

Development of Green and Facile Sample Preparation Method for Determination of Seven Neonicotinoids in Fresh Vegetables, and Dissipation and Risk Assessment of Imidacloprid and Dinotefuran

 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Standard Solutions
2.3. Sample Preparation
2.4. LC-MS/MS
2.5. Method Development and Validation
2.6. Blank and Actual Market Samples
2.7. Field Trial
2.8. Statistical Analysis
3. Results and Discussion
3.1. Method Development and Validation
3.2. A Comparison of the Developed Method with the Previous Studies
3.3. Real Samples
3.4. Dissipation of Imidacloprid and Dinotefuran
3.5. Risk Assessment of Residues of Field Trial
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Watson, G.B.; Chouinard, S.W.; Cook, K.R.; Geng, C.; Gifford, J.M.; Gustafson, G.D.; Hasler, J.M.; Larrinua, I.M.; Letherer, T.J.; Mitchell, J.C. A spinosyn-sensitive Drosophila melanogaster nicotinic acetylcholine receptor identified through chemically induced target site resistance, resistance gene identification, and heterologous expression. Insect Biochem. Mol. Biol. 2010, 40, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Tomizawa, M.; Casida, J.E. Neonicotinoid insecticide toxicology: Mechanisms of selective action. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 247–268. [Google Scholar] [CrossRef] [PubMed]
- Cartereau, A.; Martin, C.; Thany, S.H. Neonicotinoid insecticides differently modulate acetycholine-induced currents on mammalian α7 nicotinic acetylcholine receptors. Br. J. Pharmacol. 2018, 175, 1987–1998. [Google Scholar] [CrossRef] [PubMed]
- Main, A.R.; Michel, N.L.; Headley, J.V.; Peru, K.M.; Morrissey, C.A. Ecological and landscape drivers of neonicotinoid insecticide detections and concentrations in Canada’s prairie wetlands. Environ. Sci. Technol. 2015, 49, 8367–8376. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Lu, X.; Yu, B.; Wang, D.; Zhao, C.; Yang, Q.; Zhang, Q.; Tan, Y.; Wang, X.; Guo, J. Comparison of neonicotinoid residues in soils of different land use types. Sci. Total Environ. 2021, 782, 146803. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Anadón, A.; Wu, Q.; Qiao, F.; Ares, I.; Martínez-Larrañaga, M.-R.; Yuan, Z.; Martínez, M.-A. Mechanism of neonicotinoid toxicity: Impact on oxidative stress and metabolism. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 471–507. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, P.; Chang, J.; Li, W.; Yang, L.; Tian, H. Unraveling the toxic effects of neonicotinoid insecticides on the thyroid endocrine system of lizards. Environ. Pollut. 2020, 258, 113731. [Google Scholar] [CrossRef] [PubMed]
- Lundin, O.; Rundlöf, M.; Smith, H.G.; Fries, I.; Bommarco, R. Neonicotinoid insecticides and their impacts on bees: A systematic review of research approaches and identification of knowledge gaps. PLoS ONE 2015, 10, e0136928. [Google Scholar] [CrossRef]
- Monteiro, S.H.; Lehotay, S.J.; Sapozhnikova, Y.; Ninga, E.; Lightfield, A.R. High-throughput mega-method for the analysis of pesticides, veterinary drugs, and environmental contaminants by ultra-high-performance liquid chromatography–tandem mass spectrometry and robotic mini-solid-phase extraction cleanup + low-pressure gas chromatography–tandem mass spectrometry, part 1: Beef. J. Agric. Food Chem. 2020, 69, 1159–1168. [Google Scholar]
- Monteiro, S.H.; Lehotay, S.J.; Sapozhnikova, Y.; Ninga, E.; Moura Andrade, G.C.; Lightfield, A.R. Validation of the QuEChERSER mega-method for the analysis of pesticides, veterinary drugs, and environmental contaminants in tilapia (Oreochromis Niloticus). Food Addit. Contam. Part A 2022, 39, 699–709. [Google Scholar] [CrossRef]
- Abdallah, O.; Abdel Ghani, S.; Hrouzková, S. Development of validated LC-MS/MS method for imidacloprid and acetamiprid in parsley and rocket and evaluation of their dissipation dynamics. J. Liq. Chromatogr. Relat. Technol. 2017, 40, 392–399. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, L.; Hu, H.; Wu, R.; Ling, J.; Yue, S.; Yang, D.; Yu, W.; Du, W.; Shen, G. Neonicotinoid pollution in marine sediments of the East China Sea. Sci. Total Environ. 2022, 842, 156658. [Google Scholar] [CrossRef] [PubMed]
- Distefano, G.G.; Zangrando, R.; Basso, M.; Panzarin, L.; Gambaro, A.; Ghirardini, A.V.; Picone, M. The ubiquity of neonicotinoid contamination: Residues in seabirds with different trophic habits. Environ. Res. 2022, 206, 112637. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.J.; Zhang, G.; O’Neal, M.E.; Bradbury, S.P.; Coats, J.R. Quantifying neonicotinoid insecticide residues in milkweed and other forbs sampled from prairie strips established in maize and soybean fields. Agric. Ecosyst. Environ. 2022, 325, 107723. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, J.; Wang, Y.; Wang, Y.; Yang, R.; Zhou, X. Simultaneous determination of ten neonicotinoid insecticides and a metabolite in human whole blood by QuEChERS coupled with UPLC-Q Exactive orbitrap high-resolution mass spectrometry. J. Chromatogr. B 2023, 1222, 123689. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, O.I.; Abd El-Hamid, R.M.; Abdel Raheem, E.H. Clothianidin residues in green bean, pepper and watermelon crops and dietary exposure evaluation based on dispersive liquid-liquid microextraction and LC–MS/MS. J. Consum. Prot. Food Saf. 2019, 14, 293–300. [Google Scholar] [CrossRef]
- Abdallah, O.I.; Malhat, F.M. Thiacloprid residues in green onion (Allium cepa) using micro liquid–liquid extraction and liquid chromatography–tandem mass spectrometry. Agric. Res. 2020, 9, 340–348. [Google Scholar] [CrossRef]
- Abdallah, O.; Soliman, H.; El-Hefny, D.; Abd El-Hamid, R.; Malhat, F. Dissipation profile of sulfoxaflor on squash under Egyptian field conditions: A prelude to risk assessment. Int. J. Environ. Anal. Chem. 2023, 103, 3820–3834. [Google Scholar] [CrossRef]
- Pook, C.; Gritcan, I. Validation and application of a modified QuEChERS method for extracting neonicotinoid residues from New Zealand maize field soil reveals their persistence at nominally hazardous concentrations. Environ. Pollut. 2019, 255, 113075. [Google Scholar] [CrossRef]
- Luke, M.A.; Froberg, J.E.; Masumoto, H.T. Extraction and cleanup of organochlorine, organophosphate, organonitrogen, and hydrocarbon pesticides in produce for determination by gas-liquid chromatography. J. Assoc. Off. Anal. Chem. 1975, 58, 1020–1026. [Google Scholar] [CrossRef]
- Anastassiades, M.; Lehotay, S.J.; Štajnbaher, D.; Schenck, F.J. Fast and easy multiresidue method employing acetonitrile extraction/partitioning and “dispersive solid-phase extraction” for the determination of pesticide residues in produce. J. AOAC Int. 2003, 86, 412–431. [Google Scholar] [CrossRef]
- Tobiszewski, M. Metrics for green analytical chemistry. Anal. Methods 2016, 8, 2993–2999. [Google Scholar] [CrossRef]
- AGREE. Analytical Greenness Calculator Software. Available online: https://mostwiedzy.pl/wojciech-wojnowski,174235-1/AGREE (accessed on 1 February 2024).
- Stanadard Method EN 15662; Food of Plant Origin–Determination of Pesticide Residues Using GC–MS and/or LC–MS/MS following Acetonitrile Extraction/Partitioning and Clean-Up by Dispersive SPE-QuEChERS Method. European Committee for Standardization (CEN): Brussels, Belgium, 2008.
- FDA. Analytical Procedures and Methods Validation for Drugs and Biologics Guidance for Industry. 2015. Available online: https://www.fda.gov/media/87801/download (accessed on 15 February 2024).
- EU-MRL Database. Available online: https://ec.europa.eu/food/plants/pesticides/eu-pesticides-database_hu (accessed on 15 February 2024).
- European Communities. Commission directive 2002/63/EC of 11 July 2002—Establishing community methods of sampling for the official control of pesticide residues in and on products of plant and animal origin and repealing directive 79/700. Off. J. Eur. Communities 2002, 2, 30–43. [Google Scholar]
- EU. Commission Regulation (EC) No 178/2006 of 1 February 2006 amending Regulation (EC) No 396/2005 of the European Parliament and of the Council to establish Annex I listing the food and feed products to which maximum levels for pesticide residues apply. Off. J. 2006, 29, 3–25. [Google Scholar]
- Pihlström, T.; Fernández-Alba, A.R.; Amate, C.F.; Poulsen, M.E.; Hardebusch, B.; Anastassiades, M.; Lippold, R.; Cabrera, L.C.; de Kok, A.; ORegan, F. Analytical quality control and method validation procedures for pesticide residues analysis in food and feed SANTE 11312/2021. Sante 2021, 11312, v2. [Google Scholar]
- Obana, H.; Okihashi, M.; Akutsu, K.; Kitagawa, Y.; Hori, S. Determination of neonicotinoid pesticide residues in vegetables and fruits with solid phase extraction and liquid chromatography mass spectrometry. J. Agric. Food Chem. 2003, 51, 2501–2505. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xu, J.; Dong, F.; Liu, X.; Li, X.; Li, Y.; Wu, X.; Liang, X.; Zheng, Y. Simultaneous determination of four neonicotinoid insecticides residues in cereals, vegetables and fruits using ultra-performance liquid chromatography/tandem mass spectrometry. Anal. Methods 2013, 5, 1449–1455. [Google Scholar] [CrossRef]
- Singh, S.B.; Foster, G.D.; Khan, S.U. Microwave-assisted extraction for the simultaneous determination of thiamethoxam, imidacloprid, and carbendazim residues in fresh and cooked vegetable samples. J. Agric. Food Chem. 2004, 52, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Zywitz, D.; Anastassiades, M.; Scherbaum, E. Simultaneous determination of neonicotinoid insecticides in fruits and vegetables by LC-MS and LC-MS-MS—Methodology and residue data. Dtsch. Lebensm. -Rundsch. 2003, 99, 188–196. [Google Scholar]
- Wu, Q.; Li, Z.; Wang, C.; Wu, C.; Wang, W.; Wang, Z. Dispersive solid-phase extraction clean-up combined with dispersive liquid–liquid microextraction for the determination of neonicotinoid insecticides in vegetable samples by high-performance liquid chromatography. Food Anal. Methods 2011, 4, 559–566. [Google Scholar] [CrossRef]
- Zhang, F.; Li, Y.; Yu, C.; Pan, C. Determination of six neonicotinoid insecticides residues in spinach, cucumber, apple and pomelo by QuEChERS method and LC–MS/MS. Bull. Environ. Contam. Toxicol. 2012, 88, 885–890. [Google Scholar] [CrossRef]
- Watanabe, E.; Kobara, Y.; Baba, K.; Eun, H. Determination of seven neonicotinoid insecticides in cucumber and eggplant by water-based extraction and high-performance liquid chromatography. Anal. Lett. 2015, 48, 213–220. [Google Scholar] [CrossRef]
- Rawat, K.; Srivastava, A.; Tandon, S.; Singh, G.P. Method validation for simultaneous determination of four neonicotinoids in vegetables by liquid chromatography. Anal. Sci. 2023, 39, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Colzani, L.; Forni, C.; Clerici, L.; Barreca, S.; Dellavedova, P. Determination of pollutants, antibiotics, and drugs in surface water in Italy as required by the third EU Water Framework Directive Watch List: Method development, validation, and assessment. Environ. Sci. Pollut. Res. 2024, 31, 14791–14803. [Google Scholar] [CrossRef] [PubMed]
- Pena-Pereira, F.; Wojnowski, W.; Tobiszewski, M. AGREE—Analytical GREEnness metric approach and software. Anal. Chem. 2020, 92, 10076–10082. [Google Scholar] [CrossRef]
- Abdallah, O.I.; Abd El-Hamid, R.M.; Ahmed, N.S.; Saleh, S.M.; Alminderej, F.M. Terminal Residues and Risk Assessment of Spiromesifen and Spirodiclofen in Tomato Fruits. Plants 2023, 12, 1493. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, O.I.; Ahmed, N.S.; Abd El-Hamid, R.M.; Alhewairini, S.S. Residues of difenoconazole in various ready premixes with propiconazole, cyflufenamid, and mandipropamid in/on tomato fruits. Acta Chromatogr. 2023. [Google Scholar] [CrossRef]
- Algethami, J.S.; Alhamami, M.A.; Ramadan, M.F.; Abdallah, O.I. Residues of the Acaricides Abamectin, Hexythiazox, and Spiromesifen in Eggplant (Solanum melongena L.) Fruits Grown under Field Conditions in Najran, Saudi Arabia. Agriculture 2022, 13, 116. [Google Scholar] [CrossRef]
- Fantke, P.; Juraske, R. Variability of pesticide dissipation half-lives in plants. Environ. Sci. Technol. 2013, 47, 3548–3562. [Google Scholar] [CrossRef]
- Lewis, K.A.; Tzilivakis, J.; Warner, D.J.; Green, A. An international database for pesticide risk assessments and management. Hum. Ecol. Risk Assess. Int. J. 2016, 22, 1050–1064. [Google Scholar] [CrossRef]
- Fenoll, J.; Hellín, P.; Camacho, M.d.M.; Lopez, J.; González, A.; Lacasa, A.; Flores, P. Dissipation rates of procymidone and azoxystrobin in greenhouse grown lettuce and under cold storage conditions. Int. J. Environ. Anal. Chem. 2008, 88, 737–746. [Google Scholar] [CrossRef]
- CODEX Alimentarius Commission (CODEX). Available online: https://www.fao.org/fao-who-codexalimentarius/codex-texts/dbs/pestres/pesticides/en/ (accessed on 15 February 2024).
- Zhou, J.; Dong, C.; An, W.; Zhao, Q.; Zhang, Y.; Li, Z.; Jiao, B. Dissipation of imidacloprid and its metabolites in Chinese prickly ash (Zanthoxylum) and their dietary risk assessment. Ecotoxicol. Environ. Saf. 2021, 225, 112719. [Google Scholar] [CrossRef] [PubMed]
- Mohapatra, S.; Siddamallaiah, L.; Matadha, N.Y.; Udupi, V.R.; Raj, D.P.; Gadigeppa, S. Dissipation of neonicotinoid insecticides imidacloprid, indoxacarb and thiamethoxam on pomegranate (Punica granatum L.). Ecotoxicol. Environ. Saf. 2019, 171, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Hanafi, A.; Dasenaki, M.; Bletsou, A.; Thomaidis, N.S. Dissipation rate study and pre-harvest intervals calculation of imidacloprid and oxamyl in exported Egyptian green beans and chili peppers after pestigation treatment. Food Chem. 2018, 240, 1047–1054. [Google Scholar] [CrossRef]
- Héraud, F.; Barraj, L.M.; Moy, G.G. GEMS/Food consumption cluster diets. In Total Diet Studies; Springer: Berlin/Heidelberg, Germany, 2013; pp. 427–434. Available online: https://web.archive.org/web/20220305193456/http://www.who.int/foodsafety/chem/gems_regional_diet.pdf (accessed on 15 February 2024).
- EU Active Substances Database, ARfD of Imidacloprid. 2023. Available online: https://ec.europa.eu/food/plant/pesticides/eu-pesticides-database/start/screen/active-substances/details/688 (accessed on 15 February 2024).
- EPA. ARfD of Dinotefuran. Available online: https://www3.epa.gov/pesticides/chem_search/reg_actions/registration/fs_PC-044312_01-Sep-04.pdf (accessed on 15 February 2024).
- Prabhaker, N.; Castle, S.; Henneberry, T.; Toscano, N. Assessment of cross-resistance potential to neonicotinoid insecticides in Bemisia tabaci (Hemiptera: Aleyrodidae). Bull. Entomol. Res. 2005, 95, 535–543. [Google Scholar] [CrossRef]
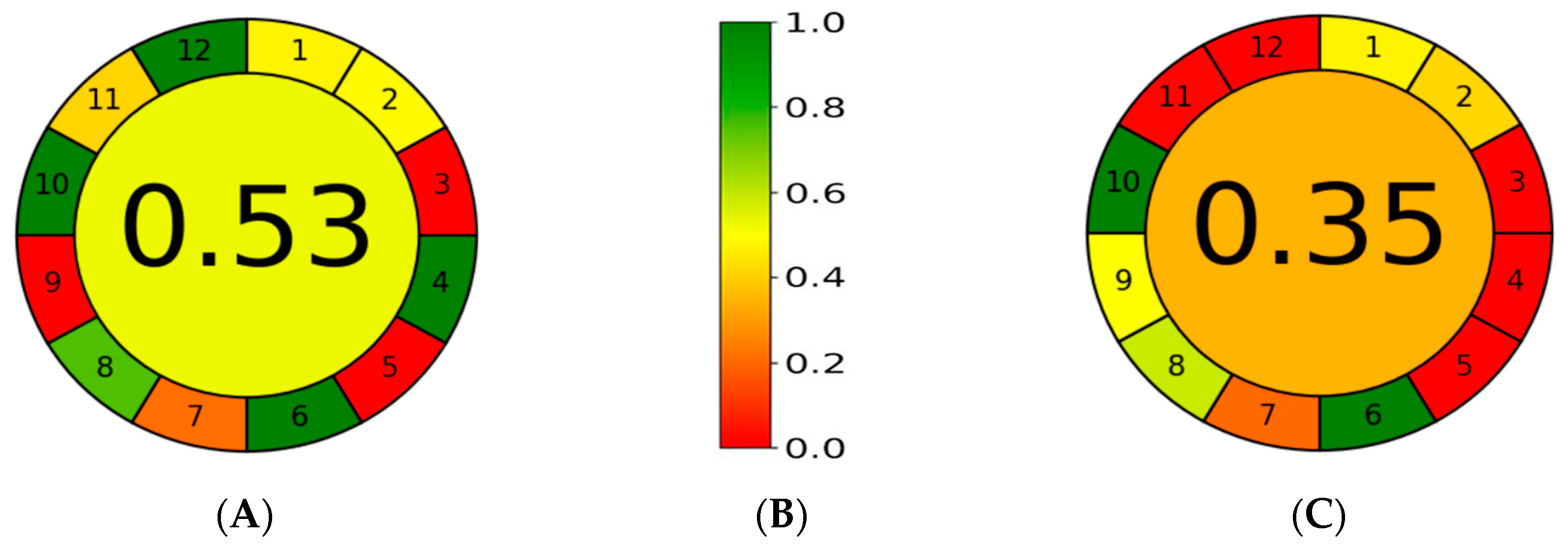

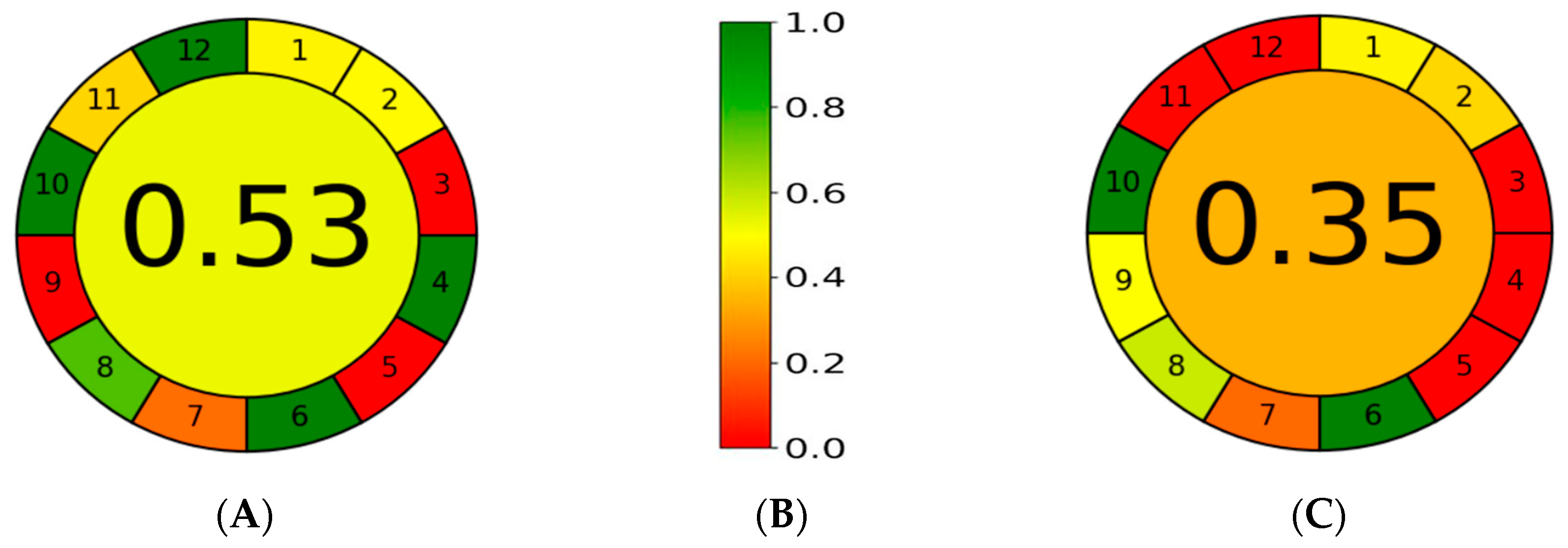{kind=link}
| R2 | Matrix Effect % | LOQ a (mg/kg) | Precision at 0.01 mg/kg | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Tomato | Lettuce | Cucumber | Tomato | Lettuce | Cucumber | %R b | RSDr c | RSDR d | ||
| Dinotefuran | 0.9999 | 0.9999 | 0.9999 | −10.8 | −15.8 | −20.9 | 0.01 | 103.0 | 6.1 | 6.9 |
| Nitenpyram | 0.9997 | 0.9998 | 0.9998 | 10.5 | −1.5 | −5.6 | 0.01 | 102.2 | 5.2 | 5.8 |
| Thiamethoxam | 0.9997 | 0.9998 | 0.9998 | −0.2 | −1.6 | −4.3 | 0.01 | 99.8 | 5.3 | 2.6 |
| Clothianidin | 0.9996 | 0.9997 | 0.9998 | −0.6 | 0.4 | −0.8 | 0.01 | 106.8 | 5.1 | 6.4 |
| Imidacloprid | 0.9999 | 0.9999 | 0.9999 | −1.8 | 3.2 | −1.7 | 0.01 | 103.2 | 5.7 | 3.1 |
| Acetamiprid | 0.9999 | 0.9999 | 0.9998 | −11.8 | −16.8 | −19.2 | 0.01 | 100.4 | 4.4 | 4.0 |
| Thiacloprid | 0.9998 | 0.9999 | 0.9999 | −12.2 | −13.7 | −15.9 | 0.01 | 101.4 | 4.8 | 4.6 |
| Tomato | Lettuce | Cucumber | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Spiking Levels (mg/kg) | Spiking Levels (mg/kg) | Spiking Levels (mg/kg) | |||||||
| 0.01 | 0.1 | 1 | 0.01 | 0.1 | 1 | 0.01 | 0.1 | 1 | |
| Dinotefuran | 107.1 (7.5) | 107.0 (2.3) | 103.0(1.1) | 106.0 (4.8) | 102.6 (7.5) | 113.2 (13.1) | 101.7 (8.3) | 91.9 (4.7) | 94.8 (5.3) |
| Nitenpyram | 104.6 (6.1) | 108.8 (2.7) | 102.1(1.3) | 106.2 (8.9) | 94.9 (5.5) | 107.0 (10.2) | 92.2 (4.6) | 109.7 (5.1) | 94.5 (2.7) |
| Thiamethoxam | 97.5 (1.2) | 102.1(0.4) | 100.9(2.3) | 99.5 (5.2) | 108.3 (11.4) | 101.9 (12.9) | 97.5 (2.3) | 94.1 (4.6) | 96.4 (7.6) |
| Clothianidin | 113.0 (3.2) | 101.3 (1.8) | 107.6(4.1) | 104.0 (8.1) | 109.6 (13.5) | 105.8 (0.4) | 97.9 (3.6) | 113.2 (4.7) | 108.5 (6.2) |
| Imidacloprid | 102.6 (5.1) | 114.4 (2.9) | 101.3(1.9) | 101.9 (4.7) | 109.5 (9.6) | 105.0 (14.6) | 93.4 (1.5) | 100.1 (5.7) | 100.3 (5.6) |
| Acetamiprid | 103.7 (3.8) | 104.7 (4.1) | 102.2(1.3) | 98.3 (5.4) | 107.2 (7.4) | 98.7 (5.3) | 94.5 (4.0) | 93.8 (3.0) | 100.3 (5.3) |
| Thiacloprid | 103.3 (2.7) | 105.0 (1.2) | 100.3(0.4) | 113.7 (8.1) | 107.8 (10.7) | 103.7 (9.1) | 92.8 (1.5) | 92.0 (5.5) | 93.7 (3.9) |
| Samples | No. | Thiamethoxam | Imidacloprid | Acetamiprid | Thiacloprid | ||||
|---|---|---|---|---|---|---|---|---|---|
| DM | Q EN | DM | Q EN | DM | Q EN | DM | Q EN | ||
| Tomato | 1 | - | - | - | - | 0.24 | 0.28 | - | - |
| 2 | - | - | 0.29 | 0.24 | - | - | - | - | |
| 3 | - | - | 0.66 | 0.69 | - | - | - | - | |
| 4 | - | - | - | - | - | - | 0.12 | 0.11 | |
| 5 | - | - | - | - | 0.13 | 0.11 | - | - | |
| Lettuce | 1 | - | - | 0.81 | 0.77 | - | - | - | - |
| 2 | - | - | 0.39 | 0.42 | - | - | - | - | |
| 3 | - | - | - | - | 0.12 | 0.15 | - | - | |
| 4 | 0.71 | 0.63 | - | - | 1.34 | 1.11 | - | - | |
| 5 | - | - | - | - | 0.13 | 0.12 | - | - | |
| Cucumber | 1 | 0.69 | 0.77 | 0.52 | 0.43 | - | - | ||
| 2 | 0.30 | 0.22 | - | - | - | - | - | - | |
| 3 | - | - | 0.93 | 0.84 | - | - | - | - | |
| 4 | - | - | 0.44 | 0.49 | - | - | - | - | |
| 5 | 0.15 | 0.13 | - | - | - | - | - | - | |
| Imidacloprid | Dinotefuran | |
|---|---|---|
| Intercept a | −0.1733 | −0.014 |
| Slope b | −0.2013 | −0.2781 |
| K | 0.2013 | 0.2781 |
| R2 c | 0.9561 | 0.9689 |
| T½ d | 3.4 days | 2.5 days |
| PHI, EU | 7 days | 14 days |
| PHI, Codex | 3 days | 3 days |
| Imidacloprid | Dinotefuran | |
|---|---|---|
| ADI/person a, mg | 3.6 | 12 |
| Tomato consumption/day/person, g [50] | 44.1 | 44.1 |
| Total food consumption/day/person, g [50] | 1342.5 | 1342.5 |
| Tomato percentage of total food consumption b | 3.3% | 3.3% |
| Maximum found residues, mg/kg c | 0.75 | 1.18 |
| EDI via tomato per person, mg d | 0.03 | 0.05 |
| Maximum allowed daily intake of tomato per person, mg e | 0.12 | 0.4 |
| Chronic risk factor % f | 25 | 13 |
| ARfD per person, mg | 4.8 g | 75 h |
| Acute risk factor % i | 0.63 | 0.07 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdallah, O.I.; Abd El-Hamid, R.M.; Ahmed, N.S.; Alhewairini, S.S.; Abdel Ghani, S.B. Development of Green and Facile Sample Preparation Method for Determination of Seven Neonicotinoids in Fresh Vegetables, and Dissipation and Risk Assessment of Imidacloprid and Dinotefuran. Foods 2024, 13, 1106. https://doi.org/10.3390/foods13071106
Abdallah OI, Abd El-Hamid RM, Ahmed NS, Alhewairini SS, Abdel Ghani SB. Development of Green and Facile Sample Preparation Method for Determination of Seven Neonicotinoids in Fresh Vegetables, and Dissipation and Risk Assessment of Imidacloprid and Dinotefuran. Foods. 2024; 13(7):1106. https://doi.org/10.3390/foods13071106
Chicago/Turabian StyleAbdallah, Osama I., Rania M. Abd El-Hamid, Nevein S. Ahmed, Saleh S. Alhewairini, and Sherif B. Abdel Ghani. 2024. "Development of Green and Facile Sample Preparation Method for Determination of Seven Neonicotinoids in Fresh Vegetables, and Dissipation and Risk Assessment of Imidacloprid and Dinotefuran" Foods 13, no. 7: 1106. https://doi.org/10.3390/foods13071106
APA StyleAbdallah, O. I., Abd El-Hamid, R. M., Ahmed, N. S., Alhewairini, S. S., & Abdel Ghani, S. B. (2024). Development of Green and Facile Sample Preparation Method for Determination of Seven Neonicotinoids in Fresh Vegetables, and Dissipation and Risk Assessment of Imidacloprid and Dinotefuran. Foods, 13(7), 1106. https://doi.org/10.3390/foods13071106