
Uncovering Evidence for Endocrine-Disrupting Chemicals That Elicit Differential Susceptibility through Gene-Environment Interactions





Abstract
:1. Introduction
2. Biology of Endocrine-Disrupting Chemicals and Implications for Diverse Populations
2.1. History and Exposure
2.2. Molecular Biology of EDCs
2.3. Effects on Biological Pathways
2.4. Implications for Population Diversity
3. Evidence for Differential Susceptibility to Endocrine-Disrupting Chemicals
4. Characterizing GxE Effects Associated with Endocrine-Disrupting Chemicals
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Diamanti-Kandarakis, E.; Bourguignon, J.P.; Giudice, L.C.; Hauser, R.; Prins, G.S.; Soto, A.M.; Zoeller, R.T.; Gore, A.C. Endocrine-Disrupting Chemicals: An Endocrine Society Scientific Statement. Endocr. Rev. 2009, 30, 293–342. [Google Scholar] [CrossRef] [PubMed]
- Attina, T.M.; Hauser, R.; Sathyanarayana, S.; Hunt, P.A.; Bourguignon, J.P.; Myers, J.P.; DiGangi, J.; Zoeller, R.T.; Trasande, L. Exposure to endocrine-disrupting chemicals in the USA: A population-based disease burden and cost analysis. Lancet Diabetes Endocrinol. 2016, 4, 996–1003. [Google Scholar] [CrossRef]
- Gore, A.C.; Chappell, V.A.; Fenton, S.E.; Flaws, J.A.; Nadal, A.; Prins, G.S.; Toppari, J.; Zoeller, R.T. EDC-2: The Endocrine Society’s Second Scientific Statement on Endocrine-Disrupting Chemicals. Endocr. Rev. 2015, 36, E1–E150. [Google Scholar] [CrossRef] [PubMed]
- Darbre, P.D. Endocrine Disruptors and Obesity. Curr. Obes. Rep. 2017, 6, 18–27. [Google Scholar] [CrossRef] [Green Version]
- Boas, M.; Feldt-Rasmussen, U.; Main, K.M. Thyroid effects of endocrine-disrupting chemicals. Mol. Cell. Endocrinol. 2012, 355, 240–248. [Google Scholar] [CrossRef]
- Gutleb, A.C.; Cambier, S.; Serchi, T. Impact of Endocrine Disruptors on the Thyroid Hormone System. Horm. Res. Paediatr. 2016, 86, 271–278. [Google Scholar] [CrossRef]
- Alaee, M. An overview of commercially used brominated flame retardants, their applications, their use patterns in different countries/regions and possible modes of release. Environ. Int. 2003, 29, 683–689. [Google Scholar] [CrossRef]
- Groh, K.J.; Backhaus, T.; Carney-Almroth, B.; Geueke, B.; Inostroza, P.A.; Lennquist, A.; Leslie, H.A.; Maffini, M.; Slunge, D.; Trasande, L.; et al. Overview of known plastic packaging-associated chemicals and their hazards. Sci. Total Environ. 2019, 651, 3253–3268. [Google Scholar] [CrossRef] [PubMed]
- Ewence, A.; Brescia, S.; Johnson, I.; Rumsby, P.C. An approach to the identification and regulation of endocrine-disrupting pesticides. Food Chem. Toxicol. 2015, 78, 214–220. [Google Scholar] [CrossRef]
- Witorsch, R.J.; Thomas, J.A. Personal care products and endocrine disruption: A critical review of the literature. Crit. Rev. Toxicol. 2010, 40, 1–30. [Google Scholar] [CrossRef]
- Wong, K.H.; Durrani, T.S. Exposures to Endocrine-disrupting Chemicals in Consumer Products—A Guide for Pediatricians. Curr. Probl. Pediatr. Adolesc. Health Care 2017, 47, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Carlstedt, F.; Jönsson, B.A.G.; Bornehag, C.-G. PVC flooring is related to human uptake of phthalates in infants: Phthalate uptake in infants. Indoor Air 2013, 23, 32–39. [Google Scholar] [CrossRef]
- Koo, H.J.; Lee, B.M. Estimated exposure to phthalates in cosmetics and risk assessment. J. Toxicol. Environ. Health A 2004, 67, 1901–1914. [Google Scholar] [CrossRef] [PubMed]
- Fall, T.; Ingelsson, E. Genome-wide association studies of obesity and metabolic syndrome. Mol. Cell. Endocrinol. 2014, 382, 740–757. [Google Scholar] [CrossRef] [PubMed]
- Sladek, R.; Rocheleau, G.; Rung, J.; Dina, C.; Shen, L.; Serre, D.; Boutin, P.; Vincent, D.R.; Belisle, A.; Hadjadj, S.; et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 2007, 445, 881–885. [Google Scholar] [CrossRef]
- Axelsson, J.; Bonde, J.P.; Giwercman, Y.L.; Rylander, L.; Giwercman, A. Gene-environment interaction and male reproductive function. Asian J. Androl. 2010, 12, 298–307. [Google Scholar] [CrossRef] [Green Version]
- Colletti, J.A.; Leland-Wavrin, K.M.; Kurz, S.G.; Hickman, M.P.; Seiler, N.L.; Samanas, N.B.; Eckert, Q.A.; Dennison, K.L.; Ding, L.; Schaffer, B.S.; et al. Validation of Six Genetic Determinants of Susceptibility to Estrogen-Induced Mammary Cancer in the Rat and Assessment of Their Relevance to Breast Cancer Risk in Humans. Genes Genomes Genet. 2014, 4, 1385–1394. [Google Scholar] [CrossRef] [Green Version]
- Jerry, D.J.; Shull, J.D.; Hadsell, D.L.; Rijnkels, M.; Dunphy, K.A.; Schneider, S.S.; Vandenberg, L.N.; Majhi, P.D.; Byrne, C.; Trentham-Dietz, A. Genetic variation in sensitivity to estrogens and breast cancer risk. Mamm. Genome 2018, 29, 24–37. [Google Scholar] [CrossRef] [Green Version]
- Colter, B.T.; Garber, H.F.; Fleming, S.M.; Fowler, J.P.; Harding, G.D.; Hooven, M.K.; Howes, A.A.; Infante, S.K.; Lang, A.L.; MacDougall, M.C.; et al. Ahr and Cyp1a2 genotypes both affect susceptibility to motor deficits following gestational and lactational exposure to polychlorinated biphenyls. Neurotoxicology 2018, 65, 125–134. [Google Scholar] [CrossRef]
- Alam, G.; Jones, B.C. Toxicogenetics: In search of host susceptibility to environmental toxicants. Front. Genet. 2014, 5, 327. [Google Scholar] [CrossRef] [Green Version]
- Qin, C.; Tanis, K.Q.; Podtelezhnikov, A.A.; Glaab, W.E.; Sistare, F.D.; DeGeorge, J.J. Toxicogenomics in drug development: A match made in heaven? Expert Opin. Drug Metab. Toxicol. 2016, 12, 847–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Futran Fuhrman, V.; Tal, A.; Arnon, S. Why endocrine-disrupting chemicals (EDCs) challenge traditional risk assessment and how to respond. J. Hazard. Mater. 2015, 286, 589–611. [Google Scholar] [CrossRef] [PubMed]
- Herbst, A.L.; Ulfelder, H.; Poskanzer, D.C. Association of Maternal Stilbestrol Therapy with Tumor Appearance in Young Women. N. Engl. J. Med. 1971, 284, 878–881. [Google Scholar] [CrossRef]
- Kiyama, R.; Wada-Kiyama, Y. Estrogenic endocrine disruptors: Molecular mechanisms of action. Environ. Int. 2015, 83, 11–40. [Google Scholar] [CrossRef]
- Elshan, N.G.R.D.; Rettig, M.B.; Jung, M.E. Molecules targeting the androgen receptor (AR) signaling axis beyond the AR-Ligand binding domain. Med. Res. Rev. 2019, 39, 910–960. [Google Scholar] [CrossRef]
- Luccio-Camelo, D.C.; Prins, G.S. Disruption of androgen receptor signaling in males by environmental chemicals. J. Steroid Biochem. Mol. Biol. 2011, 127, 74–82. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Yang, Y.; Liu, W.; Schlenk, D.; Liu, J. Glucocorticoid and mineralocorticoid receptors and corticosteroid homeostasis are potential targets for endocrine-disrupting chemicals. Environ. Int. 2019, 133, 105133. [Google Scholar] [CrossRef]
- Kamstra, J.H.; Hruba, E.; Blumberg, B.; Janesick, A.; Mandrup, S.; Hamers, T.; Legler, J. Transcriptional and Epigenetic Mechanisms Underlying Enhanced in Vitro Adipocyte Differentiation by the Brominated Flame-Retardant BDE-47. Environ. Sci. Technol. 2014, 48, 4110–4119. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Zhou, B. Thyroid endocrine system disruption by pentachlorophenol: An in vitro and in vivo assay. Aquat. Toxicol. 2013, 142–143, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Mangelsdorf, D.J.; Thummel, C.; Beato, M.; Herrlich, P.; Schütz, G.; Umesono, K.; Blumberg, B.; Kastner, P.; Mark, M.; Chambon, P.; et al. The nuclear receptor superfamily: The second decade. Cell 1995, 83, 835–839. [Google Scholar] [CrossRef] [Green Version]
- Helsen, C.; Kerkhofs, S.; Clinckemalie, L.; Spans, L.; Laurent, M.; Boonen, S.; Vanderschueren, D.; Claessens, F. Structural basis for nuclear hormone receptor DNA binding. Mol. Cell. Endocrinol. 2012, 348, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Näär, A.M.; Boutin, J.-M.; Lipkin, S.M.; Yu, V.C.; Holloway, J.M.; Glass, C.K.; Rosenfeld, M.G. The orientation and spacing of core DNA-binding motifs dictate selective transcriptional responses to three nuclear receptors. Cell 1991, 65, 1267–1279. [Google Scholar] [CrossRef]
- Denayer, S.; Helsen, C.; Thorrez, L.; Haelens, A.; Claessens, F. The Rules of DNA Recognition by the Androgen Receptor. Mol. Endocrinol. 2010, 24, 898–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malo, M.S.; Zhang, W.; Alkhoury, F.; Pushpakaran, P.; Abedrapo, M.A.; Mozumder, M.; Fleming, E.; Siddique, A.; Henderson, J.W.; Hodin, R.A. Thyroid Hormone Positively Regulates the Enterocyte Differentiation Marker Intestinal Alkaline Phosphatase Gene via an Atypical Response Element. Mol. Endocrinol. 2004, 18, 1941–1962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toyoda, N.; Zavacki, A.M.; Maia, A.L.; Harney, J.W.; Larsen, P.R. A novel retinoid X receptor-independent thyroid hormone response element is present in the human type 1 deiodinase gene. Mol. Cell. Biol. 1995, 15, 5100–5112. [Google Scholar] [CrossRef] [Green Version]
- Umesono, K.; Murakami, K.K.; Thompson, C.C.; Evans, R.M. Direct repeats as selective response elements for the thyroid hormone, retinoic acid, and vitamin D3 receptors. Cell 1991, 65, 1255–1266. [Google Scholar] [CrossRef]
- Verrijdt, G.; Schoenmakers, E.; Haelens, A.; Peeters, B.; Verhoeven, G.; Rombauts, W.; Claessens, F. Change of Specificity Mutations in Androgen-selective Enhancers: Evidence for a Role of Differential DNA Binding by the Androgen Receptor. J. Biol. Chem. 2000, 275, 12298–12305. [Google Scholar] [CrossRef] [Green Version]
- Verrijdt, G. Selective DNA recognition by the androgen receptor as a mechanism for hormone-specific regulation of gene expression. Mol. Genet. Metab. 2003, 78, 175–185. [Google Scholar] [CrossRef]
- Umesono, K.; Evans, R.M. Determinants of target gene specificity for steroid/thyroid hormone receptors. Cell 1989, 57, 1139–1146. [Google Scholar] [CrossRef]
- Yamamoto, K.R. Steroid receptor regulated transcription of specific genes and gene networks. Annu. Rev. Genet. 1985, 19, 209–252. [Google Scholar] [CrossRef]
- Lemaire, G.; Mnif, W.; Mauvais, P.; Balaguer, P.; Rahmani, R. Activation of α- and β-estrogen receptors by persistent pesticides in reporter cell lines. Life Sci. 2006, 79, 1160–1169. [Google Scholar] [CrossRef] [PubMed]
- Kleinstreuer, N.C.; Ceger, P.; Watt, E.D.; Martin, M.T.; Houck, K.A.; Browne, P.; Thomas, R.S.; Casey, W.M.; Dix, D.J.; Allen, D.; et al. Development and Validation of a Computational Model for Androgen Receptor Activity. Chem. Res. Toxicol. 2017, 30, 946–964. [Google Scholar] [CrossRef] [PubMed]
- Judson, R.S.; Magpantay, F.M.; Chickarmane, V.; Haskell, C.; Tania, N.; Taylor, J.; Xia, M.; Huang, R.; Rotroff, D.M.; Filer, D.L.; et al. Integrated Model of Chemical Perturbations of a Biological Pathway Using 18 In Vitro High-Throughput Screening Assays for the Estrogen Receptor. Toxicol. Sci. 2015, 148, 137–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diel, P.; Olff, S.; Schmidt, S.; Michna, H. Molecular Identification of Potential Selective Estrogen Receptor Modulator (SERM) Like Properties of Phytoestrogens in the Human Breast Cancer Cell Line MCF-7. Planta Med. 2001, 67, 510–514. [Google Scholar] [CrossRef]
- Katzenellenbogen, S.; Montano, M.; Ediger, R. Estrogen Receptors: Selective Ligands, Partners, and Distinctive Pharmacology. Recent Prog. Horm. Res. 2000, 55, 163–195. [Google Scholar]
- Katzenellenbogen, B.S.; Katzenellenbogen, J.A.; Mordecai, D. Zearalenones: Characterization of the estrogenic potencies and receptor interactions of a series of fungal beta-resorcylic acid lactones. Endocrinology 1979, 105, 33–40. [Google Scholar] [CrossRef]
- Combarnous, Y.; Nguyen, T.M.D. Comparative Overview of the Mechanisms of Action of Hormones and Endocrine Disruptor Compounds. Toxics 2019, 7, 5. [Google Scholar] [CrossRef] [Green Version]
- Katzenellenbogen, B.S.; Choi, I.; Delage-Mourroux, R.; Ediger, T.R.; Martini, P.G.; Montano, M.; Sun, J.; Weis, K.; A Katzenellenbogen, J. Molecular mechanisms of estrogen action: Selective ligands and receptor pharmacology. J. Steroid Biochem. Mol. Biol. 2000, 74, 279–285. [Google Scholar] [CrossRef]
- Kurz, S.G.; Hansen, K.K.; McLaughlin, M.T.; Shivaswamy, V.; Schaffer, B.S.; Gould, K.A.; McComb, R.D.; Meza, J.L.; Shull, J.D. Tissue-Specific Actions of the Ept1, Ept2, Ept6, and Ept9 Genetic Determinants of Responsiveness to Estrogens in the Female Rat. Endocrinology 2008, 149, 3850–3859. [Google Scholar] [CrossRef] [Green Version]
- Dutertre, M.; Smith, C.L. Molecular Mechanisms of Selective Estrogen Receptor Modulator (SERM) Action. Sel. Estrogen Recept. Modul. 2000, 295, 7. [Google Scholar]
- Heldring, N.; Pike, A.; Andersson, S.; Matthews, J.; Cheng, G.; Hartman, J.; Tujague, M.; Ström, A.; Treuter, E.; Warner, M.; et al. Estrogen Receptors: How Do They Signal and What Are Their Targets. Physiol. Rev. 2007, 87, 905–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustafsson, J.-Å. What pharmacologists can learn from recent advances in estrogen signalling. Trends Pharmacol. Sci. 2003, 24, 479–485. [Google Scholar] [CrossRef]
- Hall, J.M.; Couse, J.F.; Korach, K.S. The Multifaceted Mechanisms of Estradiol and Estrogen Receptor Signaling. J. Biol. Chem. 2001, 276, 36869–36872. [Google Scholar] [CrossRef] [Green Version]
- Jänne, O.A.; Shan, L.-X. Structure and Function of the Androgen Receptor. Ann. N. Y. Acad. Sci. 1991, 626, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Shiina, H.; Kawano, H.; Sato, T.; Kato, S. Androgen receptor functions in male and female physiology. J. Steroid Biochem. Mol. Biol. 2008, 109, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Wyce, A.; Bai, Y.; Nagpal, S.; Thompson, C.C. Research Resource: The Androgen Receptor Modulates Expression of Genes with Critical Roles in Muscle Development and Function. Mol. Endocrinol. 2010, 24, 1665–1674. [Google Scholar] [CrossRef] [Green Version]
- Mhaouty-Kodja, S. Role of the androgen receptor in the central nervous system. Mol. Cell. Endocrinol. 2018, 465, 103–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Hara, L.; Smith, L.B. Androgen receptor roles in spermatogenesis and infertility. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 595–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mooradian, A.D.; Morley, J.E.; Korenman, S.G. Biological Actions of Androgens. Endocr. Rev. 1987, 8, 1–28. [Google Scholar] [CrossRef]
- Lynch, C.; Sakamuru, S.; Huang, R.; Stavreva, D.A.; Varticovski, L.; Hager, G.L.; Judson, R.S.; Houck, K.A.; Kleinstreuer, N.C.; Casey, W.; et al. Identifying environmental chemicals as agonists of the androgen receptor by using a quantitative high-throughput screening platform. Toxicology 2017, 385, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.; Liang, T.; Fang, S.; Castañeda, E.; Shao, T.-C. Steroid structure and androgenic activity specificities involved in the receptor binding and nuclear retention of various androgens. J. Biol. Chem. 1973, 248, 6154–6162. [Google Scholar] [CrossRef]
- Janne, O.A.; US Department of Health and Human Services; National Institute on Drug Abuse. Androgen Interaction through Multiple Steroid Receptors: (496332006-010). In PsycEXTRA Dataset; American Psychological Association (APA): Washington, DC, USA, 1990. [Google Scholar] [CrossRef]
- Shaffer, P.L.; Jivan, A.; Dollins, D.E.; Claessens, F.; Gewirth, D.T. Structural basis of androgen receptor binding to selective androgen response elements. Proc. Natl. Acad. Sci. USA 2004, 101, 4758–4763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brent, G.A. Mechanisms of thyroid hormone action. J. Clin. Investig. 2012, 122, 3035–3043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, S.-Y.; Leonard, J.L.; Davis, P.J. Molecular aspects of thyroid hormone actions. Endocr. Rev. 2010, 31, 139–170. [Google Scholar] [CrossRef] [Green Version]
- Anyetei-Anum, C.S.; Roggero, V.R.; Allison, L.A. Thyroid hormone receptor localization in target tissues. J. Endocrinol. 2018, 237, R19–R34. [Google Scholar] [CrossRef] [PubMed]
- Mitsuhashi, T.; Tennyson, G.E.; Nikodem, V.M. Alternative splicing generates messages encoding rat c-erbA proteins that do not bind thyroid hormone. Proc. Natl. Acad. Sci. USA 1988, 85, 5804–5808. [Google Scholar] [CrossRef] [Green Version]
- Williams, G.R. Cloning and Characterization of Two Novel Thyroid Hormone Receptor Isoforms. Mol. Cell. Biol. 2000, 20, 14. [Google Scholar] [CrossRef] [Green Version]
- Astapova, I.; Hollenberg, A.N. The in vivo role of nuclear receptor corepressors in thyroid hormone action. Biochim. Biophys. Acta BBA Gen. Subj. 2013, 1830, 3876–3881. [Google Scholar] [CrossRef] [Green Version]
- Hong, H.; Kohli, K.; Garabedian, M.J.; Stallcup, M.R. GRIP1, a transcriptional coactivator for the AF-2 transactivation domain of steroid, thyroid, retinoid, and vitamin D receptors. Mol. Cell. Biol. 1997, 17, 2735–2744. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.-Y.; Brent, G.A. Thyroid hormone crosstalk with nuclear receptor signaling in metabolic regulation. Trends Endocrinol. Metab. 2010, 21, 166–173. [Google Scholar] [CrossRef] [Green Version]
- Williams, G.R. Neurodevelopmental and Neurophysiological Actions of Thyroid Hormone. J. Neuroendocrinol. 2008, 20, 784–794. [Google Scholar] [CrossRef] [PubMed]
- Mullur, R.; Liu, Y.-Y.; Brent, G.A. Thyroid Hormone Regulation of Metabolism. Physiol. Rev. 2014, 94, 355–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oetting, A.; Yen, P.M. New insights into thyroid hormone action. Best Pract. Res. Clin. Endocrinol. Metab. 2007, 21, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Braun, J.M. Early-life exposure to EDCs: Role in childhood obesity and neurodevelopment. Nat. Rev. Endocrinol. 2017, 13, 161–173. [Google Scholar] [CrossRef] [Green Version]
- Marí-Bauset, S.; Donat-Vargas, C.; Llópis-González, A.; Marí-Sanchis, A.; Peraita-Costa, I.; Llopis-Morales, J.; Morales-Suárez-Varela, M. Endocrine Disruptors and Autism Spectrum Disorder in Pregnancy: A Review and Evaluation of the Quality of the Epidemiological Evidence. Children 2018, 5, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howard, S.G. Developmental Exposure to Endocrine-disrupting Chemicals and Type 1 Diabetes Mellitus. Front. Endocrinol. 2018, 9, 513. [Google Scholar] [CrossRef]
- Lind, P.M.; Lind, L. Endocrine-disrupting chemicals and risk of diabetes: An evidence-based review. Diabetologia 2018, 61, 1495–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kajta, M.; Wójtowicz, A.K. Impact of endocrine-disrupting chemicals on neural development and the onset of neurological disorders. Pharmacol. Rep. PR 2013, 65, 1632–1639. [Google Scholar] [CrossRef]
- Webb, E.; Moon, J.; Dyrszka, L.; Rodriguez, B.; Cox, C.; Patisaul, H.; Bushkin, S.; London, E. Neurodevelopmental and neurological effects of chemicals associated with unconventional oil and natural gas operations and their potential effects on infants and children. Rev. Environ. Health 2018, 33, 3–29. [Google Scholar] [CrossRef] [Green Version]
- Zhu, B.; Zhao, G.; Yang, L.; Zhou, B. Tetrabromobisphenol A caused neurodevelopmental toxicity via disrupting thyroid hormones in zebrafish larvae. Chemosphere 2018, 197, 353–361. [Google Scholar] [CrossRef]
- Tyagi, P.; James-Todd, T.; Mínguez-Alarcón, L.; Ford, J.B.; Keller, M.; Petrozza, J.; Calafat, A.M.; Hauser, R.; Williams, P.L.; Bellavia, A. Identifying Windows of Susceptibility to Endocrine Disrupting Chemicals in Relation to Gestational Weight Gain among Pregnant Women Attending a Fertility Clinic. Environ. Res. 2021, 194, 110638. [Google Scholar] [CrossRef] [PubMed]
- Predieri, B.; Bruzzi, P.; Bigi, E.; Ciancia, S.; Madeo, S.F.; Lucaccioni, L.; Iughetti, L. Endocrine Disrupting Chemicals and Type 1 Diabetes. IJMS 2020, 21, 2937. [Google Scholar] [CrossRef] [PubMed]
- Terry, M.B.; Michels, K.B.; Brody, J.G.; Byrne, C.; Chen, S.; Jerry, D.J.; Malecki, K.M.C.; Martin, M.B.; Miller, R.L.; Neuhausen, S.L.; et al. Environmental Exposures during Windows of Susceptibility for Breast Cancer: A Framework for Prevention Research. Breast Cancer Res. 2019, 21, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.-C.; Chen, C.-H.; Guo, Y.L. Phthalate esters and childhood asthma: A systematic review and congener-specific meta-analysis. Environ. Pollut. 2017, 229, 655–660. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, S.; Masai, E.; Kamimura, N.; Takahashi, K.; Anderson, R.C.; Faisal, P.A. Phthalates impact human health: Epidemiological evidences and plausible mechanism of action. J. Hazard. Mater. 2017, 340, 360–383. [Google Scholar] [CrossRef] [PubMed]
- Paciência, I.; Rufo, J.C.; Silva, D.; Martins, C.; Mendes, F.; Farraia, M.; Delgado, L.; Fernandes, E.D.O.; Padrão, P.; Moreira, P.; et al. Exposure to indoor endocrine-disrupting chemicals and childhood asthma and obesity. Allergy 2019, 74, 1277–1291. [Google Scholar] [CrossRef] [PubMed]
- Omran, G.A.; Gaber, H.D.; Mostafa, N.A.M.; Abdel-Gaber, R.M.; Salah, E.A. Potential hazards of bisphenol A exposure to semen quality and sperm DNA integrity among infertile men. Reprod. Toxicol. 2018, 81, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Humblet, O.; Birnbaum, L.; Rimm, E.; Mittleman, M.A.; Hauser, R. Dioxins and Cardiovascular Disease Mortality. Environ. Health Perspect. 2008, 116, 1443–1448. [Google Scholar] [CrossRef]
- Hu, W.-Y.; Shi, G.-B.; Hu, D.-P.; Nelles, J.L.; Prins, G.S. Actions of estrogens and endocrine-disrupting chemicals on human prostate stem/progenitor cells and prostate cancer risk. Mol. Cell. Endocrinol. 2012, 354, 63–73. [Google Scholar] [CrossRef] [Green Version]
- Rachoń, D. Endocrine-disrupting chemicals (EDCs) and female cancer: Informing the patients. Rev. Endocr. Metab. Disord. 2015, 16, 359–364. [Google Scholar] [CrossRef] [Green Version]
- Tan, M.E.; Li, J.; Xu, H.E.; Melcher, K.; Yong, E. Androgen receptor: Structure, role in prostate cancer and drug discovery. Acta Pharmacol. Sin. 2015, 36, 3–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moeller, L.C.; Führer, D. Thyroid hormone, thyroid hormone receptors, and cancer: A clinical perspective. Endocr. Relat. Cancer 2013, 20, R19–R29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calsolaro, V.; Pasqualetti, G.; Niccolai, F.; Caraccio, N.; Monzani, F. Thyroid Disrupting Chemicals. IJMS 2017, 18, 2583. [Google Scholar] [CrossRef] [Green Version]
- Okamura, K.; Taurog, A.; Krulich, L. Strain Differences among Rats in Response to Remington Iodine-Deficient Diets. Endocrinology 1981, 109, 458–463. [Google Scholar] [CrossRef] [PubMed]
- Penhale, W.J.; Farmer, A.; Irvine, W.J. Thyroiditis in T cell-depleted rats. Influence of strain, radiation dose, adjuvants and antilymphocyte serum. Clin. Exp. Immunol. 1975, 21, 362–375. [Google Scholar]
- Larsen, M.C.; Brake, P.B.; Parmar, D.; Jefcoate, C.R. The Induction of Five Rat Hepatic P450 Cytochromes by Phenobarbital and Similarly Acting Compounds Is Regulated by a Sexually Dimorphic, Dietary-Dependent Endocrine Factor That Is Highly Strain Specific. Arch. Biochem. Biophys. 1994, 315, 24–34. [Google Scholar] [CrossRef]
- Ikegwuonu, F.I.; Ganem, L.G.; Larsen, M.C.; Shen, X.; Jefcoate, C.R. The Regulation by Gender, Strain, Dose, and Feeding Status of the Induction of Multiple Forms of Cytochrome P450 Isozymes in Rat Hepatic Microsomes by 2,4,5,2′,4′,5′-Hexachlorobiphenyl. Toxicol. Appl. Pharmacol. 1996, 139, 33–41. [Google Scholar] [CrossRef]
- Simanainen, U. Adult 2,3,7,8-Tetrachlorodibenzo-p-Dioxin (TCDD) Exposure and Effects on Male Reproductive Organs in Three Differentially TCDD-Susceptible Rat Lines. Toxicol. Sci. 2004, 81, 401–407. [Google Scholar] [CrossRef]
- Simanainen, U.; Haavisto, T.; Tuomisto, J.T.; Paranko, J.; Toppari, J.; Tuomisto, J.; Peterson, R.E.; Viluksela, M. Pattern of Male Reproductive System Effects After in Utero and Lactational 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) Exposure in Three Differentially TCDD-Sensitive Rat Lines. Toxicol. Sci. 2004, 80, 101–108. [Google Scholar] [CrossRef] [Green Version]
- Spearow, J.L. Genetic Variation in Susceptibility to Endocrine Disruption by Estrogen in Mice. Science 1999, 285, 1259–1261. [Google Scholar] [CrossRef]
- Spearow, J.L.; O’Henley, P.; Doemeny, P.; Sera, R.; Leffler, R.; Sofos, T.; Barkley, M. Genetic variation in physiological sensitivity to estrogen in mice. APMIS 2001, 109, S289–S297. [Google Scholar] [CrossRef]
- Cummings, A.M.; Kavlock, R.J. Gene–Environment Interactions: A Review of Effects on Reproduction and Development. Crit. Rev. Toxicol. 2004, 34, 461–485. [Google Scholar] [CrossRef] [PubMed]
- Duan, Q.L.; Du, R.; Lasky-Su, J.; Klanderman, B.J.; Partch, A.B.; Peters, S.P.; Irvin, C.G.; Hanrahan, J.P.; Lima, J.J.; Blake, K.V.; et al. A polymorphism in the thyroid hormone receptor gene is associated with bronchodilator response in asthmatics. Pharmacogenomics J. 2013, 13, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Kalikiri, M.K.; Mamidala, M.P.; Rao, A.N.; Rajesh, V. Analysis and functional characterization of sequence variations in ligand binding domain of thyroid hormone receptors in autism spectrum disorder (ASD) patients: Thyroid Hormone Receptor Mutations in ASD Patients. Autism Res. 2017, 10, 1919–1928. [Google Scholar] [CrossRef] [PubMed]
- Alberobello, A.T.; Congedo, V.; Liu, H.; Cochran, C.; Skarulis, M.C.; Forrest, D.; Celi, F.S. An intronic SNP in the thyroid hormone receptor b gene is associated with pituitary cell-specific over-expression of a mutant thyroid hormone receptor b2 (R338W) in the index case of pituitary-selective resistance to thyroid hormone. J. Transl. Med. 2011, 7, 144. [Google Scholar] [CrossRef] [Green Version]
- Safer, J.D. The Thyroid Hormone Receptor-Gene Mutation R383H Is Associated with Isolated Central Resistance to Thyroid Hormone. J. Clin. Endocrinol. Metab. 1999, 84, 3099–3109. [Google Scholar] [CrossRef]
- Kiss-Toth, E.; Harlock, E.; Lath, D.; Quertermous, T.; Wilkinson, J.M. A TNF Variant that Associates with Susceptibility to Musculoskeletal Disease Modulates Thyroid Hormone Receptor Binding to Control Promoter Activation. PLoS ONE 2013, 8, e76034. [Google Scholar] [CrossRef] [Green Version]
- Puzianowska-Kuznicka, M.; Krystyniak, A.; Madej, A.; Cheng, S.-Y.; Nauman, J. Functionally Impaired TR Mutants Are Present in Thyroid Papillary Cancer. J. Clin. Endocrinol. Metab. 2002, 87, 1120–1128. [Google Scholar] [CrossRef]
- Szymański, Ł.; Matak, D.; Bartnik, E.; Szczylik, C.; Czarnecka, A.M. Thyroid Hormones as Renal Cell Cancer Regulators. J. Signal Transduct. 2016, 2016, 1362407. [Google Scholar] [CrossRef]
- Lin, K.-H.; Shieh, H.-Y.; Chen, S.-L.; Hsu, H.-C. Expression of mutant thyroid hormone nuclear receptors in human hepatocellular carcinoma cells. Mol. Carcinog. 1999, 26, 53–61. [Google Scholar] [CrossRef]
- Erf, G.F.; Marsh, J.A. Effect of dietary triiodothyronine on mixed-lymphocyte responsiveness in young male chickens. Dev. Comp. Immunol. 1989, 13, 177–186. [Google Scholar] [CrossRef]
- Liu, Y.; Qu, K.; Hai, Y.; Li, X.; Zhao, L.; Zhao, C. SNP mutations occurring in thyroid hormone receptor influenced individual susceptibility to triiodothyronine: Molecular dynamics and site-directed mutagenesis approaches. J. Cell. Biochem. 2018, 119, 2604–2616. [Google Scholar] [CrossRef]
- Darnerud, P.O. Brominated flame retardants as possible endocrine disrupters. Int. J. Androl. 2008, 31, 152–160. [Google Scholar] [CrossRef]
- Legler, J. Are brominated flame retardants endocrine disruptors? Environ. Int. 2003, 29, 879–885. [Google Scholar] [CrossRef]
- Eggesbø, M.; Thomsen, C.; Jørgensen, J.V.; Becher, G.; Odland, J.Ø.; Longnecker, M.P. Associations between brominated flame retardants in human milk and thyroid-stimulating hormone (TSH) in neonates. Environ. Res. 2011, 111, 737–743. [Google Scholar] [CrossRef] [Green Version]
- Kronborg, T.M.; Hansen, J.F.; Rasmussen, Å.K.; Vorkamp, K.; Nielsen, C.H.; Frederiksen, M.; Hofman-Bang, J.; Hahn, C.H.; Ramhøj, L.; Feldt-Rasmussen, U. The flame-retardant DE-71 (a mixture of polybrominated diphenyl ethers) inhibits human differentiated thyroid cell function in vitro. PLoS ONE 2017, 12, e0179858. [Google Scholar] [CrossRef] [Green Version]
- Truong, L.; Marvel, S.; Reif, D.M.; Thomas, D.G.; Pande, P.; Dasgupta, S.; Simonich, M.T.; Waters, K.M.; Tanguay, R.L. The multi-dimensional embryonic zebrafish platform predicts flame retardant bioactivity. Reprod. Toxicol. 2020, 96, 359–369. [Google Scholar] [CrossRef]
- Balik-Meisner, M.; Truong, L.; Scholl, E.H.; La Du, J.K.; Tanguay, R.L.; Reif, D.M. Elucidating Gene-by-Environment Interactions Associated with Differential Susceptibility to Chemical Exposure. Environ. Health Perspect. 2018, 126, 067010. [Google Scholar] [CrossRef]
- Gelissen, I.C.; McLachlan, A.J. The pharmacogenomics of statins. Pharmacol. Res. 2014, 88, 99–106. [Google Scholar] [CrossRef]
- Kitzmiller, J.; Mikulik, E.; Dauki, A.; Mukherjee, C.; Luzum, J. Pharmacogenomics of statins: Understanding susceptibility to adverse effects. Pharm. Pers. Med. 2016, 9, 97–106. [Google Scholar] [CrossRef] [Green Version]
- Davis, A.P.; Grondin, C.J.; Johnson, R.J.; Sciaky, D.; McMorran, R.; Wiegers, J.; Wiegers, T.C.; Mattingly, C.J. The Comparative Toxicogenomics Database: Update 2019. Nucleic Acids Res. 2019, 47, D948–D954. [Google Scholar] [CrossRef]
- Crump, D.; Porter, E.; Egloff, C.; Williams, K.L.; Letcher, R.J.; Gauthier, L.T.; Kennedy, S.W. 1,2-Dibromo-4-(1,2-dibromoethyl)-cyclohexane and tris(methylphenyl) phosphate cause significant effects on development, mRNA expression, and circulating bile acid concentrations in chicken embryos. Toxicol. Appl. Pharmacol. 2014, 277, 279–287. [Google Scholar] [CrossRef]
- Kitamura, S.; Kato, T.; Iida, M.; Jinno, N.; Suzuki, T.; Ohta, S.; Fujimoto, N.; Hanada, H.; Kashiwagi, K.; Kashiwagi, A. Anti-thyroid hormonal activity of tetrabromobisphenol A, a flame retardant, and related compounds: Affinity to the mammalian thyroid hormone receptor, and effect on tadpole metamorphosis. Life Sci. 2005, 76, 1589–1601. [Google Scholar] [CrossRef]
- Thomas, M.B.; Stapleton, H.M.; Dills, R.L.; Violette, H.D.; Christakis, D.A.; Sathyanarayana, S. Demographic and dietary risk factors in relation to urinary metabolites of organophosphate flame retardants in toddlers. Chemosphere 2017, 185, 918–925. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Lam, J.C.; Han, J.; Wang, X.; Guo, Y.; Lam, P.K.; Zhou, B. Developmental exposure to the organophosphorus flame-retardant tris(1,3-dichloro-2-propyl) phosphate: Estrogenic activity, endocrine disruption and reproductive effects on zebrafish. Aquat. Toxicol. 2015, 160, 163–171. [Google Scholar] [CrossRef]
- Ma, Z.; Yu, Y.; Tang, S.; Liu, H.; Su, G.; Xie, Y.; Giesy, J.P.; Hecker, M.; Yu, H. Differential modulation of expression of nuclear receptor mediated genes by tris(2-butoxyethyl) phosphate (TBOEP) on early life stages of zebrafish (Danio rerio). Aquat. Toxicol. 2015, 169, 196–203. [Google Scholar] [CrossRef]
- Sun, H.; Xu, L.-C.; Chen, J.-F.; Song, L.; Wang, X.-R. Effect of bisphenol a, tetrachlorobisphenol A and pentachlorophenol on the transcriptional activities of androgen receptor-mediated reporter gene. Food Chem. Toxicol. 2006, 44, 1916–1921. [Google Scholar] [CrossRef]
- Zhao, G.; Marceau, R.; Zhang, D.; Tzeng, J.-Y. Assessing Gene-Environment Interactions for Common and Rare Variants with Binary Traits Using Gene-Trait Similarity Regression. Genetics 2015, 199, 695–710. [Google Scholar] [CrossRef]
- Zhu, Y.; Ma, X.; Su, G.; Yu, L.; Letcher, R.J.; Hou, J.; Yu, H.; Giesy, J.P.; Liu, C. Environmentally Relevant Concentrations of the Flame-Retardant Tris(1,3-dichloro-2-propyl) Phosphate Inhibit Growth of Female Zebrafish and Decrease Fecundity. Environ. Sci. Technol. 2015, 49, 14579–14587. [Google Scholar] [CrossRef]
- Dishaw, L.V.; JMacaulay, L.; Roberts, S.C.; Stapleton, H.M. Exposures, mechanisms, and impacts of endocrine-active flame retardants. Curr. Opin. Pharmacol. 2014, 19, 125–133. [Google Scholar] [CrossRef] [Green Version]
- Kojima, H.; Takeuchi, S.; Itoh, T.; Iida, M.; Kobayashi, S.; Yoshida, T. In vitro endocrine disruption potential of organophosphate flame retardants via human nuclear receptors. Toxicology 2013, 314, 76–83. [Google Scholar] [CrossRef]
- National Toxicology Program. NTP Toxicology and Carcinogenesis Studies of Tetrakis(hydroxymethyl)phosphonium sulfate (THPS) (CAS No. 55566-30-8) and Tetrakis(hydroxymethyl)phosphonium chloride (THPC) (CAS No. 124-64-1) in F344/N Rats and B6C3F1 Mice (Gavage Studies). Natl. Toxicol. Program Tech. Rep. Ser. 1987, 296, 1–290. [Google Scholar]
- Böckers, M.; Paul, N.W.; Efferth, T. Organophosphate ester tri-o-cresyl phosphate interacts with estrogen receptor α in MCF-7 breast cancer cells promoting cancer growth. Toxicol. Appl. Pharmacol. 2020, 395, 114977. [Google Scholar] [CrossRef]
- Chupeau, Z.; Bonvallot, N.; Mercier, F.; Le Bot, B.; Chevrier, C.; Glorennec, P. Organophosphorus Flame Retardants: A Global Review of Indoor Contamination and Human Exposure in Europe and Epidemiological Evidence. Int. J. Environ. Res. Public. Health 2020, 17, 6713. [Google Scholar] [CrossRef]
- Xu, Q.; Wu, D.; Dang, Y.; Yu, L.; Liu, C.; Wang, J. Reproduction impairment and endocrine disruption in adult zebrafish (Danio rerio) after waterborne exposure to TBOEP. Aquat. Toxicol. 2017, 182, 163–171. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, W.; Li, A.; Song, M. Tetrachlorobisphenol A induced immunosuppression and uterine injury in mice. Ecotoxicol. Environ. Saf. 2021, 207, 111527. [Google Scholar] [CrossRef] [PubMed]
- Reers, A.R.; Eng, M.L.; Williams, T.D.; Elliott, J.E.; Cox, M.E.; Beischlag, T.V. The Flame-Retardant Tris(1,3-dichloro-2-propyl) Phosphate Represses Androgen Signaling in Human Prostate Cancer Cell Lines: EFFECT OF TDCIPP ON ANDROGEN SIGNALING. J. Biochem. Mol. Toxicol. 2016, 30, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, Y.; Dong, S.; Han, L.; Guo, R.; Fu, Y.; Zhang, S.; Chen, J. The risk and impact of organophosphate esters on the development of female-specific cancers: Comparative analysis of patients with benign and malignant tumors. J. Hazard. Mater. 2021, 404, 124020. [Google Scholar] [CrossRef]
- Sugeng, E.J.; de Cock, M.; Leonards, P.E.G.; van de Bor, M. Toddler behavior, the home environment, and flame-retardant exposure. Chemosphere 2020, 252, 126588. [Google Scholar] [CrossRef]
- Hu, L.; Peng, T.; Huang, J.; Su, T.; Luo, R.; Zheng, Y.; Zhong, Z.; Yu, P.; Nie, K.; Zheng, L. Tri-ortho-cresyl phosphate (TOCP) induced ovarian failure in mice is related to the Hippo signaling pathway disruption. Reprod. Toxicol. 2019, 83, 21–27. [Google Scholar] [CrossRef]
- Egloff, C.; Crump, D.; Porter, E.; Williams, K.L.; Letcher, R.J.; Gauthier, L.T.; Kennedy, S.W. Tris(2-butoxyethyl)phosphate and triethyl phosphate alter embryonic development, hepatic mRNA expression, thyroid hormone levels, and circulating bile acid concentrations in chicken embryos. Toxicol. Appl. Pharmacol. 2014, 279, 303–310. [Google Scholar] [CrossRef] [PubMed]
- U.S. Environmental Protection Agency. CompTox Chemicals Dashboard. Available online: https://comptox.epa.gov/dashboard/DTXSID0021331 (accessed on 23 June 2020).
- U.S. Environmental Protection Agency. CompTox Chemicals Dashboard. Available online: https://comptox.epa.gov/dashboard/DTXSID4021391 (accessed on 23 June 2020).
- U.S. Environmental Protection Agency. CompTox Chemicals Dashboard. Available online: https://comptox.epa.gov/dashboard/DTXSID5021330 (accessed on 23 June 2020).
- U.S. Environmental Protection Agency. CompTox Chemicals Dashboard. Available online: https://comptox.epa.gov/dashboard/DTXSID5021413 (accessed on 23 June 2020).
- U.S. Environmental Protection Agency. CompTox Chemicals Dashboard. Available online: https://comptox.epa.gov/dashboard/DTXSID5021758 (accessed on 23 June 2020).
- U.S. Environmental Protection Agency. CompTox Chemicals Dashboard. Available online: https://comptox.epa.gov/dashboard/DTXSID3021770 (accessed on 23 June 2020).
- U.S. Environmental Protection Agency. CompTox Chemicals Dashboard. Available online: https://comptox.epa.gov/dashboard/DTXSID3024861 (accessed on 23 June 2020).
- U.S. Environmental Protection Agency. CompTox Chemicals Dashboard. Available online: https://comptox.epa.gov/dashboard/DTXSID4028880 (accessed on 23 June 2020).
- U.S. Environmental Protection Agency. CompTox Chemicals Dashboard. Available online: https://comptox.epa.gov/dashboard/DTXSID4030047 (accessed on 23 June 2020).
- U.S. Environmental Protection Agency. CompTox Chemicals Dashboard. Available online: https://comptox.epa.gov/dashboard/DTXSID6032192 (accessed on 23 June 2020).
- Huang, T.; Hu, F.B. Gene-environment interactions and obesity: Recent developments and future directions. BMC Med. Genom. 2015, 8, S2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.-U.; Kim, J.D.; Park, C.-S. Gene-Environment Interactions in Asthma: Genetic and Epigenetic Effects. Yonsei Med. J. 2015, 56, 877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovely, C.; Rampersad, M.; Fernandes, Y.; Eberhart, J. Gene-environment interactions in development and disease: Gene-environment interactions in development and disease. Wiley Interdiscip. Rev. Dev. Biol. 2017, 6, e247. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-W.; Park, K.; Kang, R.J.; Gonzales, E.L.; Oh, H.A.; Seung, H.; Ko, M.J.; Cheong, J.H.; Chung, C.; Shin, C.Y. Gene-environment interaction counterbalances social impairment in mouse models of autism. Sci. Rep. 2019, 9, 11490. [Google Scholar] [CrossRef]
- Heianza, Y.; Qi, L. Impact of Genes and Environment on Obesity and Cardiovascular Disease. Endocrinology 2019, 160, 81–100. [Google Scholar] [CrossRef] [PubMed]
- Krauss, R.S.; Hong, M. Gene–Environment Interactions and the Etiology of Birth Defects. In Current Topics in Developmental Biology; Elsevier: Amsterdam, The Netherlands, 2016; Volume 116, pp. 569–580. [Google Scholar]
- Rudolph, A.; Chang-Claude, J.; Schmidt, M.K. Gene–environment interaction and risk of breast cancer. Br. J. Cancer 2016, 114, 125–133. [Google Scholar] [CrossRef] [Green Version]
- Simonds, N.I.; Ghazarian, A.A.; Pimentel, C.B.; Schully, S.D.; Ellison, G.L.; Gillanders, E.M.; Mechanic, L.E. Review of the Gene-Environment Interaction Literature in Cancer: What Do We Know?: Gene-Environment Interaction Literature Review. Genet. Epidemiol. 2016, 40, 356–365. [Google Scholar] [CrossRef] [Green Version]
- Zafarmand, M.H.; Tajik, P.; Spijker, R.; Agyemang, C. Gene-environment interaction on the risk of type 2 diabetes among ethnic minority populations living in Europe and North America: A systematic review. Curr. Diabetes Rev. 2020, 16, 457–470. [Google Scholar] [CrossRef] [PubMed]
- McAllister, K.; Mechanic, L.E.; Amos, C.; Aschard, H.; Blair, I.A.; Chatterjee, N.; Conti, D.; Gauderman, W.J.; Hsu, L.; Hutter, C.M.; et al. Current Challenges and New Opportunities for Gene-Environment Interaction Studies of Complex Diseases. Am. J. Epidemiol. 2017, 186, 753–761. [Google Scholar] [CrossRef] [Green Version]
- Simon, P.H.G.; Sylvestre, M.-P.; Tremblay, J.; Hamet, P. Key Considerations and Methods in the Study of Gene–Environment Interactions. Am. J. Hypertens. 2016, 29, 891–899. [Google Scholar] [CrossRef] [Green Version]
- Gauderman, W.J.; Mukherjee, B.; Aschard, H.; Hsu, L.; Lewinger, J.P.; Patel, C.J.; Witte, J.S.; Amos, C.; Tai, C.G.; Conti, D.; et al. Update on the State of the Science for Analytical Methods for Gene-Environment Interactions. Am. J. Epidemiol. 2017, 186, 762–770. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Lewinger, J.P.; Conti, D.; Morrison, J.L.; Gauderman, W.J. Detecting Gene-Environment Interactions for a Quantitative Trait in a Genome-Wide Association Study: Detecting Gene-Environment Interactions for a Quantitative. Genet. Epidemiol. 2016, 40, 394–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basson, J.; Sung, Y.J.; de Las Fuentes, L.; Schwander, K.L.; Vazquez, A.; Rao, D.C. Three Approaches to Modeling Gene-Environment Interactions in Longitudinal Family Data: Gene-Smoking Interactions in Blood Pressure: Gene-Smoking Interactions in Blood Pressure. Genet. Epidemiol. 2016, 40, 73–80. [Google Scholar] [CrossRef] [Green Version]
- Sitlani, C.M.; Dupuis, J.; Rice, K.M.; Sun, F.; Pitsillides, A.N.; Cupples, L.A.; Psaty, B.M. Genome-wide gene–environment interactions on quantitative traits using family data. Eur. J. Hum. Genet. 2016, 24, 1022–1028. [Google Scholar] [CrossRef] [Green Version]
- Meisner, A.; Kundu, P.; Chatterjee, N. Case-Only Analysis of Gene-Environment Interactions Using Polygenic Risk Scores. Am. J. Epidemiol. 2019, 188, 2013–2020. [Google Scholar] [CrossRef] [PubMed]
- Thompson, E.A. Genetic epidemiology: A review of the statistical basis. Stat. Med. 1986, 5, 291–302. [Google Scholar] [CrossRef]
- Wu, C.; Jiang, Y.; Ren, J.; Cui, Y.; Ma, S. Dissecting gene-environment interactions: A penalized robust approach accounting for hierarchical structures. Stat. Med. 2018, 37, 437–456. [Google Scholar] [CrossRef]
- Moore, R.; Casale, F.P.; Bonder, M.J.; Horta, D.; Franke, L.; Barroso, I.; Stegle, O. A linear mixed-model approach to study multivariate gene–environment interactions. Nat. Genet. 2019, 51, 180–186. [Google Scholar] [CrossRef]
- Coombes, B.J.; Biernacka, J.M. Application of the parametric bootstrap for gene-set analysis of gene–environment interactions. Eur. J. Hum. Genet. 2018, 26, 1679–1686. [Google Scholar] [CrossRef]
- Shi, G.; Nehorai, A. Robustness of meta-analyses in finding gene × environment interactions. PLoS ONE 2017, 12, e0171446. [Google Scholar]
- Piegorsch, W.W.; Weinberg, C.R.; Taylor, J.A. Non-hierarchical logistic models and case-only designs for assessing susceptibility in population-based case-control studies. Stat. Med. 1994, 13, 153–162. [Google Scholar] [CrossRef]
- Chatterjee, N.; Carroll, R.J. Semiparametric maximum likelihood estimation exploiting gene-environment independence in case-control studies. Biometrika 2005, 92, 399–418. [Google Scholar] [CrossRef]
- Murcray, C.E.; Lewinger, J.P.; Gauderman, W.J. Gene-Environment Interaction in Genome-Wide Association Studies. Am. J. Epidemiol. 2008, 169, 219–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, B.; Chatterjee, N. Exploiting Gene-Environment Independence for Analysis of Case-Control Studies: An Empirical Bayes-Type Shrinkage Estimator to Trade-Off between Bias and Efficiency. Biometrics 2008, 64, 685–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umbach, D.M.; Weinberg, C.R. Designing and analysing case-control studies to exploit independence of genotype and exposure. Stat. Med. 1997, 16, 13. [Google Scholar] [CrossRef]
- Paré, G.; Cook, N.R.; Ridker, P.M.; Chasman, D.I. On the Use of Variance per Genotype as a Tool to Identify Quantitative Trait Interaction Effects: A Report from the Women’s Genome Health Study. PLoS Genet. 2010, 6, e1000981. [Google Scholar] [CrossRef] [PubMed]
- Kooperberg, C.; LeBlanc, M. Increasing the power of identifying gene × gene interactions in genome-wide association studies. Genet. Epidemiol. 2008, 32, 255–263. [Google Scholar] [CrossRef] [Green Version]
- Biernacka, J.M.; Jenkins, G.D.; Wang, L.; Moyer, A.M.; Fridley, B.L. Use of the gamma method for self-contained gene-set analysis of SNP data. Eur. J. Hum. Genet. 2012, 20, 565–571. [Google Scholar] [CrossRef]
- Fridley, B.L.; Biernacka, J.M. Gene set analysis of SNP data: Benefits, challenges, and future directions. Eur. J. Hum. Genet. 2011, 19, 837–843. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. Analysing biological pathways in genome-wide association studies. Nat. Rev. Genet. 2010, 11, 843–854. [Google Scholar] [CrossRef] [PubMed]
- De Leeuw, C.A.; Mooij, J.M.; Heskes, T.; Posthuma, D. MAGMA: Generalized Gene-Set Analysis of GWAS Data. PLoS Comput. Biol. 2015, 11, e1004219. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Lee, S.; Christiani, D.C.; Lin, X. Test for interactions between a genetic marker set and environment in generalized linear models. Biostatistics 2013, 14, 667–681. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Li, M.; Bucan, M. Pathway-Based Approaches for Analysis of Genomewide Association Studies. Am. J. Hum. Genet. 2007, 81, 1278–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzeng, J.-Y.; Zhang, D.; Pongpanich, M.; Smith, C.; McCarthy, M.I.; Sale, M.M.; Worrall, B.B.; Hsu, F.-C.; Thomas, D.C.; Sullivan, P.F. Studying Gene and Gene-Environment Effects of Uncommon and Common Variants on Continuous Traits: A Marker-Set Approach Using Gene-Trait Similarity Regression. Am. J. Hum. Genet. 2011, 89, 277–288. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.D.; Davis, J.R.; Aschard, H.; Battle, A.; Conti, D.; Du, M.; Eskin, E.; Fallin, M.D.; Hsu, L.; Kraft, P.; et al. Incorporation of Biological Knowledge into the Study of Gene-Environment Interactions. Am. J. Epidemiol. 2017, 186, 771–777. [Google Scholar] [CrossRef] [Green Version]
- Garcia, G.R.; Noyes, P.D.; Tanguay, R.L. Advancements in zebrafish applications for 21st century toxicology. Pharmacol. Ther. 2016, 161, 11–21. [Google Scholar] [CrossRef] [Green Version]
- Falconer, D.S. Introduction to Quantitative Genetics; Longman, Scientific; Technical: Burnt Mill, Harlow, UK; Wiley: New York, NY, USA, 1989. [Google Scholar]
- Tenesa, A.; Haley, C.S. The heritability of human disease: Estimation, uses and abuses. Nat. Rev. Genet. 2013, 14, 139–149. [Google Scholar] [CrossRef]
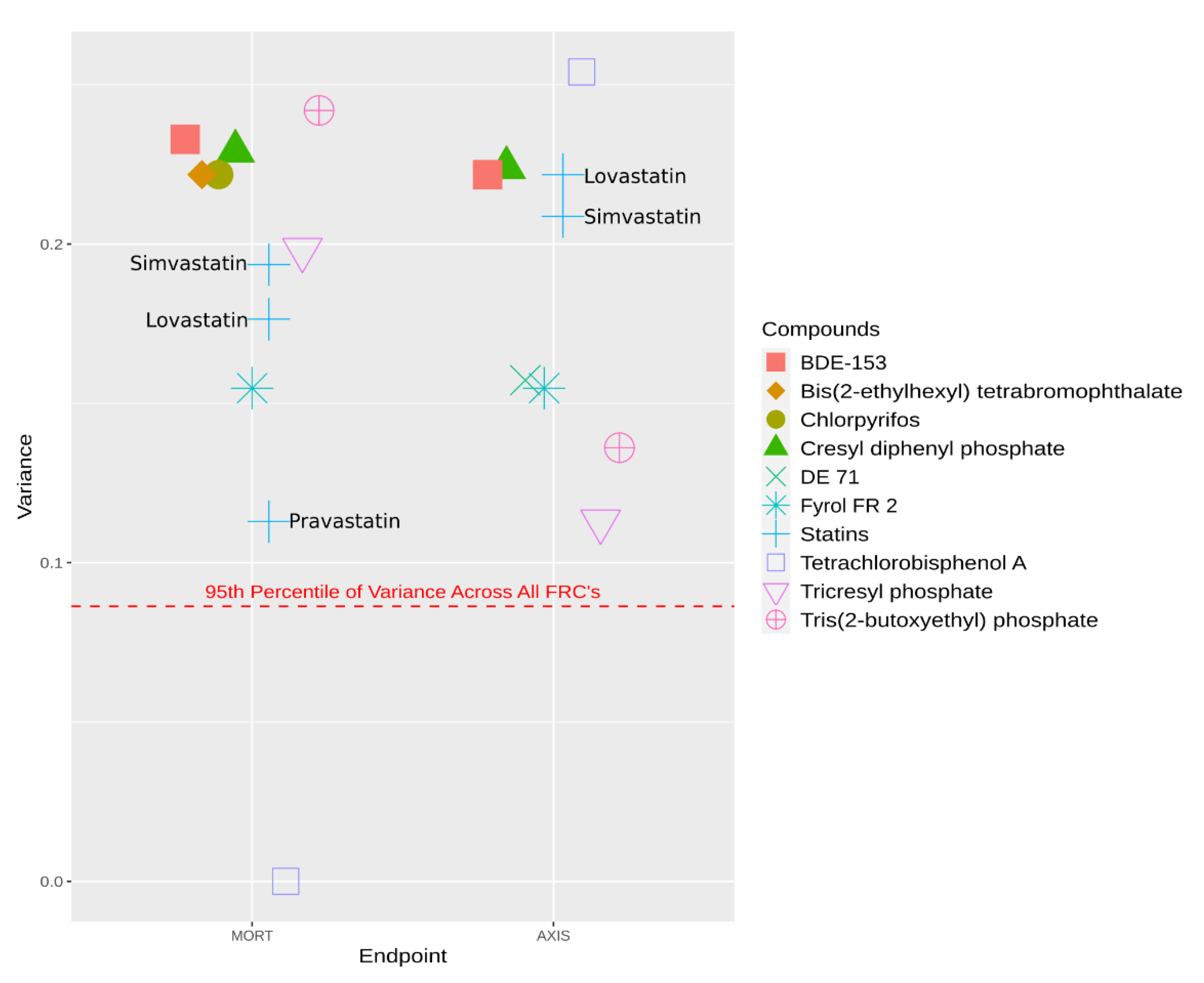


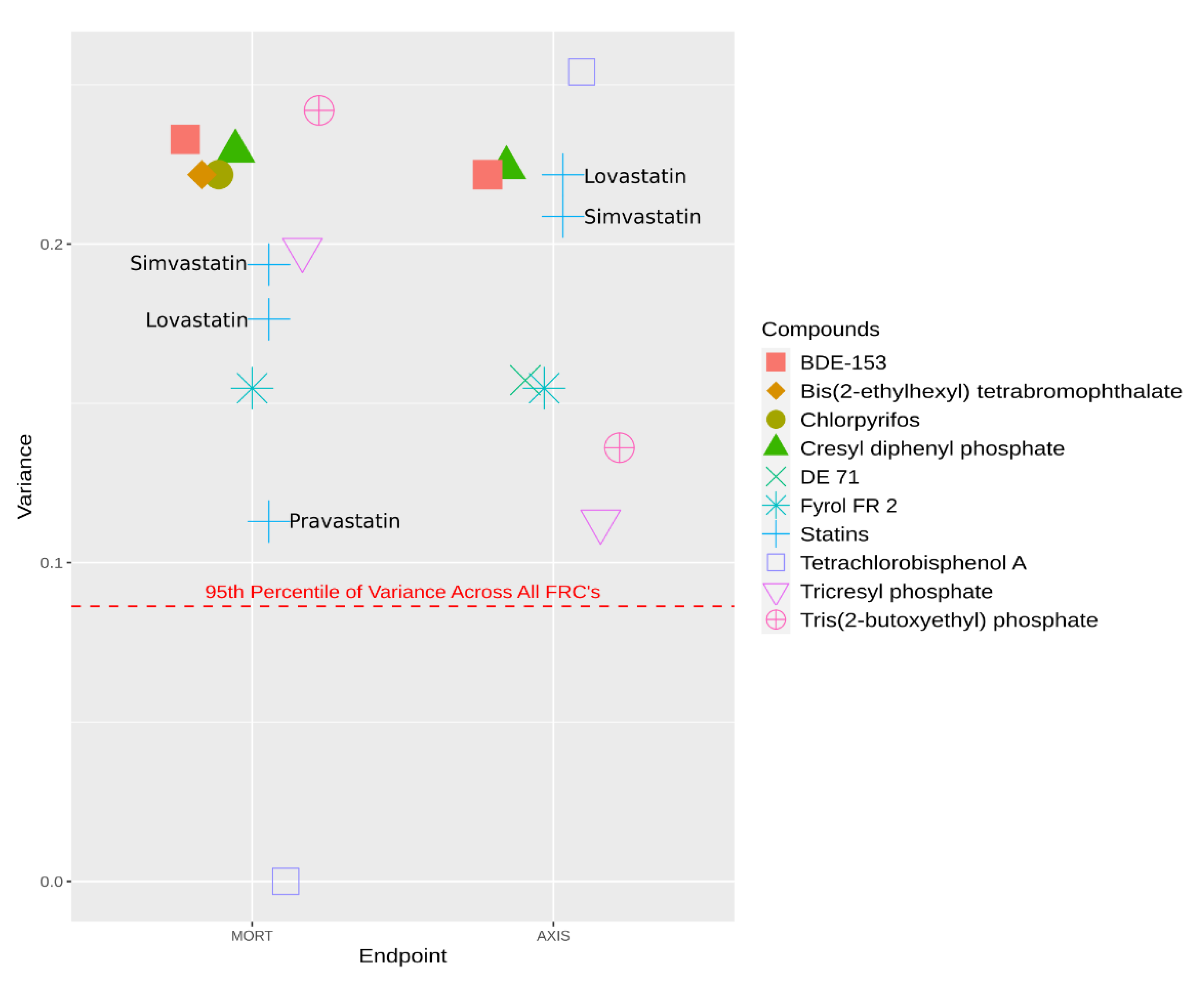{kind=link}
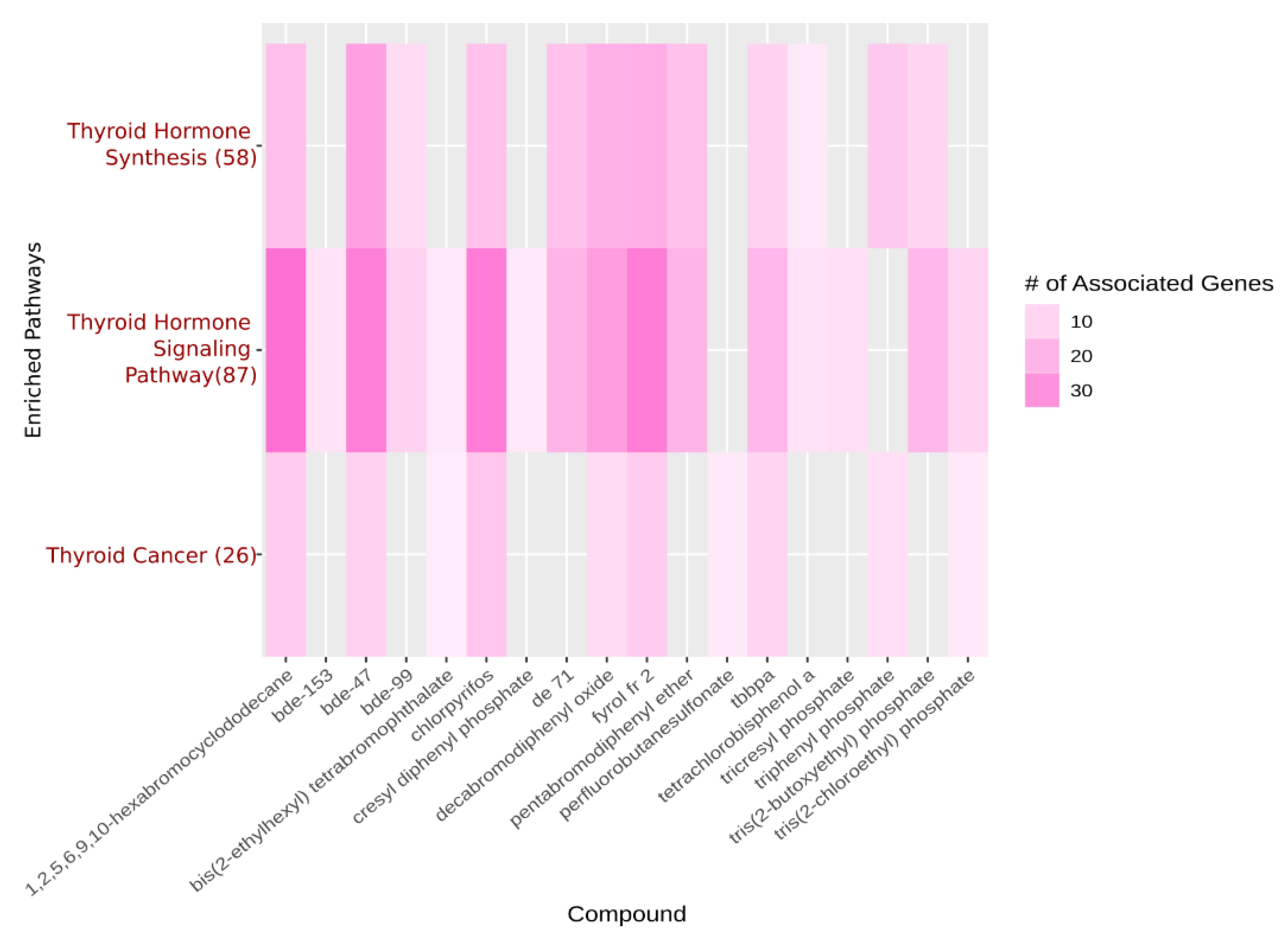{kind=link}
| Approach | Sub-Approaches | Use Cases | Citation |
|---|---|---|---|
| FAMILY-BASED | generalized estimating equations | Pedigree data is available and exposure mis-specification is a concern | Basson et al. (2016) [161], Sitlani et al. (2016) [162] |
| heirarchical linear model | Pedigree data is available and type I error is a concern | ||
| linear mixed effects model | Pedigree data is available and type I error is a concern | ||
| CASE-CONTROL | Penalized method with least absolute deviation loss function | When large genome-wide data is available and hierarchical “main effects, interactions” structure is a concern | Wu et al. (2018) [165] |
| Similarity-based regression | When large genome-wide data is available and rare-variants with binary phenotypes are being investigated | Zhao et al. (2015) [124] | |
| linear mixed model | When large genome-wide data is available and multiple exposure are being investigated | BIOS Consortium (2016) [166] | |
| Parametric bootstrap | Removes need for permutation tests when large genome-wide data is available | Gauderman et al. (2017) [159], Coombes et al. (2018) [167] | |
| CASE-ONLY | Traditional | Increases precision when independence between exposure and genetics can be assumed | Piegorsch et al. (1994) [169] |
| Multiple maximum-likelihood | Increases precision and relaxes independence assumption | Umbach and Weinberg (1997) [170], Chatterjee and Carrol (2005) [173], Mukherjee and Chatterjee (2008) [171] | |
| Bayesian | |||
| 2-STEP | Likelihood ratio to traditional | Increases power and reduces multiple testing correction in situations where traditional case-control or case-case only approaches would be appropriate | Murcray et al. (2008) [171], Pare (2010) [174], Kooperburg and LeBlanc (2008) [175] |
| Levene’s test to traditional | |||
| Marginal effects to traditional | |||
| Modified Pare et al. | Robust in situations with with multiple exposure and reduce type I error versus other 2-step approaches | Zhang et al. (2016) [160] | |
| Combined Pare and Kooperburg | |||
| GENE-SET ANALYSIS (GSA) | Traditional | Increases power versus more traditional approaches | Biernacka et al. (2012) [176] |
| With similarity regression | GSA when there are multiple covariates and opposite effects that may cancel each other out are a concern | Tzeng et al. (2013) [180] | |
| GESAT | Established method for user friendly GSA | Lin et al. (2013) [180] | |
| META-ANALYSIS | NA | Situations where investigators want to combine data from multiple studies to identify possible gene-environment interactions | Shi et al. (2017) [168] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wallis, D.J.; Truong, L.; La Du, J.; Tanguay, R.L.; Reif, D.M. Uncovering Evidence for Endocrine-Disrupting Chemicals That Elicit Differential Susceptibility through Gene-Environment Interactions. Toxics 2021, 9, 77. https://doi.org/10.3390/toxics9040077
Wallis DJ, Truong L, La Du J, Tanguay RL, Reif DM. Uncovering Evidence for Endocrine-Disrupting Chemicals That Elicit Differential Susceptibility through Gene-Environment Interactions. Toxics. 2021; 9(4):77. https://doi.org/10.3390/toxics9040077
Chicago/Turabian StyleWallis, Dylan J., Lisa Truong, Jane La Du, Robyn L. Tanguay, and David M. Reif. 2021. "Uncovering Evidence for Endocrine-Disrupting Chemicals That Elicit Differential Susceptibility through Gene-Environment Interactions" Toxics 9, no. 4: 77. https://doi.org/10.3390/toxics9040077
APA StyleWallis, D. J., Truong, L., La Du, J., Tanguay, R. L., & Reif, D. M. (2021). Uncovering Evidence for Endocrine-Disrupting Chemicals That Elicit Differential Susceptibility through Gene-Environment Interactions. Toxics, 9(4), 77. https://doi.org/10.3390/toxics9040077