

Hepatic Transcriptome Comparative In Silico Analysis Reveals Similar Pathways and Targets Altered by Legacy and Alternative Per- and Polyfluoroalkyl Substances in Mice
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. ToxPrint Clustering Analysis
2.2. Data Acquisition
2.3. BaseSpace Correlation Engine
2.4. Principal Component Analysis
2.5. Ingenuity Pathway Analysis
2.6. FACTORIAL Activation Assay
2.7. Heatmap Construction
3. Results
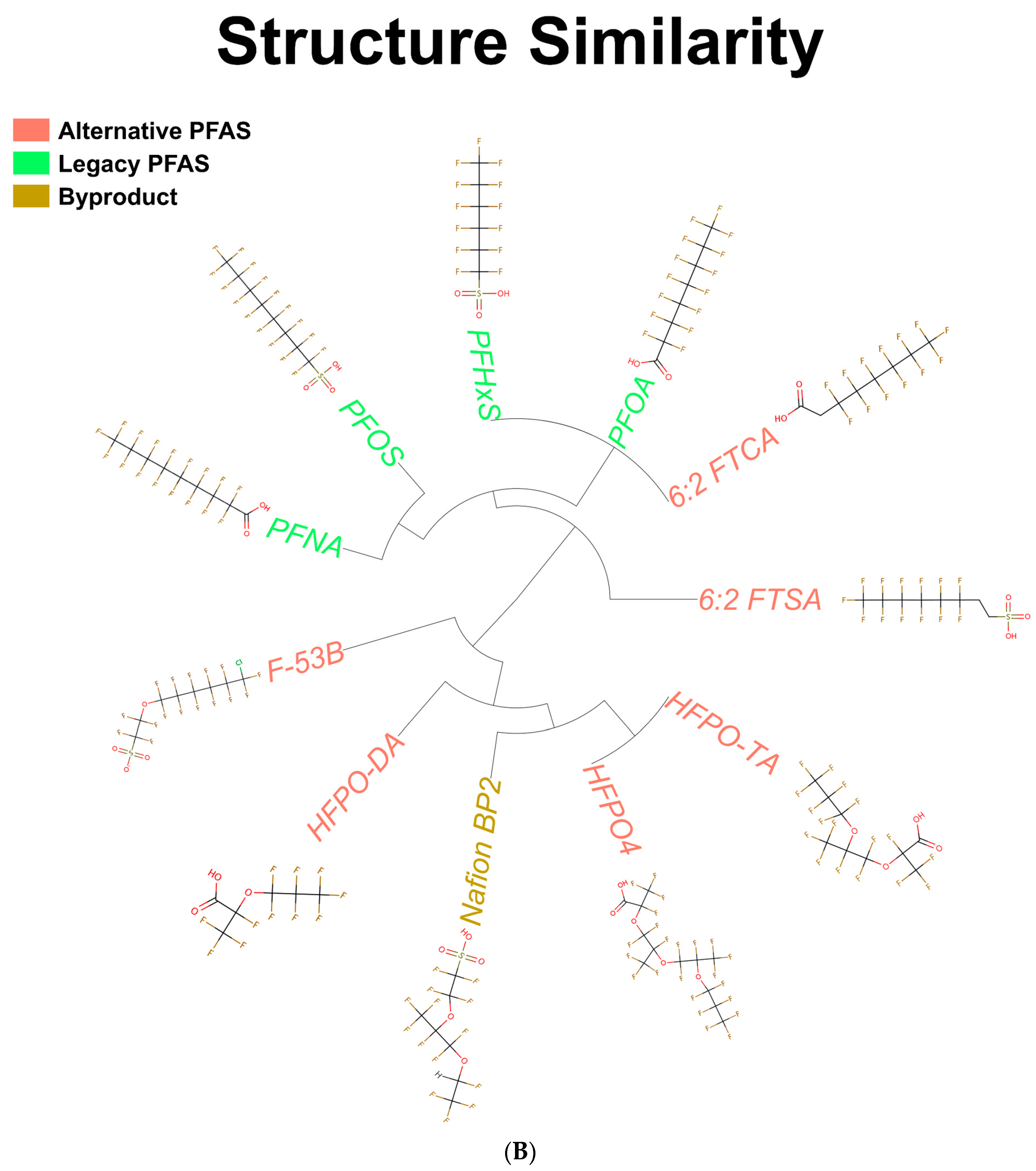
3.1. Structures of PFAS Chemicals Examined in the Study
3.2. Identification of Genes Modulated by PFAS in the Mouse Liver
3.3. Altered Canonical Pathways by PPARα Activators and PFAS
3.4. Transcriptional Regulator and Factor Activation Status in PFAS and PPARα Activator Treated Mice
4. Discussion
4.1. Effects of PFAS on Bile Acid Metabolism
4.2. Effects of PFAS on PPARα Activity
4.3. Effects of PFAS on CAR, PXR, and Nrf2 Activity
4.4. Effects of PFAS on SREBP1/2 Activity
4.5. Effects of PFAS on STAT5b Activity
4.6. Study Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Reardon, A.J.F.; Rowan-Carroll, A.; Ferguson, S.S.; Leingartner, K.; Gagne, R.; Kuo, B.; Williams, A.; Lorusso, L.; Bourdon-Lacombe, J.A.; Carrier, R.; et al. Potency Ranking of Per- and Polyfluoroalkyl Substances Using High-Throughput Transcriptomic Analysis of Human Liver Spheroids. Toxicol. Sci. 2021, 184, 154–169. [Google Scholar] [CrossRef] [PubMed]
- Blake, B.E.; Fenton, S.E. Early life exposure to per- and polyfluoroalkyl substances (PFAS) and latent health outcomes: A review including the placenta as a target tissue and possible driver of peri- and postnatal effects. Toxicology 2020, 443, 152565. [Google Scholar] [CrossRef] [PubMed]
- DeWitt, J.C.; Blossom, S.J.; Schaider, L.A. Exposure to per-fluoroalkyl and polyfluoroalkyl substances leads to immunotoxicity: Epidemiological and toxicological evidence. J. Expo. Sci. Environ. Epidemiol. 2019, 29, 148–156. [Google Scholar] [CrossRef]
- Fenton, S.E.; Ducatman, A.; Boobis, A.; DeWitt, J.C.; Lau, C.; Ng, C.; Smith, J.S.; Roberts, S.M. Per- and Polyfluoroalkyl Substance Toxicity and Human Health Review: Current State of Knowledge and Strategies for Informing Future Research. Environ. Toxicol. Chem. 2021, 40, 606–630. [Google Scholar] [CrossRef]
- Calafat, A.M.; Kato, K.; Hubbard, K.; Jia, T.; Botelho, J.C.; Wong, L.Y. Legacy and alternative per- and polyfluoroalkyl substances in the U.S. general population: Paired serum-urine data from the 2013–2014 National Health and Nutrition Examination Survey. Environ. Int. 2019, 131, 105048. [Google Scholar] [CrossRef] [PubMed]
- Rogers, R.D.; Reh, C.M.; Breysse, P. Advancing per- and polyfluoroalkyl substances (PFAS) research: An overview of ATSDR and NCEH activities and recommendations. J. Expo. Sci. Environ. Epidemiol. 2021, 31, 961–971. [Google Scholar] [CrossRef]
- CDC. Fourth National Report on Human Exposure to Environmental Chemicals; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2021; Volume 2, pp. 253–285. [Google Scholar]
- DeLuca, N.M.; Angrish, M.; Wilkins, A.; Thayer, K.; Cohen Hubal, E.A. Human exposure pathways to poly- and perfluoroalkyl substances (PFAS) from indoor media: A systematic review protocol. Environ. Int. 2021, 146, 106308. [Google Scholar] [CrossRef] [PubMed]
- Bassler, J.; Ducatman, A.; Elliott, M.; Wen, S.; Wahlang, B.; Barnett, J.; Cave, M.C. Environmental perfluoroalkyl acid exposures are associated with liver disease characterized by apoptosis and altered serum adipocytokines. Environ. Pollut. 2019, 247, 1055–1063. [Google Scholar] [CrossRef]
- Jin, R.; McConnell, R.; Catherine, C.; Xu, S.; Walker, D.I.; Stratakis, N.; Jones, D.P.; Miller, G.W.; Peng, C.; Conti, D.V.; et al. Perfluoroalkyl substances and severity of nonalcoholic fatty liver in Children: An untargeted metabolomics approach. Environ. Int. 2020, 134, 105220. [Google Scholar] [CrossRef]
- Armstrong, L.E.; Guo, G.L. Understanding Environmental Contaminants’ Direct Effects on Non-alcoholic Fatty Liver Disease Progression. Curr. Environ. Health Rep. 2019, 6, 95–104. [Google Scholar] [CrossRef]
- Andersen, M.E.; Hagenbuch, B.; Apte, U.; Corton, J.C.; Fletcher, T.; Lau, C.; Roth, W.L.; Staels, B.; Vega, G.L.; Clewell, H.J., 3rd; et al. Why is elevation of serum cholesterol associated with exposure to perfluoroalkyl substances (PFAS) in humans? A workshop report on potential mechanisms. Toxicology 2021, 459, 152845. [Google Scholar] [CrossRef] [PubMed]
- Kotlarz, N.; McCord, J.; Collier, D.; Lea, C.S.; Strynar, M.; Lindstrom, A.B.; Wilkie, A.A.; Islam, J.Y.; Matney, K.; Tarte, P.; et al. Measurement of Novel, Drinking Water-Associated PFAS in Blood from Adults and Children in Wilmington, North Carolina. Environ. Health Perspect. 2020, 128, 77005. [Google Scholar] [CrossRef] [PubMed]
- Guillette, T.C.; McCord, J.; Guillette, M.; Polera, M.E.; Rachels, K.T.; Morgeson, C.; Kotlarz, N.; Knappe, D.R.U.; Reading, B.J.; Strynar, M.; et al. Elevated levels of per- and polyfluoroalkyl substances in Cape Fear River Striped Bass (Morone saxatilis) are associated with biomarkers of altered immune and liver function. Environ. Int. 2020, 136, 105358. [Google Scholar] [CrossRef] [PubMed]
- Sheng, N.; Zhou, X.; Zheng, F.; Pan, Y.; Guo, X.; Guo, Y.; Sun, Y.; Dai, J. Comparative hepatotoxicity of 6:2 fluorotelomer carboxylic acid and 6:2 fluorotelomer sulfonic acid, two fluorinated alternatives to long-chain perfluoroalkyl acids, on adult male mice. Arch. Toxicol. 2017, 91, 2909–2919. [Google Scholar] [CrossRef] [PubMed]
- Conley, J.M.; Lambright, C.S.; Evans, N.; Medlock-Kakaley, E.; Hill, D.; McCord, J.; Strynar, M.J.; Wehmas, L.C.; Hester, S.; MacMillan, D.K.; et al. Developmental toxicity of Nafion byproduct 2 (NBP2) in the Sprague-Dawley rat with comparisons to hexafluoropropylene oxide-dimer acid (HFPO-DA or GenX) and perfluorooctane sulfonate (PFOS). Environ. Int. 2022, 160, 107056. [Google Scholar] [CrossRef]
- Sen, P.; Qadri, S.; Luukkonen, P.K.; Ragnarsdottir, O.; McGlinchey, A.; Jantti, S.; Juuti, A.; Arola, J.; Schlezinger, J.J.; Webster, T.F.; et al. Exposure to environmental contaminants is associated with altered hepatic lipid metabolism in non-alcoholic fatty liver disease. J. Hepatol. 2022, 76, 283–293. [Google Scholar] [CrossRef]
- Klaunig, J.E.; Babich, M.A.; Baetcke, K.P.; Cook, J.C.; Corton, J.C.; David, R.M.; DeLuca, J.G.; Lai, D.Y.; McKee, R.H.; Peters, J.M.; et al. PPARalpha agonist-induced rodent tumors: Modes of action and human relevance. Crit. Rev. Toxicol. 2003, 33, 655–780. [Google Scholar] [CrossRef]
- Corton, J.C.; Peters, J.M.; Klaunig, J.E. The PPARalpha-dependent rodent liver tumor response is not relevant to humans: Addressing misconceptions. Arch. Toxicol. 2018, 92, 83–119. [Google Scholar] [CrossRef]
- Klaunig, J.E.; Hocevar, B.A.; Kamendulis, L.M. Mode of Action analysis of perfluorooctanoic acid (PFOA) tumorigenicity and Human Relevance. Reprod. Toxicol. 2012, 33, 410–418. [Google Scholar] [CrossRef]
- Thompson, C.M.; Fitch, S.E.; Ring, C.; Rish, W.; Cullen, J.M.; Haws, L.C. Development of an oral reference dose for the perfluorinated compound GenX. J. Appl. Toxicol. 2019, 39, 1267–1282. [Google Scholar] [CrossRef]
- Oshida, K.; Vasani, N.; Jones, C.; Moore, T.; Hester, S.; Nesnow, S.; Auerbach, S.; Geter, D.R.; Aleksunes, L.M.; Thomas, R.S.; et al. Identification of chemical modulators of the constitutive activated receptor (CAR) in a gene expression compendium. Nucl. Recept. Signal 2015, 13, e002. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Vallanat, B.; Nelson, D.M.; Yeung, L.W.Y.; Guruge, K.S.; Lam, P.K.S.; Lehman-McKeeman, L.D.; Corton, J.C. Evidence for the involvement of xenobiotic-responsive nuclear receptors in transcriptional effects upon perfluoroalkyl acid exposure in diverse species. Reprod. Toxicol. 2009, 27, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Rooney, J.P.; Oshida, K.; Kumar, R.; Baldwin, W.S.; Corton, J.C. Chemical Activation of the Constitutive Androstane Receptor Leads to Activation of Oxidant-Induced Nrf2. Toxicol. Sci. 2019, 167, 172–189. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Takahashi, M.; Kano, M.; Amaike, Y.; Ishii, C.; Maeda, K.; Kudoh, Y.; Morishita, T.; Hosaka, T.; Sasaki, T.; et al. Activation of nuclear receptor CAR by an environmental pollutant perfluorooctanoic acid. Arch. Toxicol. 2017, 91, 2365–2374. [Google Scholar] [CrossRef] [PubMed]
- Alharthy, S.A.; Hardej, D. The role of transcription factor Nrf2 in the toxicity of perfluorooctane sulfonate (PFOS) and perfluorooctanoic acid (PFOA) in C57BL/6 mouse astrocytes. Environ. Toxicol. Pharmacol. 2021, 86, 103652. [Google Scholar] [CrossRef] [PubMed]
- Pfohl, M.; Ingram, L.; Marques, E.; Auclair, A.; Barlock, B.; Jamwal, R.; Anderson, D.; Cummings, B.S.; Slitt, A.L. Perfluorooctanesulfonic Acid and Perfluorohexanesulfonic Acid Alter the Blood Lipidome and the Hepatic Proteome in a Murine Model of Diet-Induced Obesity. Toxicol. Sci. 2020, 178, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Corton, J.C. Frequent Modulation of the Sterol Regulatory Element Binding Protein (SREBP) by Chemical Exposure in the Livers of Rats. Comput. Toxicol. 2019, 10, 113–129. [Google Scholar] [CrossRef]
- Yan, S.; Wang, J.; Dai, J. Activation of sterol regulatory element-binding proteins in mice exposed to perfluorooctanoic acid for 28 days. Arch. Toxicol. 2015, 89, 1569–1578. [Google Scholar] [CrossRef]
- Rooney, J.; Chorley, B.; Corton, J.C. A gene expression biomarker identifies factors that modulate sterol regulatory element binding protein. Comput. Toxicol. 2019, 10, 63–77. [Google Scholar] [CrossRef]
- Rosen, M.B.; Schmid, J.R.; Corton, J.C.; Zehr, R.D.; Das, K.P.; Abbott, B.D.; Lau, C. Gene Expression Profiling in Wild-Type and PPARalpha-Null Mice Exposed to Perfluorooctane Sulfonate Reveals PPARalpha-Independent Effects. PPAR Res. 2010, 2010, 794739. [Google Scholar] [CrossRef]
- Rosen, M.B.; Abbott, B.D.; Wolf, D.C.; Corton, J.C.; Wood, C.R.; Schmid, J.E.; Das, K.P.; Zehr, R.D.; Blair, E.T.; Lau, C. Gene profiling in the livers of wild-type and PPARalpha-null mice exposed to perfluorooctanoic acid. Toxicol. Pathol. 2008, 36, 592–607. [Google Scholar] [CrossRef] [PubMed]
- Rosen, M.B.; Das, K.P.; Rooney, J.; Abbott, B.; Lau, C.; Corton, J.C. PPARalpha-independent transcriptional targets of perfluoroalkyl acids revealed by transcript profiling. Toxicology 2017, 387, 95–107. [Google Scholar] [CrossRef]
- Wang, J.; Wang, X.; Sheng, N.; Zhou, X.; Cui, R.; Zhang, H.; Dai, J. RNA-sequencing analysis reveals the hepatotoxic mechanism of perfluoroalkyl alternatives, HFPO2 and HFPO4, following exposure in mice. J. Appl. Toxicol. 2017, 37, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Sheng, N.; Pan, Y.; Guo, Y.; Sun, Y.; Dai, J. Hepatotoxic Effects of Hexafluoropropylene Oxide Trimer Acid (HFPO-TA), A Novel Perfluorooctanoic Acid (PFOA) Alternative, on Mice. Environ. Sci. Technol. 2018, 52, 8005–8015. [Google Scholar] [CrossRef] [PubMed]
- Kane, C.D.; Stevens, K.A.; Fischer, J.E.; Haghpassand, M.; Royer, L.J.; Aldinger, C.; Landschulz, K.T.; Zagouras, P.; Bagley, S.W.; Hada, W.; et al. Molecular characterization of novel and selective peroxisome proliferator-activated receptor alpha agonists with robust hypolipidemic activity in vivo. Mol. Pharmacol. 2009, 75, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, L.M.; de Groot, P.J.; Hooiveld, G.J.; Koppen, A.; Kalkhoven, E.; Muller, M.; Kersten, S. Effect of synthetic dietary triglycerides: A novel research paradigm for nutrigenomics. PLoS ONE 2008, 3, e1681. [Google Scholar] [CrossRef]
- Oshida, K.; Vasani, N.; Thomas, R.S.; Applegate, D.; Gonzalez, F.J.; Aleksunes, L.M.; Klaassen, C.D.; Corton, J.C. Screening a mouse liver gene expression compendium identifies modulators of the aryl hydrocarbon receptor (AhR). Toxicology 2015, 336, 99–112. [Google Scholar] [CrossRef]
- Rooney, J.P.; Ryan, N.; Chorley, B.N.; Hester, S.D.; Kenyon, E.M.; Schmid, J.E.; George, B.J.; Hughes, M.F.; Sey, Y.M.; Tennant, A.; et al. From the Cover: Genomic Effects of Androstenedione and Sex-Specific Liver Cancer Susceptibility in Mice. Toxicol. Sci. 2017, 160, 15–29. [Google Scholar] [CrossRef]
- Kupershmidt, I.; Su, Q.J.; Grewal, A.; Sundaresh, S.; Halperin, I.; Flynn, J.; Shekar, M.; Wang, H.; Park, J.; Cui, W.; et al. Ontology-based meta-analysis of global collections of high-throughput public data. PLoS ONE 2010, 5, e13066. [Google Scholar] [CrossRef]
- Oshida, K.; Waxman, D.J.; Corton, J.C. Chemical and Hormonal Effects on STAT5b-Dependent Sexual Dimorphism of the Liver Transcriptome. PLoS ONE 2016, 11, e0150284. [Google Scholar] [CrossRef]
- Oshida, K.; Vasani, N.; Thomas, R.S.; Applegate, D.; Rosen, M.; Abbott, B.; Lau, C.; Guo, G.; Aleksunes, L.M.; Klaassen, C.; et al. Identification of modulators of the nuclear receptor peroxisome proliferator-activated receptor alpha (PPARalpha) in a mouse liver gene expression compendium. PLoS ONE 2015, 10, e0112655. [Google Scholar] [CrossRef] [PubMed]
- Robarts, D.R.; McGreal, S.R.; Umbaugh, D.S.; Parkes, W.S.; Kotulkar, M.; Abernathy, S.; Lee, N.; Jaeschke, H.; Gunewardena, S.; Whelan, S.A.; et al. Regulation of Liver Regeneration by Hepatocyte O-GlcNAcylation in Mice. Cell Mol. Gastroenterol. Hepatol. 2022, 13, 1510–1529. [Google Scholar] [CrossRef]
- Houck, K.A.; Patlewicz, G.; Richard, A.M.; Williams, A.J.; Shobair, M.A.; Smeltz, M.; Clifton, M.S.; Wetmore, B.; Medvedev, A.; Makarov, S. Bioactivity profiling of per- and polyfluoroalkyl substances (PFAS) identifies potential toxicity pathways related to molecular structure. Toxicology 2021, 457, 152789. [Google Scholar] [CrossRef] [PubMed]
- Gunewardena, S.; Huck, I.; Walesky, C.; Robarts, D.; Weinman, S.; Apte, U. Progressive loss of hepatocyte nuclear factor 4 alpha activity in chronic liver diseases in humans. Hepatology 2022, 76, 372–386. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, Y.; Klaassen, C.D.; Cheng, X. Alteration of Bile Acid and Cholesterol Biosynthesis and Transport by Perfluorononanoic Acid (PFNA) in Mice. Toxicol. Sci. 2018, 162, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Oshida, K.; Vasani, N.; Waxman, D.J.; Corton, J.C. Disruption of STAT5b-Regulated Sexual Dimorphism of the Liver Transcriptome by Diverse Factors Is a Common Event. PLoS ONE 2016, 11, e0148308. [Google Scholar] [CrossRef]
- Rooney, J.; Oshida, K.; Vasani, N.; Vallanat, B.; Ryan, N.; Chorley, B.N.; Wang, X.; Bell, D.A.; Wu, K.C.; Aleksunes, L.M.; et al. Activation of Nrf2 in the liver is associated with stress resistance mediated by suppression of the growth hormone-regulated STAT5b transcription factor. PLoS ONE 2018, 13, e0200004. [Google Scholar] [CrossRef]
- Chappell, G.A.; Thompson, C.M.; Wolf, J.C.; Cullen, J.M.; Klaunig, J.E.; Haws, L.C. Assessment of the Mode of Action Underlying the Effects of GenX in Mouse Liver and Implications for Assessing Human Health Risks. Toxicol. Pathol. 2020, 48, 494–508. [Google Scholar] [CrossRef]
- Martin, M.T.; Dix, D.J.; Judson, R.S.; Kavlock, R.J.; Reif, D.M.; Richard, A.M.; Rotroff, D.M.; Romanov, S.; Medvedev, A.; Poltoratskaya, N.; et al. Impact of environmental chemicals on key transcription regulators and correlation to toxicity end points within EPA’s ToxCast program. Chem. Res. Toxicol. 2010, 23, 578–590. [Google Scholar] [CrossRef]
- Guo, H.; Chen, J.; Zhang, H.; Yao, J.; Sheng, N.; Li, Q.; Guo, Y.; Wu, C.; Xie, W.; Dai, J. Exposure to GenX and Its Novel Analogs Disrupts Hepatic Bile Acid Metabolism in Male Mice. Environ. Sci. Technol. 2022, 56, 6133–6143. [Google Scholar] [CrossRef]
- Conley, J.M.; Lambright, C.S.; Evans, N.; Strynar, M.J.; McCord, J.; McIntyre, B.S.; Travlos, G.S.; Cardon, M.C.; Medlock-Kakaley, E.; Hartig, P.C.; et al. Adverse Maternal, Fetal, and Postnatal Effects of Hexafluoropropylene Oxide Dimer Acid (GenX) from Oral Gestational Exposure in Sprague-Dawley Rats. Environ. Health Perspect. 2019, 127, 37008. [Google Scholar] [CrossRef]
- Conley, J.M.; Lambright, C.S.; Evans, N.; McCord, J.; Strynar, M.J.; Hill, D.; Medlock-Kakaley, E.; Wilson, V.S.; Gray, L.E., Jr. Hexafluoropropylene oxide-dimer acid (HFPO-DA or GenX) alters maternal and fetal glucose and lipid metabolism and produces neonatal mortality, low birthweight, and hepatomegaly in the Sprague-Dawley rat. Environ. Int. 2021, 146, 106204. [Google Scholar] [CrossRef] [PubMed]
- Tien, E.S.; Negishi, M. Nuclear receptors CAR and PXR in the regulation of hepatic metabolism. Xenobiotica 2006, 36, 1152–1163. [Google Scholar] [CrossRef] [PubMed]
- Behr, A.C.; Plinsch, C.; Braeuning, A.; Buhrke, T. Activation of human nuclear receptors by perfluoroalkylated substances (PFAS). Toxicol. In Vitro 2020, 62, 104700. [Google Scholar] [CrossRef] [PubMed]
- Bijland, S.; Rensen, P.C.; Pieterman, E.J.; Maas, A.C.; van der Hoorn, J.W.; van Erk, M.J.; Havekes, L.M.; Willems van Dijk, K.; Chang, S.C.; Ehresman, D.J.; et al. Perfluoroalkyl sulfonates cause alkyl chain length-dependent hepatic steatosis and hypolipidemia mainly by impairing lipoprotein production in APOE*3-Leiden CETP mice. Toxicol. Sci. 2011, 123, 290–303. [Google Scholar] [CrossRef] [PubMed]
- Lille-Langoy, R.; Goldstone, J.V.; Rusten, M.; Milnes, M.R.; Male, R.; Stegeman, J.J.; Blumberg, B.; Goksoyr, A. Environmental contaminants activate human and polar bear (Ursus maritimus) pregnane X receptors (PXR, NR1I2) differently. Toxicol. Appl. Pharmacol. 2015, 284, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Shimano, H. Sterol regulatory element-binding proteins (SREBPs): Transcriptional regulators of lipid synthetic genes. Prog. Lipid Res. 2001, 40, 439–452. [Google Scholar] [CrossRef] [PubMed]
- Pettinelli, P.; Del Pozo, T.; Araya, J.; Rodrigo, R.; Araya, A.V.; Smok, G.; Csendes, A.; Gutierrez, L.; Rojas, J.; Korn, O.; et al. Enhancement in liver SREBP-1c/PPAR-alpha ratio and steatosis in obese patients: Correlations with insulin resistance and n-3 long-chain polyunsaturated fatty acid depletion. Biochim. Biophys. Acta 2009, 1792, 1080–1086. [Google Scholar] [CrossRef]
- Das, K.P.; Wood, C.R.; Lin, M.T.; Starkov, A.A.; Lau, C.; Wallace, K.B.; Corton, J.C.; Abbott, B.D. Perfluoroalkyl acids-induced liver steatosis: Effects on genes controlling lipid homeostasis. Toxicology 2017, 378, 37–52. [Google Scholar] [CrossRef]
- Knight, B.L.; Hebbachi, A.; Hauton, D.; Brown, A.M.; Wiggins, D.; Patel, D.D.; Gibbons, G.F. A role for PPARalpha in the control of SREBP activity and lipid synthesis in the liver. Biochem. J. 2005, 389, 413–421. [Google Scholar] [CrossRef]
- Pan, Z.; Miao, W.; Wang, C.; Tu, W.; Jin, C.; Jin, Y. 6:2 Cl-PFESA has the potential to cause liver damage and induce lipid metabolism disorders in female mice through the action of PPAR-gamma. Environ. Pollut. 2021, 287, 117329. [Google Scholar] [CrossRef] [PubMed]
- Lorbek, G.; Perse, M.; Horvat, S.; Bjorkhem, I.; Rozman, D. Sex differences in the hepatic cholesterol sensing mechanisms in mice. Molecules 2013, 18, 11067–11085. [Google Scholar] [CrossRef] [PubMed]
- Waxman, D.J.; O’Connor, C. Growth hormone regulation of sex-dependent liver gene expression. Mol. Endocrinol. 2006, 20, 2613–2629. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical Name | Abbreviation | DTXSID | PMID | Dose | Timepoint |
|---|---|---|---|---|---|
| Perfluorooctane sulfonate | PFOS | DTXSID3031864 | 20936131 | Daily 3 mg/kg or 10 mg/kg | 7 days |
| Perfluorooctanoic acid | PFOA | DTXSID8031865 | 18281256 | Daily 3 mg/kg | 7 days |
| Perfluorononanoic acid | PFNA | DTXSID8031863 | 28558994 | Daily 1 mg/kg or 3 mg/kg | 7 days |
| perfluorohexane sulfonate | PFHxS | DTXSID7040150 | 28558994 | Daily 3 mg/kg or 10 mg/kg | 7 days |
| Perfluoro-2-([perfluoro-3-(perfluoroethoxy)-2-propanyl]oxy)ethanesulfonic acid | Nafion BP2 | DTXSID10892352 | # | Daily 0.03, 0.3, 3, or 6 mg/kg | 7 days |
| Ammonium perfluoro-2-methyl-3-oxahexanoate | HFPO-DA (HFPO2) (GenX) | DTXSID40108559 | 27553808 32138627 | Daily 1 mg/kg Daily 0.1, 0.5, or 5 mg/kg | 28 days 90 days |
| Perfluoro-(2,5,8-trimethyl-3,6,9-trioxadodecanoic)acid | HFPO4 | DTXSID70276659 | 27553808 | Daily 1 mg/kg | 28 days |
| Perfluoro-2,5-dimethyl-3,6-dioxanonanoic acid | HFPO-TA | DTXSID00892442 | 29927593 | Daily 0.02, 0.1, 0.5 mg/kg | 28 days |
| Potassium 9-chlorohexadecafluoro-3-oxanonane-1-sulfonate | F-53B | DTXSID60881236 | # | Daily 5 mg/kg | 28 days |
| 6:2 Fluorotelomer sulfonic acid | 6:2 FTSA | DTXSID6067331 | 28032147 | Daily 5 mg/kg | 28 days |
| 2-Perfluorohexyl ethanoic acid | 6:2 FTCA | DTXSID50472556 | 28032147 | Daily 5 mg/kg | 28 days |
| (S)-2-methyl-2-(3-(1-(2-(4-(trifluoromethoxy)phenyl)acetyl)piperidin-3-yl)phenoxy)propanoic acid sodium salt | CP-865520 | DTXSID4044032 | 18971326 | Daily 1 mg/kg | 5 days |
| (S)-2-(3-(1-(2-(4-isopropylphenyl)acetyl)piperidin-3-yl)phenoxy)-2-methylpropanoic acid sodium salt | CP-775146 | DTXSID9044033 | 18971326 | Daily 1 mg/kg | 5 days |
| (S)-2-(3-(1-((4-isopropylbenzyloxy)carbonyl)piperidin-3-yl)phenoxy)-2-methylpropanoic acid sodium salt | CP-868388 | DTXSID4044034 | 18971326 | Daily 1 mg/kg | 5 days |
| Propan-2-yl 2-[4-(4-chlorobenzoyl)phenoxy]-2-methylpropanoate | Fenofibrate | DTXSID2029874 | 18301758 | Single 4 mg/mL | 6 h |
| ([4-Chloro-6-(2,3-dimethylanilino)pyrimidin-2-yl]sulfanyl)acetic acid | WY-14,643 | DTXSID4020290 | 26215100 | Single 250 mg/kg | 8 h |
| Transcription Factor Activation or Suppression | |||||
|---|---|---|---|---|---|
| PPARα | NRF2 | CAR | SREBP1/2 | STAT5b | |
| PFOS | + * | + * | + * | + * | - * |
| PFOA | + * | + | + * | + * | - * |
| PFNA | + * | + | + * | + * | - * |
| PFHxS | + * | + | + * | + * | - * |
| Nafion BP2 | NA | + | + * | + | - |
| HFPO-DA | + * | + | NA | + | - |
| HFPO4 | + * | + | + | + | - |
| HFPO-TA | + * | + | + | + | - |
| F-53B | + * | NA | + | + | - |
| 6:2 FTSA | NA | + | NA | NA | - |
| 6:2 FTCA | NA | NA | NA | NA | NA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Robarts, D.R.; Dai, J.; Lau, C.; Apte, U.; Corton, J.C. Hepatic Transcriptome Comparative In Silico Analysis Reveals Similar Pathways and Targets Altered by Legacy and Alternative Per- and Polyfluoroalkyl Substances in Mice. Toxics 2023, 11, 963. https://doi.org/10.3390/toxics11120963
Robarts DR, Dai J, Lau C, Apte U, Corton JC. Hepatic Transcriptome Comparative In Silico Analysis Reveals Similar Pathways and Targets Altered by Legacy and Alternative Per- and Polyfluoroalkyl Substances in Mice. Toxics. 2023; 11(12):963. https://doi.org/10.3390/toxics11120963
Chicago/Turabian StyleRobarts, Dakota R., Jiayin Dai, Christopher Lau, Udayan Apte, and J. Christopher Corton. 2023. "Hepatic Transcriptome Comparative In Silico Analysis Reveals Similar Pathways and Targets Altered by Legacy and Alternative Per- and Polyfluoroalkyl Substances in Mice" Toxics 11, no. 12: 963. https://doi.org/10.3390/toxics11120963
APA StyleRobarts, D. R., Dai, J., Lau, C., Apte, U., & Corton, J. C. (2023). Hepatic Transcriptome Comparative In Silico Analysis Reveals Similar Pathways and Targets Altered by Legacy and Alternative Per- and Polyfluoroalkyl Substances in Mice. Toxics, 11(12), 963. https://doi.org/10.3390/toxics11120963