
The Integrated Analysis of miRNome and Degradome Sequencing Reveals the Regulatory Mechanisms of Seed Development and Oil Biosynthesis in Pecan (Carya illinoinensis)

 , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Sample Collection
2.2. An Analysis of the Oil Content and Components and the Soluble Sugar Content
2.3. Small RNA Library Construction and Sequencing
2.4. Analysis of Small RNA Sequences
2.5. RNA Isolation and Transcriptome Sequencing
2.6. Differentially Expressed miRNAs and Target Prediction
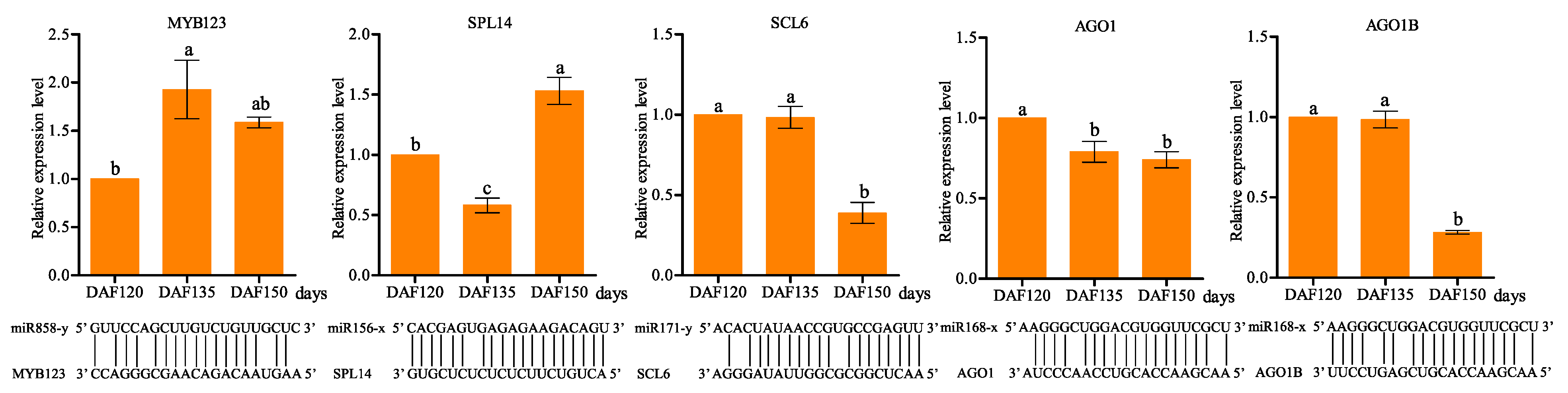
2.7. Validation of the Expression Patterns of miRNAs and Target Genes
2.8. Statistical Analysis
3. Results
3.1. Oil and Sugar Contents and Fatty Acid Composition Analyses
3.2. Sequence Analysis of Small RNAs
3.3. Identification of Known and Novel MicroRNAs in Developing Pecan Seeds
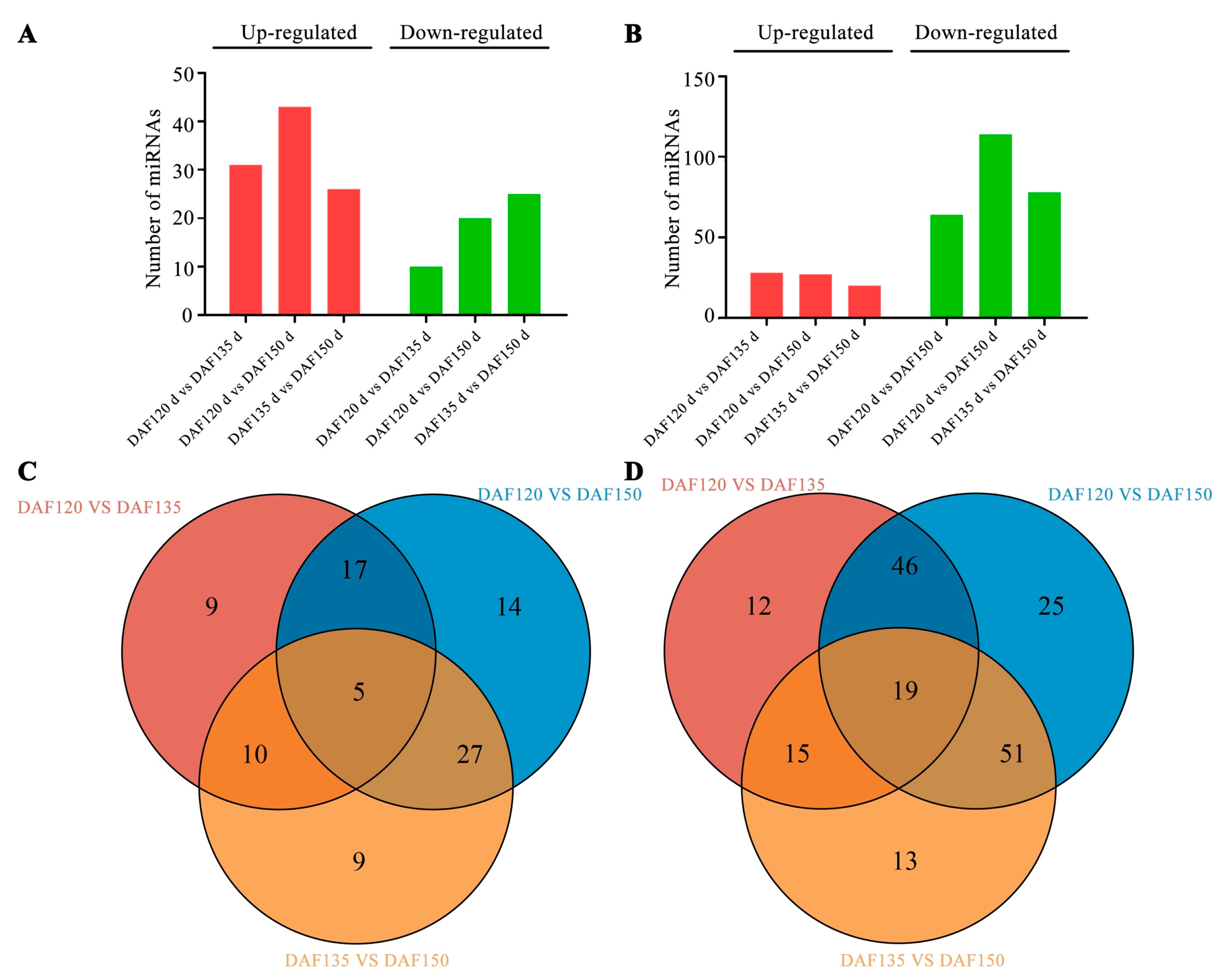
3.4. Differentially Expressed miRNAs during Pecan Seed Development
3.5. Expression Trends of DEMs during Seed Development and Oil Biosynthesis in Pecan
3.6. Identification and Characterization of miRNA Targets
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Grauke, L.J.; Wood, B.W.; Harris, M.K. Crop Vulnerability: Carya. Hortscience 2016, 51, 653–663. [Google Scholar] [CrossRef]
- Zhu, K.; Fan, P.; Liu, H.; Tan, P.; Ma, W.; Mo, Z.; Zhao, J.; Chu, G.; Peng, F. Insight into the CBL and CIPK Gene Families in Pecan (Carya illinoinensis): Identification, Evolution and Expression Patterns in Drought Response. BMC Plant Biol. 2022, 22, 221. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Ni, J.; Shah, F.A.; Wang, Q.; Wang, Z.; Wu, L.; Fu, S. Transcriptome Analysis of Pecan Seeds at Different Developing Stages and Identification of Key Genes Involved in Lipid Metabolism. PLoS ONE 2018, 13, e0195913. [Google Scholar] [CrossRef]
- Atanasov, A.G.; Sabharanjak, S.M.; Zengin, G.; Mollica, A.; Szostak, A.; Simirgiotis, M.; Huminiecki, L.; Horbanczuk, O.K.; Nabavi, S.M.; Mocan, A. Pecan Nuts: A Review of Reported Bioactivities and Health Effects. Trends Food Sci. Technol. 2018, 71, 246–257. [Google Scholar] [CrossRef]
- Zhang, C.; Yao, X.; Ren, H.; Wang, K.; Chang, J. Genome-Wide Identification and Characterization of the Phenylalanine Ammonia-Lyase Gene Family in Pecan (Carya illinoinensis). Sci. Hortic. 2022, 295, 110800. [Google Scholar] [CrossRef]
- Thelen, J.J.; Ohlrogge, J.B. Metabolic Engineering of Fatty Acid Biosynthesis in Plants. Metab. Eng. 2002, 4, 12–21. [Google Scholar] [CrossRef]
- Huang, R.; Huang, Y.; Sun, Z.; Huang, J.; Wang, Z. Transcriptome Analysis of Genes Involved in Lipid Biosynthesis in the Developing Embryo of Pecan (Carya illinoinensis). J. Agric. Food Chem. 2017, 65, 4223–4236. [Google Scholar] [CrossRef]
- Chen, C.; Zeng, Z.; Liu, Z.; Xia, R. Small RNAs, Emerging Regulators Critical for the Development of Horticultural Traits. Hortic. Res. 2018, 5, 63. [Google Scholar] [CrossRef] [PubMed]
- Budak, H.; Akpinar, B.A. Plant miRNAs: Biogenesis, Organization and Origins. Funct. Integr. Genom. 2015, 15, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.; Zhao, Q.; Zhu, D.; Yu, J. Characterization of microRNAs Expression during Maize Seed Development. BMC Genom. 2012, 13, 360. [Google Scholar] [CrossRef]
- Parreira, J.R.; Cappuccio, M.; Balestrazzi, A.; Fevereiro, P.; de Sousa Araujo, S. MicroRNAs Expression Dynamics Reveal Post-Transcriptional Mechanisms Regulating Seed Development in Phaseolus vulgaris L. Hortic. Res. 2021, 8, 18. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhao, X.; Dai, Z.; Ma, F.; Miao, X.; Shi, Z. OsmiR396/Growth Regulating Factor Modulate Rice Grain Size through Direct Regulation of Embryo-Specific miR408. Plant Physiol. 2021, 186, 519–533. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Guo, R.; Jiang, Y.; Ye, X.; Yang, Z.; Meng, Y.; Shao, C. Genome-Wide Identification and Characterization of Novel microRNAs in Seed Development of Soybean. Biosci. Biotechnol. Biochem. 2019, 83, 233–242. [Google Scholar] [CrossRef]
- Zhao, X.; Yang, G.; Liu, X.; Yu, Z.; Peng, S. Integrated Analysis of Seed microRNA and mRNA Transcriptome Reveals Important Functional Genes and microRNA-Targets in the Process of Walnut (Juglans regia) Seed Oil Accumulation. Int. J. Mol. Sci. 2020, 21, 9093. [Google Scholar] [CrossRef]
- Lyu, Y.-Z.; Jiang, H.; Sun, H.-N.; Yang, Y.; Chao, Y.; Huang, L.-B.; Dong, X.-Y. Lipidomic and Comparative Transcriptomic Analysis of Fatty Acid Synthesis Pathway in Carya illinoinensis Embryo. Tree Physiol. 2023, 43, 1675–1690. [Google Scholar] [CrossRef]
- Mo, Z.; Feng, G.; Su, W.; Liu, Z.; Peng, F. Identification of miRNAs Associated with Graft Union Development in Pecan [Carya illinoinensis (Wangenh.) K. Koch]. Forests 2018, 9, 472. [Google Scholar] [CrossRef]
- Liu, Z.; Li, F.; Peng, F.; Tan, P.; Zhu, K.; Feng, G.; Mo, Z.; Li, Y. Identification of Grafting-Responsive MicroRNAs Associated with Growth Regulation in Pecan [Carya illinoinensis (Wangenh.) K. Koch]. Forests 2020, 11, 196. [Google Scholar] [CrossRef]
- Dabrowski, G.; Czaplicki, S.; Konopka, I. Composition and Quality of Poppy (Papaver somniferum L.) Seed Oil Depending on the Extraction Method. LWT-Food Sci. Technol. 2020, 134, 110167. [Google Scholar] [CrossRef]
- Zhu, J.; Li, W.; Zhou, Y.; Pei, L.; Liu, J.; Xia, X.; Che, R.; Li, H. Molecular Characterization, Expression and Functional Analysis of Acyl-CoA-Binding Protein Gene Family in Maize (Zea mays). BMC Plant Biol. 2021, 21, 94. [Google Scholar] [CrossRef]
- Huang, Y.; Xiao, L.; Zhang, Z.; Zhang, R.; Wang, Z.; Huang, C.; Huang, R.; Luan, Y.; Fan, T.; Wang, J.; et al. The Genomes of Pecan and Chinese Hickory Provide Insights into Carya Evolution and Nut Nutrition. GigaScience 2019, 8, giz036. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Ernst, J.; Bar-Joseph, Z. STEM: A Tool for the Analysis of Short Time Series Gene Expression Data. BMC Bioinformatics 2006, 7, 191. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Zhuang, Z.; Zhao, P.X. psRNATarget: A Plant Small RNA Target Analysis Server (2017 Release). Nucleic Acids Res. 2018, 46, W49–W54. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Rawsthorne, S. Carbon Flux and Fatty Acid Synthesis in Plants. Prog. Lipid Res. 2002, 41, 182–196. [Google Scholar] [CrossRef]
- Huang, C.; Li, Y.; Wang, K.; Xi, J.; Xu, Y.; Hong, J.; Si, X.; Ye, H.; Lyu, S.; Xia, G.; et al. Integrated Transcriptome and Proteome Analysis of Developing Embryo Reveals the Mechanisms Underlying the High Levels of Oil Accumulation in Carya cathayensis Sarg. Tree Physiol. 2022, 42, 684–702. [Google Scholar] [CrossRef]
- Wang, Z.; Qiao, Y.; Zhang, J.; Shi, W.; Zhang, J. Genome Wide Identification of microRNAs Involved in Fatty Acid and Lipid Metabolism of Brassica napus by Small RNA and Degradome Sequencing. Gene 2017, 619, 61–70. [Google Scholar] [CrossRef]
- He, M.; Kong, X.; Jiang, Y.; Qu, H.; Zhu, H. MicroRNAs: Emerging Regulators in Horticultural Crops. Trends Plant Sci. 2022, 27, 936–951. [Google Scholar] [CrossRef]
- Zhang, Y.-P.; Zhang, Y.-Y.; Thakur, K.; Zhang, F.; Hu, F.; Zhang, J.-G.; Wei, P.-C.; Wei, Z.-J. Integration of miRNAs, Degradome, and Transcriptome Omics Uncovers a Complex Regulatory Network and Provides Insights Into Lipid and Fatty Acid Synthesis During Sesame Seed Development. Front. Plant Sci. 2021, 12, 709197. [Google Scholar] [CrossRef]
- Zhang, T.; Li, Z.; Song, X.; Han, L.; Wang, L.; Zhang, J.; Long, Y.; Pei, X. Identification and Characterization of microRNAs in the Developing Seed of Linseed Flax (Linum usitatissimum L.). Int. J. Mol. Sci. 2020, 21, 2708. [Google Scholar] [CrossRef]
- Huan, T.; Rong, J.; Liu, C.; Zhang, X.; Tanriverdi, K.; Joehanes, R.; Chen, B.H.; Murabito, J.M.; Yao, C.; Courchesne, P.; et al. Genome-Wide Identification of microRNA Expression Quantitative Trait Loci. Nat. Commun. 2015, 6, 6601. [Google Scholar] [CrossRef]
- Riechmann, J.L.; Heard, J.; Martin, G.; Reuber, L.; Jiang, C.; Keddie, J.; Adam, L.; Pineda, O.; Ratcliffe, O.J.; Samaha, R.R.; et al. Arabidopsis Transcription Factors: Genome-Wide Comparative Analysis among Eukaryotes. Science 2000, 290, 2105–2110. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ding, J.; Yu, X.; Li, H.; Ruan, C. Identification and Expression Analysis of Critical microRNA-Transcription Factor Regulatory Modules Related to Seed Development and Oil Accumulation in Developing Hippophae Rhamnoides Seeds. Ind. Crop. Prod. 2019, 137, 33–42. [Google Scholar] [CrossRef]
- Wu, B.; Ruan, C.; Shah, A.H.; Li, D.; Li, H.; Ding, J.; Li, J.; Du, W. Identification of miRNA-mRNA Regulatory Modules Involved in Lipid Metabolism and Seed Development in a Woody Oil Tree (Camellia oleifera). Cells 2022, 11, 71. [Google Scholar] [CrossRef]
- Li, D.; Jin, C.; Duan, S.; Zhu, Y.; Qi, S.; Liu, K.; Gao, C.; Ma, H.; Zhang, M.; Liao, Y.; et al. MYB89 Transcription Factor Represses Seed Oil Accumulation. Plant Physiol. 2017, 173, 1211–1225. [Google Scholar] [CrossRef]
- Cao, Y.; Fan, T.; Wang, L.; Zhang, L.; Li, Y. Large-Scale Analysis of Putative Euphorbiaceae R2R3-MYB Transcription Factors Identifies a MYB Involved in Seed Oil Biosynthesis. BMC Plant Biol. 2023, 23, 145. [Google Scholar] [CrossRef]
- Fang, L.; Wang, Y. MicroRNAs in Woody Plants. Front. Plant Sci. 2021, 12, 686831. [Google Scholar] [CrossRef]
- Nodine, M.D.; Bartel, D.P. MicroRNAs Prevent Precocious Gene Expression and Enable Pattern Formation during Plant Embryogenesis. Genes Dev. 2010, 24, 2678–2692. [Google Scholar] [CrossRef]
- Wang, J.; Jian, H.; Wang, T.; Wei, L.; Li, J.; Li, C.; Liu, L. Identification of microRNAs Actively Involved in Fatty Acid Biosynthesis in Developing Brassica napus Seeds Using High-Throughput Sequencing. Front. Plant Sci. 2016, 7, 1570. [Google Scholar] [CrossRef]
- Yan, R.; Song, S.; Li, H.; Sun, H. Functional Analysis of the eTM-miR171-SCL6 Module Regulating Somatic Embryogenesis in Lilium pumilum DC. Fisch. Hortic. Res. 2022, 9, uhac045. [Google Scholar] [CrossRef]
- Ding, J.; Ruan, C.; Guan, Y.; Krishna, P. Identification of microRNAs Involved in Lipid Biosynthesis and Seed Size in Developing Sea Buckthorn Seeds Using High-Throughput Sequencing. Sci. Rep. 2018, 8, 4022. [Google Scholar] [CrossRef] [PubMed]
- Dhaka, N.; Sharma, R. MicroRNA-Mediated Regulation of Agronomically Important Seed Traits: A Treasure Trove with Shades of Grey! Crit. Rev. Biotechnol. 2021, 41, 594–608. [Google Scholar] [CrossRef] [PubMed]
- Na, G.; Mu, X.; Grabowski, P.; Schmutz, J.; Lu, C. Enhancing Micro RNA 167A Expression in Seed Decreases the A-linolenic Acid Content and Increases Seed Size in Camelina sativa. Plant J. 2019, 98, 346–358. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Known miRNAs | Novel miRNAs | Total |
|---|---|---|---|
| DAF120 | 152 | 317 | 469 |
| DAF135 | 189 | 320 | 509 |
| DAF150 | 185 | 314 | 499 |
| Total | 365 | 321 | 686 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, K.; Wei, L.; Ma, W.; Zhao, J.; Chen, M.; Wei, G.; Liu, H.; Tan, P.; Peng, F. The Integrated Analysis of miRNome and Degradome Sequencing Reveals the Regulatory Mechanisms of Seed Development and Oil Biosynthesis in Pecan (Carya illinoinensis). Foods 2024, 13, 2934. https://doi.org/10.3390/foods13182934
Zhu K, Wei L, Ma W, Zhao J, Chen M, Wei G, Liu H, Tan P, Peng F. The Integrated Analysis of miRNome and Degradome Sequencing Reveals the Regulatory Mechanisms of Seed Development and Oil Biosynthesis in Pecan (Carya illinoinensis). Foods. 2024; 13(18):2934. https://doi.org/10.3390/foods13182934
Chicago/Turabian StyleZhu, Kaikai, Lu Wei, Wenjuan Ma, Juan Zhao, Mengyun Chen, Guo Wei, Hui Liu, Pengpeng Tan, and Fangren Peng. 2024. "The Integrated Analysis of miRNome and Degradome Sequencing Reveals the Regulatory Mechanisms of Seed Development and Oil Biosynthesis in Pecan (Carya illinoinensis)" Foods 13, no. 18: 2934. https://doi.org/10.3390/foods13182934