. were used to specifically amplify the extracted DNA of different strains, the genes were
atpD,
fusA,
glnS,
gltB,
gyrB,
infB, and
pps, respectively. The amplification system and conditions were the same as the 16S rRNA PCR reaction. Then, the amplified product was sequenced and analyzed, and the unidirectional measurement was performed to obtain the nucleic acid sequences of the 7 housekeeping genes of the strains. The primer sequences were shown in
Table S2.
According to the 16S rRNA sequencing results and the 3036 bp nucleic acid sequence of 7 housekeeping genes spliced in sequence, 35 Cronobacter strains were analyzed by neighbor-joining phylogeny analysis using MEGA7.0 software, and the procedure was repeated 1000 times. At the same time, the type strain and reference strain were selected for comparison, and phylogenetic trees based on 16S rRNA and MLST were constructed, respectively.
2.5. Drug Resistance Evaluation
For all
Cronobacter isolates, drug susceptibility tests of 20 commonly used clinical antibiotics in 9 categories were carried out. The operation was performed according to the implementation standard of the American Clinical and Laboratory Standards Institute (CLSI) M100 antimicrobial susceptibility test, and the susceptibility results were interpreted according to its specifications. The names and doses of antibiotics and the judgment results were shown in
Table 1.
2.6. Transcriptome Sequencing of Representative Drug-Resistant Bacteria
The representative resistant strain was selected for transcriptome sequencing. The strain without antibiotic treatment was activated and cultured under normal conditions. The treatment conditions of the strain requiring antibiotic treatment were as follows: the frozen bacterial solution was removed from −20 °C and added to TSB liquid medium with 2% inoculum for overnight culture, and then reactivated in TSB liquid medium with 4 μg/mL erythromycin for 6 h. The strains before and after treatment were analyzed by transcriptome sequencing. RNA was extracted and purified from the samples and tested for concentration and purity (OD260/280 and OD260/230) along with RNA integrity number (RIN) for quality control (
Table S4). RNA of acceptable quality was subjected to subsequent cDNA synthesis and library construction and then the libraries were double-end sequenced. The raw upland data are filtered to obtain high quality sequences and the filtered sequences are tested for quality using Q20 and Q30 values (
Table S5). The high-quality filtered sequences are only then available for comparison with the reference genome of the species. The expression of each gene was calculated according to the comparison results. On this basis, the expression difference analysis, enrichment analysis, and cluster analysis were carried out.
2.7. Verification of Drug Resistance-Related Genes
According to the screening results of transcriptomics tolerance-related genes, 10 genes with a high expression ratio and high correlation with drug resistance were selected for qRT-PCR verification. According to the results of tolerance evaluation, two strains with strong and weak drug resistance were selected for quantitative real-time PCR detection of related genes before and after treatment to investigate the expression differences of each gene in different strains. RNA from strains were extracted with a simple P total RNA extraction kit (Bioer Technology Co., Ltd., Hangzhou, China). NanoDrop spectrophotometer (Thermo, Wilmington, DE, USA) was used to measure the purity and integrity of RNA samples. A reverse transcription kit (Takara Bio, Shiga, Japan) was used for reverse transcription. Additionally, RT-qPCR was performed by SYBR Premix Ex Taq (Takara Bio, Shiga, Japan) according to the manufacturer’s instructions; the reaction was conducted with a QuantStudio™ 3 system. The value of 2
−∆∆Ct was calculated to analyze and compare the changes in the expression levels of each gene before and after treatment to verify the accuracy of the omics analysis results. The target genes were matched according to the corresponding sequences in
C. sakazakii ATCC 29544
T genome in the NCBI database, and real-time quantitative primer design was performed in the software Primer Premier 5.0. The gene names and primer sequences were shown in
Table S3.
3. Results
3.1. 16S rRNA Molecular Identification Results
The 16S rRNA sequencing results of 35 isolates were compared and analyzed in the GenBank database, and the results showed that they all belonged to the genus
Cronobacter spp., and the similarity between the sequences exceeded 99%, including
C. sakazakii (
n = 27, 77.14%), C. malonaticus (
n = 5, 14.29%),
C. turicensis (
n = 2, 5.71%), and
C. dublinensis (
n = 1, 2.86%). On this basis,
C. sakazakii ATCC 29544T,
C. sakazakii ATCC 12868,
C. malonaticus LMG 23826T,
C. turicensis DSM 18703T, and
C. dublinensis DSM 18705T were used as reference strains, and the phylogenetic tree was constructed according to the 16S rRNA sequencing results, as shown in
Figure 1. It can be seen that the strains of the four species of Cronobacter were divided into two large clusters, in which
C. sakazakii and
C. malonaticus were divided into one cluster, and
C. turicensis and
C. dublinensis were divided into another cluster. The strains of the four species showed clear phylogenetic relationships, on the whole, indicating that 16S rRNA could be used for molecular identification and typing of
Cronobacter spp.
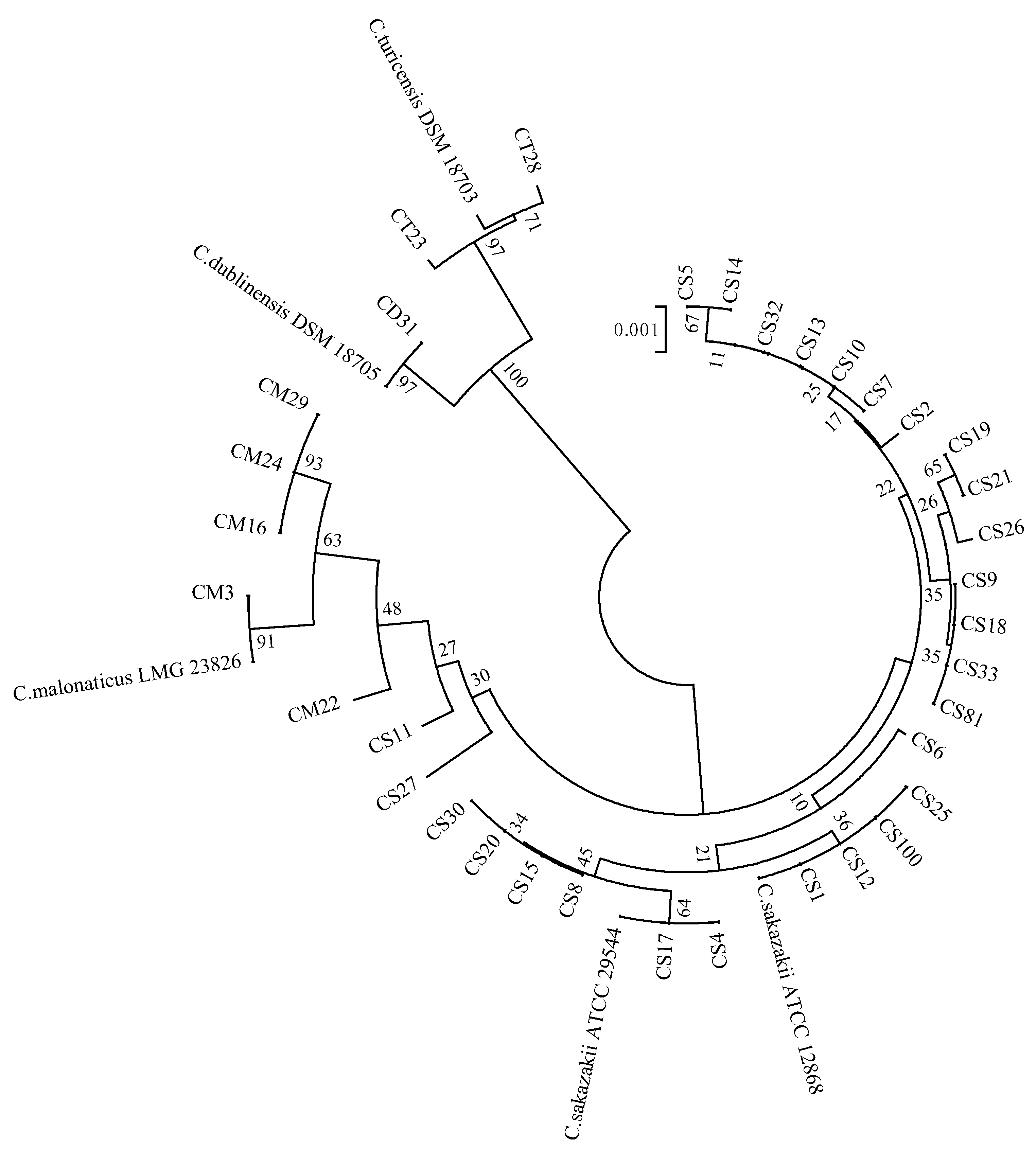
3.2. MLST Analysis of Cronobacter Strains
A total of 35 STs were obtained by MLST typing of
Cronobacter spp., mainly distributed in 15 homologous complexes. Due to the different sample collection areas and sources, the sequence types were relatively scattered, and some strains could not find the homologous complex matching them in the database, which was the unique type. In addition, three strains of Cronobacter had not been recorded in the database, which were new sequence types discovered for the first time in the world. By uploading to the Cronobacter MLST database (
http://www.pubmlst.org/cronobacter accessed on 22 March 2022), the corresponding sequence numbers were obtained, which were one strain of
C. dublinensis (CD31, ST788) and two strains of
C. sakazakii (CS32, ST789, and CS33, ST790). The allele numbers of each housekeeping gene and sequence types of all isolates were shown in
Table 2.
A phylogenetic tree was constructed based on the nucleic acid sequences of 35 sequence types of 3036 bp and 10 strains of Cronobacter were used as a reference to evaluate the genetic relationship among different sequence types of strains. The results were shown in
Figure 2. All strains were divided into five large clusters and seven species kept a certain distance from each other, among which
C. sakazakii and
C. malonaticus showed a closer relationship. Within
C. sakazakii species, ST4 and ST268, ST13 and ST789, ST64 and ST261, ST73, ST269 and ST790 had closer phylogenetic relationships. Among C. malonaticus species, ST7 and ST201 were closely related in phylogeny. According to the results of the MLST database, there was only one base difference between these similar sequence types. Therefore, the strains corresponding to each sequence type were likely to have similar phylogenetic relationships at the whole genome level.
3.3. Drug Resistance Analysis
The drug susceptibility results of 35 Cronobacter strains were shown in
Table 3. All Cronobacter isolates had the highest resistance rate to erythromycin (100%), followed by sulfamethoxazole/trimethoprim (45.71%). The others were neomycin (37.14%), cefazolin (28.57%), kanamycin (28.57%), ceftriaxone (25.71%), amikacin (25.71%), ampicillin (20%), ceftazidime (20%), cefuroxime (14.29%), polymyxin B (14.29%), gentamicin (11.43%), norfloxacin (11.43%), piperacillin (8.57%), cefoperazone (8.57%), tetracycline (8.57%), doxycycline (8.57%), ofloxacin (2.86%), and chloramphenicol (2.86%). All strains were sensitive to ciprofloxacin.
The drug resistance profile of Cronobacter strains was shown in
Table 4. There were six strains with one drug resistance spectrum (erythromycin), accounting for 17.14%. The double drug resistance spectrum showed three spectrum types and a total of five strains, accounting for 14.29%. There were six types of triple-drug resistance spectrum and seven strains in total, accounting for 20%. The quadruple drug resistance spectrum includes four spectrum types and a total of four strains, accounting for 11.43%. The five-fold drug resistance spectrum showed three spectrum types and a total of three strains, accounting for 8.57%. The six-fold drug resistance spectrum showed two spectrum types and a total of two strains, accounting for 5.71%. The seven-fold drug resistance spectrum showed six spectrum types and a total of six strains, accounting for 17.14%. There was one strain in each of the eleven and thirteen-fold drug resistance spectrums, accounting for 2.86%. According to the results, 24 strains were multidrug resistant (resistant to three or more classes of antibiotics), accounting for 68.57% of the total number of Cronobacter isolates. At the same time,
C. sakazakii ATCC 29544 was only resistant to erythromycin and sulfamethoxazole/trimethoprim, which was a double drug resistance. In conclusion, compared with the reference strains, most Cronobacter isolates showed strong drug resistance and multi-drug resistance, among which the representative drug-resistant strains were CS14 (ST42) and CS17 (ST64).
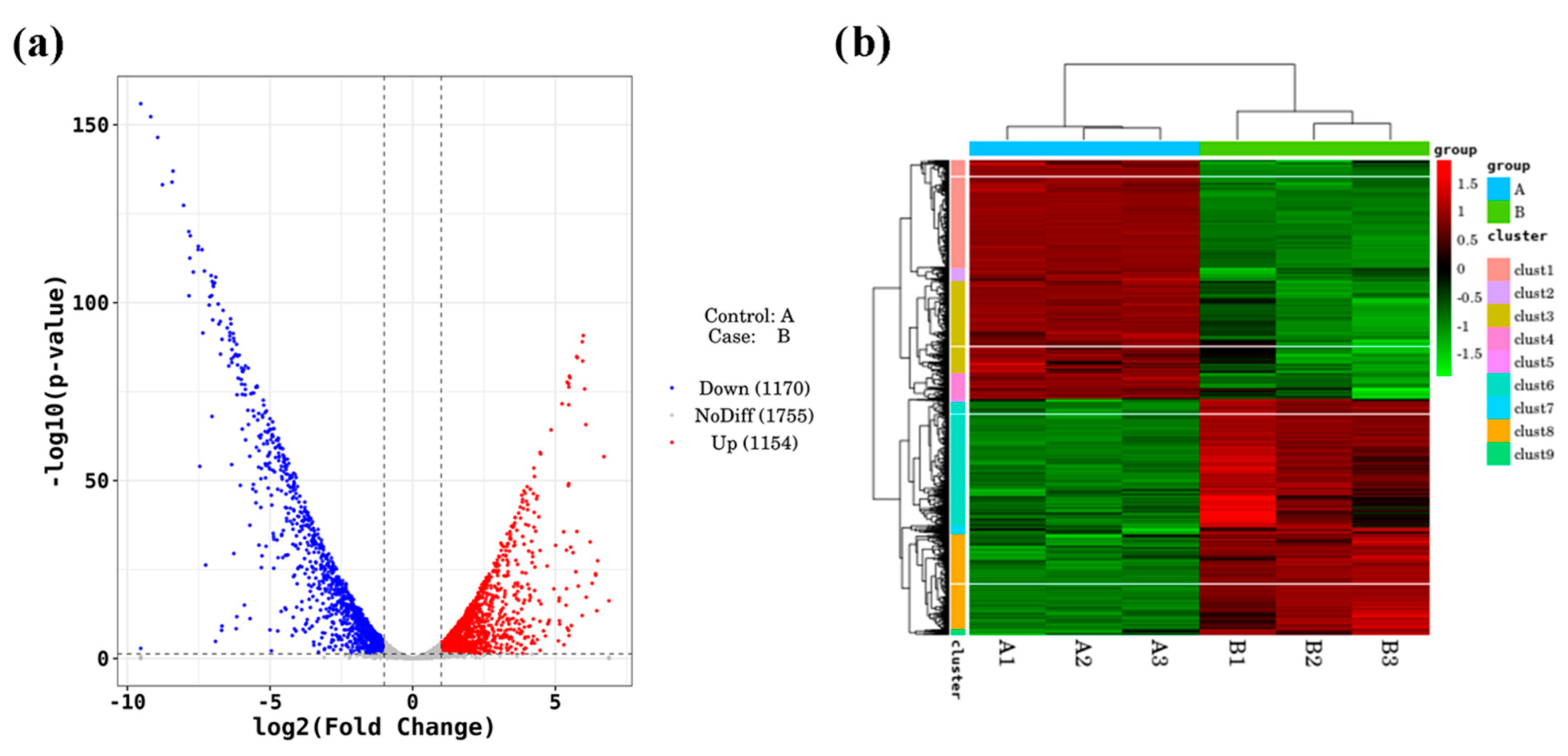
3.4. Transcriptome Analysis of Strains under Antibiotic Conditions
Based on the results of the drug resistance test, CS14, which has a high ability to survive under antibiotic conditions, was selected as the representative strain. The normal culture group (A) and the antibiotic treatment group (B) were compared with each other, and the overall situation of the screened differentially expressed genes is shown in
Figure 3.
Figure 3a as a volcano map of differentially expressed genes. Translated with
www.DeepL.com/Translator (free version, accessed on 3 April 2022), a total of 2324 differentially expressed genes were screened, among which 1154 genes were upregulated and 1170 genes were downregulated. The cluster heat map of differentially expressed genes was obtained by two-way cluster analysis on the union of screened differentially expressed genes and samples. As can be seen in
Figure 3b, the gene expression between the three duplicate samples of the control group and the treatment group was consistent, and the gene expression between the comparison groups was significantly different.
KEGG enrichment analysis was performed on differentially expressed genes and the top 30 pathways with the most significant enrichment were selected for demonstration. The results were shown in
Figure 4. Signaling pathways can be divided into three categories: environmental information processing, genetic information processing, and metabolism. The metabolic category contained 26 signaling pathways and 381 differentially expressed genes, which mainly focused on carbohydrate synthesis and metabolism, energy metabolism, amino acid metabolism, terpenoids and polyketides metabolism, lipid metabolism, degradation of heterogeneous organisms, and metabolic processes. The genetic information processing category included three signaling pathways and 75 differentially expressed genes, mainly focusing on ribosomal translation and RNA transcription signaling pathways. The environmental information processing category included one signaling pathway and 115 differentially expressed genes, focusing on membrane transport processes.
According to the results of RNA-seq analysis under antibiotic treatment, a total of 94 genes related to drug resistance were screened, among which 77 genes were differentially expressed, 35 genes were upregulated, and 42 genes were downregulated. Some differentially expressed genes were screened in
Table 5, listed in order of
p value from smallest to largest.
3.5. qRT-PCR Validation of Drug Resistance-Related Genes
Ten differentially expressed genes related to drug resistance were screened and qRT-PCR was performed on CS14 and CS17 strains with multiple drug resistance, and CS6 and CS9 strains with only one double drug resistance. The results were shown in
Table 6. The relative expression of CS14 genes was consistent with the transcriptome analysis results. Eight genes were upregulated and significantly upregulated (2
−ΔΔCt ≥ 2,
p < 0.05), and two genes were downregulated and significantly downregulated (2
−ΔΔCt ≤ 0.5,
p < 0.05). The relative gene expression of CS17 was consistent with that of CS14, among which six genes were significantly upregulated and two genes were significantly downregulated. For CS6 with weak drug resistance, eight out of ten genes were downregulated, among which four genes were significantly downregulated, and no genes were significantly upregulated. For another strain CS9 with weak drug resistance, seven out of ten genes were downregulated, including two significantly downregulated genes, and no genes that were significantly upregulated. In general, the results of qRT-PCR were consistent with the transcriptome, with higher upregulation or downregulation levels in the two resistant strains compared with the two less resistant strains.
3.6. Analysis of Drug Resistance Mechanism
When the strain was challenged with antibiotics in the bacterial chemotaxis pathway (ko02030), the genes CSK29544_01507 and CSK29544_02725 encoding methyl receptor chemotactic proteins were significantly upregulated by 2.24 and 2.66-fold, respectively. The gene CSK29544_02809 encoding the methyl receptor chemotactic citrate sensor was significantly upregulated by 2.36-fold; the gene CSK29544_00467 encoding the methyl receptor chemosensory sensor was significantly upregulated by 2.06 times; and the genes fliG and fliM encoding the flagellar motor switch protein were significantly upregulated by 1.76 and 1.83 times, respectively. At the same time, the genes CSK29544_01807 and CSK29544_02659 encoding methyl receptor chemosensory sensors were significantly downregulated by 1.57 and 2.21 times, respectively. The gene cheR encoding chemotactic protein methyltransferase was significantly downregulated by 1.79 times. The gene cheB encoding the chemotactic family, protein-glutamate methyl esterase/glutaminase, was significantly down-regulated by 2.51 times. The gene encoding purine-binding chemotactic protein cheW was significantly downregulated by 2.20 times; and the gene encoding chemotactic protein cheZ was significantly downregulated by 2.22 times. It can be seen that Cronobacter can enhance the drug resistance of bacteria as a whole by regulating the partial upregulation or downregulation of chemotaxis-related genes.
In the ABC transporter pathway (KO02010), 29 genes were significantly upregulated, and 86 genes were significantly downregulated. The gene mdlA encoding the ATP-binding protein of the multidrug efflux system was significantly upregulated by 2.05 times, and the gene yojI encoding the ATP-binding/permease protein of the multi-drug transport system was significantly upregulated by 1.96 times. The four coding genes potA, potB, potC, and potD involved in spermidine transport were significantly upregulated by 2.46, 3.10, 2.5,9 and 2.06 times, respectively. The gene proX encoding the glycine betaine/proline transport system substrate binding protein was significantly upregulated by 2.76 times; the gene proW encoding the glycine betaine/proline transport system permease protein was significantly upregulated by 2.60 times; and encoding the glycine betaine/proline gene proV of the acid transport system ATP-binding protein was significantly upregulated 4.23-fold. The genes togM and togN encoding the oligogalacturonic acid transport system permease proteins were significantly upregulated by 2.11 and 2.37 times, respectively, and the gene togB encoding the low-galacturonic acid transport system substrate-binding protein was significantly downregulated by 2.39 times. The three encoding genes mlaD, mlaE, and mlaF involved in phospholipid transport were significantly upregulated by 1.88, 2.63, and 1.88 times, respectively. The genes encoding glutamine transport glnP and glnQ were significantly upregulated; the genes involved in zinc ion transport znuB and znuC were significantly upregulated; the genes involved in lipoprotein transport lolC, lolD, and lolE were significantly upregulated; and the encoding genes lptF involved in lipopolysaccharide transport were significantly upregulated and lptG were significantly upregulated.
In the two-component signaling pathway (ko02020), 25 genes were significantly upregulated and 46 genes were significantly downregulated. The gene baeR encoding the ompR family and response regulator was significantly downregulated by 2.20 times. The gene mdtA encoding the multidrug efflux system membrane fusion protein was significantly downregulated by 1.50 times. The gene mdtD encoding MFS transporter, DHA2 family, multidrug resistance protein was significantly downregulated by 2.26 times. The gene acrD encoding the MDR efflux system was significantly downregulated by 2.01-fold. In summary, under the stimulation of antibiotics, Cronobacter spp. secretes more drug efflux proteins, enhances drug resistance by activating the multidrug efflux system, and reduces the transport activity of nucleic acids, lipids, amino acids, and other molecular substances. The damaging effect of drugs on cells.
4. Discussion
In this study, 35 STs were obtained by MSLT on all isolated
Cronobacter spp. strains, which were distributed in raw materials, semi-finished products, finished products, and the processing environment. The main sequence types were ST1 (14.42%), ST4 (18.27%), and ST64 (11.54%). This study found that ST4 was present in the finished product, and many studies have reported that ST4 was associated with infantile meningitis [
35]. ST1 has also been isolated from clinically infected infants [
36]. ST64 is one of the main contamination sequence types of PIF and its processing environment in China, enough to show its drug resistance and difficulty to disinfect [
16]. Therefore, it is very important to study different sequence types of
Cronobacter spp. to reduce the contamination of
Cronobacter spp. in PIF. In addition, three of the isolates were not recorded in the database and were new sequence types found for the first time in the world. Phylogenetic trees were constructed based on the MLST results and one representative strain for each ST was selected for follow-up studies.
The isolates showed 100% resistance to erythromycin, followed by 45.71% (16/35) to sulfamethoxazole/trimethoprim, indicating that Cronobacter spp. used in this study had a good tolerance rate to macrolides and sulfonamides antibiotics. Meanwhile, all tested isolates were sensitive to ciprofloxacin. We found that the strains were only 11.43% and 2.86% resistant to norfloxacin and ofloxacin (both belong to fluoroquinolones), so the contamination of Cronobacter spp. could be treated with fluoroquinolones. This study also found that strains with only one drug resistance spectrum (erythromycin) accounted for 17.14% (6/35), and multidrug-resistant strains accounted for 68.57% (24/35), indicating that the overall drug resistance of Cronobacter spp. was strong. Among them, CS14 (ST42) and CS17 (ST64) were the most representative drug-resistant strains, which were 13 and 11 multiple drug resistance, respectively.
In recent years, many reports have found that a variety of antibiotics, such as gentamicin, ampicillin, kanamycin, and ciprofloxacin, can kill
Cronobacter spp. [
37]. Lai et al. found that
Cronobacter spp. had consistent resistance to ampicillin, cefazolin, and extended-spectrum penicillin [
38]. Kim et al. found that
Cronobacter spp. isolated from food was resistant to ceftaroline and ampicillin [
39]. It was further confirmed that even though antibiotics can be effective in eliminating the contamination caused by this bacteria, long-term use can cause multi-drug resistance in
Cronobacter spp. Pakbin et al. showed that
C. sakazakii isolates were completely resistant to ampicillin and amoxicillin and showed multidrug resistance, and moderate resistance to ciprofloxacin and tetracycline antibiotics [
40]. Odeyemi et al. found that
Cronobacter spp. was resistant to erythromycin and sulfamethoxazole, and all bacteria were able to form biofilms [
41]. This finding is highly consistent with the findings of this experiment. In conclusion, multi-drug resistance of
Cronobacter spp. is increasing year by year, and screening for effective antibiotics is one of the urgent research priorities.
In this study, a total of 77 differentially expressed genes related to drug resistance were screened by transcriptome analysis of CS14 under antibiotic conditions, and some highly correlated genes included
emrA, CSK29544_03309,
yojI,
mdfA,
fliM,
mdlA,
emrB,
pump, CSK29544_03853, and
baeR. We found that the regulatory genes related to methyl-accepting chemotaxis were significantly upregulated by about two-fold, and the regulatory genes fliG and fliM related to flagellar motor switch protein were significantly upregulated by 1.76 and 1.83-fold, respectively. In addition, the regulatory genes
mdlA and
yojI related to the multidrug efflux system were also significantly upregulated by approximately two-fold, as were some genes involved in amino acid, sugar, and lipid transport. In summary, under the stimulation of antibiotic conditions,
Cronobacter spp. can activate the multidrug efflux system by regulating the expression of chemotaxis-related genes, thereby, secreting more drug efflux proteins to enhance drug resistance. The results of the study also revealed that
Cronobacter spp. can reduce cellular damage from drugs by reducing the transport activity of nucleic acids, lipids, amino acid, and other molecular substances. Some researchers found the multi-drug resistance operon
mar in the genome of
Cronobacter spp. Bao et al. also found that the polymyxin resistance gene
pmrA was related to biofilm formation in
Cronobacter spp., but, overall, the antibiotic resistance level of this genus was lower than that of other foodborne pathogens [
42]. However, the results of this study showed that the
Cronobacter strains had a strong drug resistance level and a multi-drug resistance spectrum, which may be due to the stronger resistance of the
Cronobacter spp. from PIF and its processing environment.





{kind=link}
{kind=link}
{kind=link}
{kind=link}