
A 3D-Printed Integrated Handheld Biosensor for the Detection of Vibrio parahaemolyticus

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. RPA-CRISPR/Cas12a Procedure
2.3. Preparation of AuNPs and ssDNA-Modified AuNPs
2.4. Lateral Flow Strip Procedure
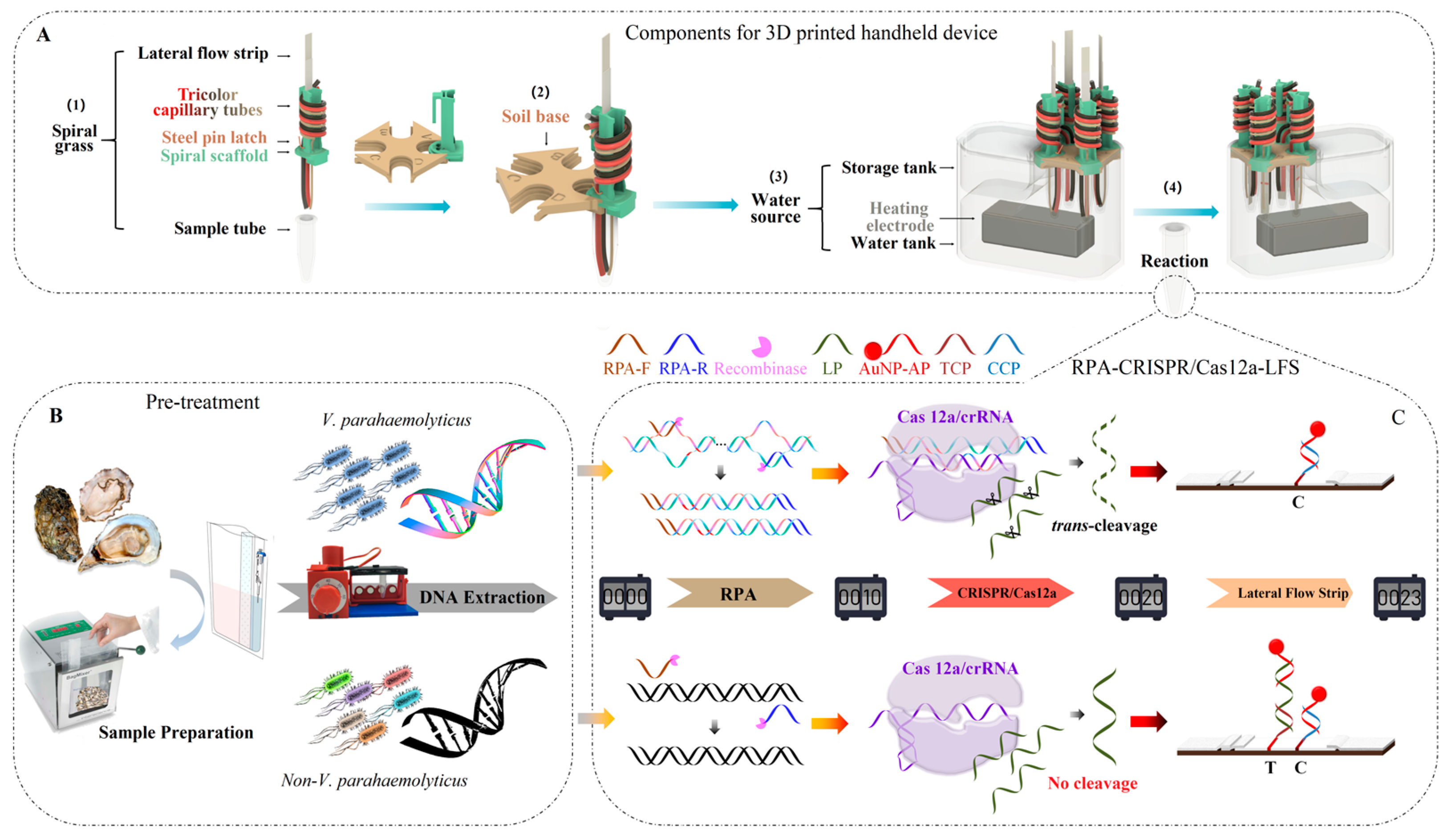
2.5. Design of the 3D-Printed Handheld Device
2.6. Application of the Integrated Biosensor in V. parahaemolyticus-Spiked Oyster Samples
3. Results and Discussion
3.1. Mechanism of the 3D-Printed Integrated Handheld Biosensor (IHB)
3.2. Development of RPA-CRISPR/Cas12a
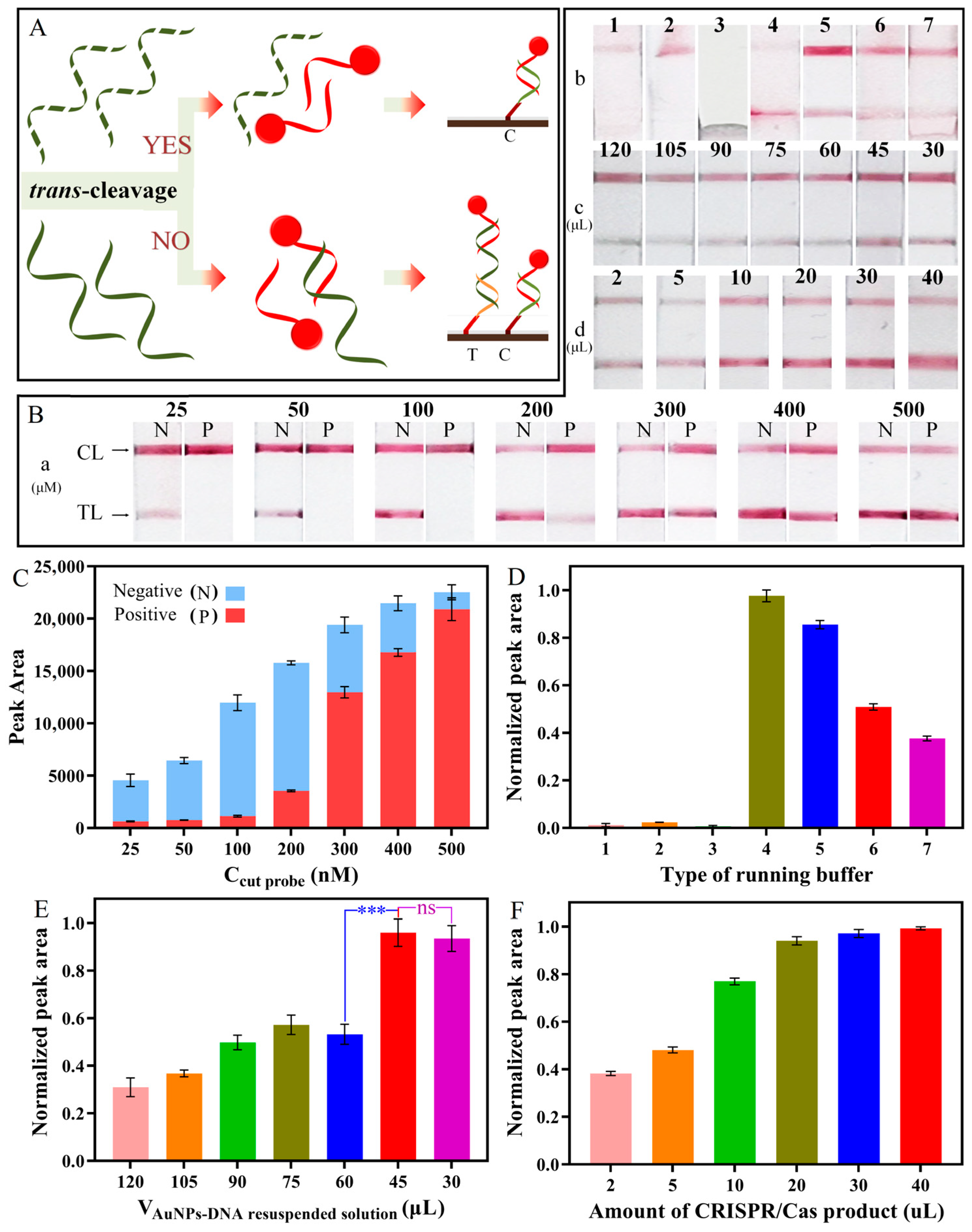
3.3. Optimization of RPA-CRISPR/Cas12a
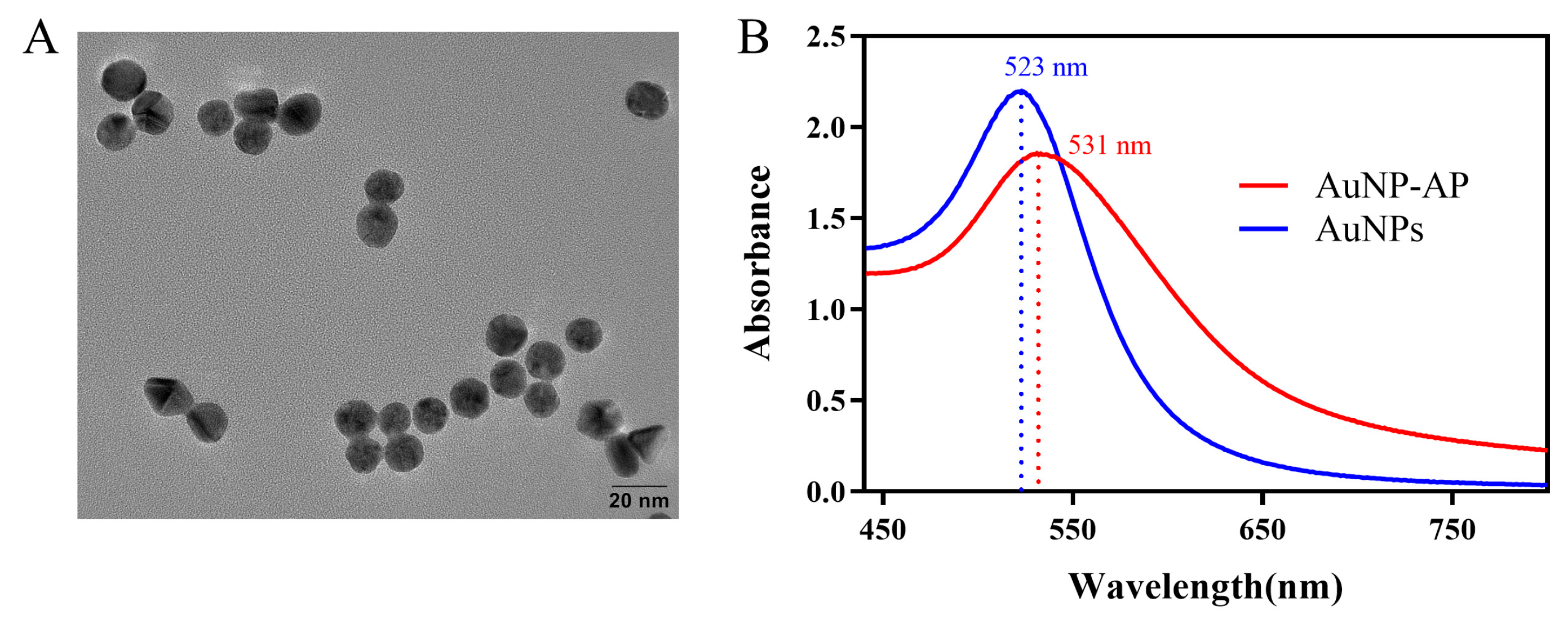
3.4. Characterization of the AuNPs and AuNP-AP
3.5. Optimization of the Lateral Flow Strip
3.6. Sensitivity of the IHB for V. parahaemolyticus Detection
3.7. Specificity of the IHB for V. parahaemolyticus Detection
3.8. Real Sample Analysis by the IHB
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Anderson, K.M.; Poosala, P.; Lindley, S.R.; Anderson, D.M. Managing the risk of Vibrio parahaemolyticus infections associated with oyster consumption: A review. Compr. Rev. Food Sci. Food Saf. 2019, 19, 1187–1217. [Google Scholar]
- Brumfield Kyle, D.; Chen Arlene, J.; Gangwar, M.; Usmani, M.; Hasan Nur, A.; Jutla Antarpreet, S.; Huq, A.; Colwell Rita, R. Environmental Factors Influencing Occurrence of Vibrio parahaemolyticus and Vibrio vulnificus. Appl. Environ. Microb. 2023, 89, e00307–e00323. [Google Scholar] [CrossRef] [PubMed]
- Noman, E.; Al-Gheethi, A.; Radin Mohamed, R.M.S.; Talip, B.; Al-Sahari, M.; Al-Shaibani, M. Quantitative microbiological risk assessment of complex microbial community in Prawn farm wastewater and applicability of nanoparticles and probiotics for eliminating of antibiotic-resistant bacteria. J. Hazard. Mater. 2021, 419, 126418. [Google Scholar] [CrossRef] [PubMed]
- Letchumanan, V.; Loo, K.-Y.; Law, J.W.-F.; Wong, S.H.; Goh, B.-H.; Ab Mutalib, N.-S.; Lee, L.-H. Vibrio parahaemolyticus: The protagonist of foodborne diseases. Prog. Microbes Mol. Biol. 2019, 2, 705. [Google Scholar] [CrossRef]
- Wang, D.; Flint, S.H.; Palmer, J.S.; Gagic, D.; Fletcher, G.C.; On, S.L.W. Global expansion of Vibrio parahaemolyticus threatens the seafood industry: Perspective on controlling its biofilm formation. LWT 2022, 158, 113182. [Google Scholar] [CrossRef]
- Bonnin-Jusserand, M.; Copin, S.; Le Bris, C.; Brauge, T.; Gay, M.; Brisabois, A.; Grard, T.; Midelet-Bourdin, G. Vibrio species involved in seafood-borne outbreaks (Vibrio cholerae, V. parahaemolyticus and V. vulnificus): Review of microbiological versus recent molecular detection methods in seafood products. Crit. Rev. Food Sci. 2019, 59, 597–610. [Google Scholar] [CrossRef]
- Xu, J.; Turner, J.W.; Idso, M.; Biryukov, S.V.; Rognstad, L.; Gong, H.; Trainer, V.L.; Wells, M.L.; Strom, M.S.; Yu, Q. In situ strain-level detection and identification of Vibrio parahaemolyticus using surface-enhanced Raman spectroscopy. Anal. Chem. 2013, 85, 2630–2637. [Google Scholar]
- Zhai, Y.; Meng, X.; Li, L.; Liu, Y.; Xu, K.; Zhao, C.; Wang, J.; Song, X.; Li, J.; Jin, M. Rapid detection of Vibrio parahaemolyticus using magnetic nanobead-based immunoseparation and quantum dot-based immunofluorescence. RSC Adv. 2021, 11, 38638–38647. [Google Scholar] [CrossRef]
- Anupama, K.P.; Chakraborty, A.; Karunasagar, I.; Karunasagar, I.; Maiti, B. Loop-mediated isothermal amplification assay as a point-of-care diagnostic tool for Vibrio parahaemolyticus: Recent developments and improvements. Expert Rev. Mol. Diagn. 2019, 19, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.Y.; Lee, K.A.; Lee, M.J.; Sekhon, S.S.; Rhee, S.K.; Cho, S.J.; Ko, J.H.; Lee, L.; Han, J.; Kim, S.Y.; et al. Surface plasmon resonance aptamer biosensor for discriminating pathogenic bacteria Vibrio parahaemolyticus. J. Nanosci. Nanotechnol. 2018, 18, 1599–1605. [Google Scholar] [CrossRef]
- Wang, K.; Du, L.; Zhang, L.; Luo, X.; Yang, T.; Zeng, H. On-site marine pathogen (Vibrio parahaemolyticus) rapid colorimetric determination based on modified-free aptamer and metal-organic frameworks with simple washing step. Sens. Actuators B-Chem. 2022, 372, 132695. [Google Scholar] [CrossRef]
- Zaman, M.A.; Wu, M.; Ren, W.; Jensen, M.A.; Davis, R.W.; Hesselink, L. Spectral tweezers: Single sample spectroscopy using optoelectronic tweezers. Appl. Phys. Lett. 2024, 124, 071104. [Google Scholar] [CrossRef]
- Zhang, J.; Huang, H.; Song, G.; Huang, K.; Luo, Y.; Liu, Q.; He, X.; Cheng, N. Intelligent biosensing strategies for rapid detection in food safety: A review. Biosens. Bioelectron. 2022, 202, 114003. [Google Scholar] [CrossRef] [PubMed]
- Deveau, H.; Garneau, J.E.; Moineau, S. CRISPR/Cas system and its role in phage-bacteria interactions. Annu. Rev. Microbiol. 2010, 64, 475–493. [Google Scholar] [CrossRef] [PubMed]
- Hirakawa, M.P.; Krishnakumar, R.; Timlin, J.A.; Carney, J.P.; Butler, K.S. Gene editing and CRISPR in the clinic: Current and future perspectives. Biosci. Rep. 2020, 40, BSR20200127. [Google Scholar] [CrossRef] [PubMed]
- Weng, Z.; You, Z.; Yang, J.; Mohammad, N.; Lin, M.; Wei, Q.; Gao, X.; Zhang, Y. CRISPR-Cas Biochemistry and CRISPR-Based Molecular Diagnostics. Angew. Chem. Int. Ed. 2023, 62, e202214987. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Guo, Z.; Zhong, K.; Osire, T.; Sun, Y.; Jiang, L. State of the art in CRISPR/Cas system-based signal conversion and amplification applied in the field of food analysis. Trends Food Sci. Technol. 2023, 135, 174–189. [Google Scholar] [CrossRef]
- Swarts, D.C.; Jinek, M. Mechanistic Insights into the cis-and trans-Acting DNase Activities of Cas12a. Mol. Cell 2019, 73, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Chen, F.; Li, Q.; Wang, L.; Fan, C. Isothermal amplification of nucleic acids. Chem. Rev. 2015, 115, 12491–12545. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Yang, H.; Cao, J.; Zhang, J.; He, Q.; Deng, R. Isothermal nucleic acid amplification for food safety analysis. TrAC, Trends Anal. Chem. 2022, 153, 116641. [Google Scholar] [CrossRef]
- Chen, X.; Wang, L.; He, F.; Chen, G.; Bai, L.; He, K.; Zhang, F.; Xu, X. Label-Free Colorimetric Method for Detection of Vibrio parahaemolyticus by Trimming the G-Quadruplex DNAzyme with CRISPR/Cas12a. Anal. Chem. 2021, 93, 14300–14306. [Google Scholar] [CrossRef]
- Pan, R.; Liu, J.; Wang, P.; Wu, D.; Chen, J.; Wu, Y.; Li, G. Ultrasensitive CRISPR/Cas12a-Driven SERS Biosensor for On-Site Nucleic Acid Detection and Its Application to Milk Authenticity Testing. J. Agric. Food. Chem. 2022, 70, 4484–4491. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Chen, Y.; Yang, Q.; Peng, C.; Wang, X.; Zhang, M.; Qian, S.; Xu, J.; Wu, J. A reversible valve-assisted chip coupling with integrated sample treatment and CRISPR/Cas12a for visual detection of Vibrio parahaemolyticus. Biosens. Bioelectron. 2021, 188, 113352. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Ye, Q.; Chen, M.; Zhou, B.; Zhang, J.; Pang, R.; Xue, L.; Wang, J.; Zeng, H.; Wu, S.; et al. An ultrasensitive CRISPR/Cas12a based electrochemical biosensor for Listeria monocytogenes detection. Biosens. Bioelectron. 2021, 179, 113073. [Google Scholar] [CrossRef] [PubMed]
- Gootenberg, J.S.; Abudayyeh, O.O.; Lee, J.W.; Essletzbichler, P.; Dy, A.J.; Joung, J.; Verdine, V.; Donghia, N.; Daringer, N.M.; Freije, C.A. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 2017, 356, 438–442. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.S.; Ma, E.; Harrington, L.B.; Da Costa, M.; Tian, X.; Palefsky, J.M.; Doudna, J.A. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 2018, 360, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, S.; Wu, N.; Wu, J.; Wang, G.; Zhao, G.; Wang, J. HOLMESv2: A CRISPR-Cas12b-assisted platform for nucleic acid detection and DNA methylation quantitation. ACS Synth. Biol. 2019, 8, 2228–2237. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wei, Y.; Cheng, N.; Huang, K.; Wang, W.; Zhang, L.; Xu, W.; Luo, Y. Nucleic acid biosensor synthesis of an all-in-one universal blocking linker recombinase polymerase amplification with a peptide nucleic acid-based lateral flow device for ultrasensitive detection of food pathogens. Anal. Chem. 2018, 90, 708–715. [Google Scholar] [CrossRef]
- Nnachi, R.C.; Sui, N.; Ke, B.; Luo, Z.; Bhalla, N.; He, D.; Yang, Z. Biosensors for rapid detection of bacterial pathogens in water, food and environment. Environ. Int. 2022, 166, 107357. [Google Scholar] [CrossRef] [PubMed]
- Ying, N.; Wang, Y.; Song, X.; Yang, L.; Qin, B.; Wu, Y.; Fang, W. Lateral flow colorimetric biosensor for detection of Vibrio parahaemolyticus based on hybridization chain reaction and aptamer. Microchim. Acta. 2021, 188, 381. [Google Scholar] [CrossRef]
- Zhuang, J.; Zhao, Z.; Lian, K.; Yin, L.; Wang, J.; Man, S.; Liu, G.; Ma, L. SERS-based CRISPR/Cas assay on microfluidic paper analytical devices for supersensitive detection of pathogenic bacteria in foods. Biosens. Bioelectron. 2022, 207, 114167. [Google Scholar] [CrossRef]
- Ren, Y.; Cao, L.; Zhang, X.; Jiao, R.; Ou, D.; Wang, Y.; Zhang, D.; Shen, Y.; Ling, N.; Ye, Y. A novel fluorescence resonance energy transfer (FRET)-based paper sensor with smartphone for quantitative detection of Vibrio parahaemolyticus. Food Control 2023, 145, 109412. [Google Scholar] [CrossRef]
- Weckman, N.E.; Ermann, N.; Gutierrez, R.; Chen, K.; Graham, J.; Tivony, R.; Heron, A.; Keyser, U.F. Multiplexed DNA Identification Using Site Specific dCas9 Barcodes and Nanopore Sensing. ACS Sens. 2019, 4, 2065–2072. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; He, Y.; Lan, T.; Zhang, J. Installing CRISPR–Cas12a sensors in a portable glucose meter for point-of-care detection of analytes. Analyst 2021, 146, 3114–3120. [Google Scholar] [CrossRef]
- Wu, H.; Qian, C.; Wu, C.; Wang, Z.; Wang, D.; Ye, Z.; Ping, J.; Wu, J.; Ji, F. End-point dual specific detection of nucleic acids using CRISPR/Cas12a based portable biosensor. Biosens. Bioelectron. 2020, 157, 112153. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wang, W.; Yang, Z.; Wang, X.; Xu, W.; Huang, K.; Luo, Y.; He, X.; Cheng, N. A portable 3D-printed biosensing device for rapid detection of genetically modified maize MON810. Sens. Actuators B-Chem. 2021, 349, 130748. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, M.; Wang, X.; Majid, Z.; Huang, K.; Xu, W.; Luo, Y.; Cheng, N. A portable 3D-printed pretreatment device combined with graded lateral flow assay for detection of S. aureus. Sens. Actuators B-Chem. 2023, 383, 133601. [Google Scholar] [CrossRef]
- Liu, B.; Liu, J. Freezing directed construction of bio/nano interfaces: Reagentless conjugation, denser spherical nucleic acids, and better nanoflares. J. Am. Chem. Soc. 2017, 139, 9471–9474. [Google Scholar] [CrossRef] [PubMed]
- Zagorovsky, K.a.C.; Warren, C.W. A plasmonic DNAzyme strategy for point-of-care genetic detection of infectious pathogens. Angew. Chem. Int. Ed. 2013, 52, 3168–3171. [Google Scholar] [CrossRef] [PubMed]
- Maye, M.M.; Han, L.; Kariuki, N.N.; Ly, N.K.; Chan, W.-B.; Luo, J.; Zhong, C.-J. Gold and alloy nanoparticles in solution and thin film assembly: Spectrophotometric determination of molar absorptivity. Anal. Chim. Acta. 2003, 496, 17–27. [Google Scholar] [CrossRef]
- Kongrueng, J.; Tansila, N.; Mitraparp-arthorn, P.; Nishibuchi, M.; Vora, G.J.; Vuddhakul, V. LAMP assay to detect Vibrio parahaemolyticus causing acute hepatopancreatic necrosis disease in shrimp. Aquacult. Int. 2015, 23, 1179–1188. [Google Scholar] [CrossRef]
- Geng, Y.; Tan, K.; Liu, L.; Sun, X.X.; Zhao, B.; Wang, J. Development and evaluation of a rapid and sensitive RPA assay for specific detection of Vibrio parahaemolyticus in seafood. BMC Microbiol. 2019, 19, 186. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Mun, H.; Kim, S.R.; Kim, M.G.; Chang, J.Y.; Shim, W.B. A colorimetric Loop-mediated isothermal amplification (LAMP) assay based on HRP-mimicking molecular beacon for the rapid detection of Vibrio parahaemolyticus. Biosens. Bioelectron. 2020, 151, 111968. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Ren, Y.; Han, X.; Xue, J.; Shan, T.; Chen, Z.; Liu, Y.; Wang, Q. Recombinase polymerase amplification-lateral flow (RPA-LF) assay combined with immunomagnetic separation for rapid visual detection of Vibrio parahaemolyticus in raw oysters. Anal. Bioanal. Chem. 2020, 412, 2903–2914. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Liu, C.; Shi, Y.; Wu, J.; Wu, J.; Chen, H. Selective endpoint visualized detection of Vibrio parahaemolyticus with CRISPR/Cas12a assisted PCR using thermal cycler for on-site application. Talanta 2020, 214, 120818. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Spiked (CFU/mL) | Measured (CFU/mL) | Recovery of the Assay (%) | RSD (%) |
|---|---|---|---|---|
| Oyster sample 1 | 0 | ND * | ||
| 102 | 106.03 ± 2.05 | 106 | 1.93 | |
| Oyster sample 2 | 0 | ND * | ||
| 103 | 1027.12 ± 31.91 | 103 | 3.11 | |
| Oyster sample 3 | 0 | ND * | ||
| 104 | 9937.49 ± 432.14 | 99 | 4.35 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Y.; Zhang, Q.; Li, Y.; Pang, X.; Cheng, N. A 3D-Printed Integrated Handheld Biosensor for the Detection of Vibrio parahaemolyticus. Foods 2024, 13, 1775. https://doi.org/10.3390/foods13111775
Xu Y, Zhang Q, Li Y, Pang X, Cheng N. A 3D-Printed Integrated Handheld Biosensor for the Detection of Vibrio parahaemolyticus. Foods. 2024; 13(11):1775. https://doi.org/10.3390/foods13111775
Chicago/Turabian StyleXu, Yuancong, Qian Zhang, Yunyi Li, Xiaoxu Pang, and Nan Cheng. 2024. "A 3D-Printed Integrated Handheld Biosensor for the Detection of Vibrio parahaemolyticus" Foods 13, no. 11: 1775. https://doi.org/10.3390/foods13111775