
Engineered Dual Antioxidant Enzyme Complexes Targeting ICAM-1 on Brain Endothelium Reduce Brain Injury-Associated Neuroinflammation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
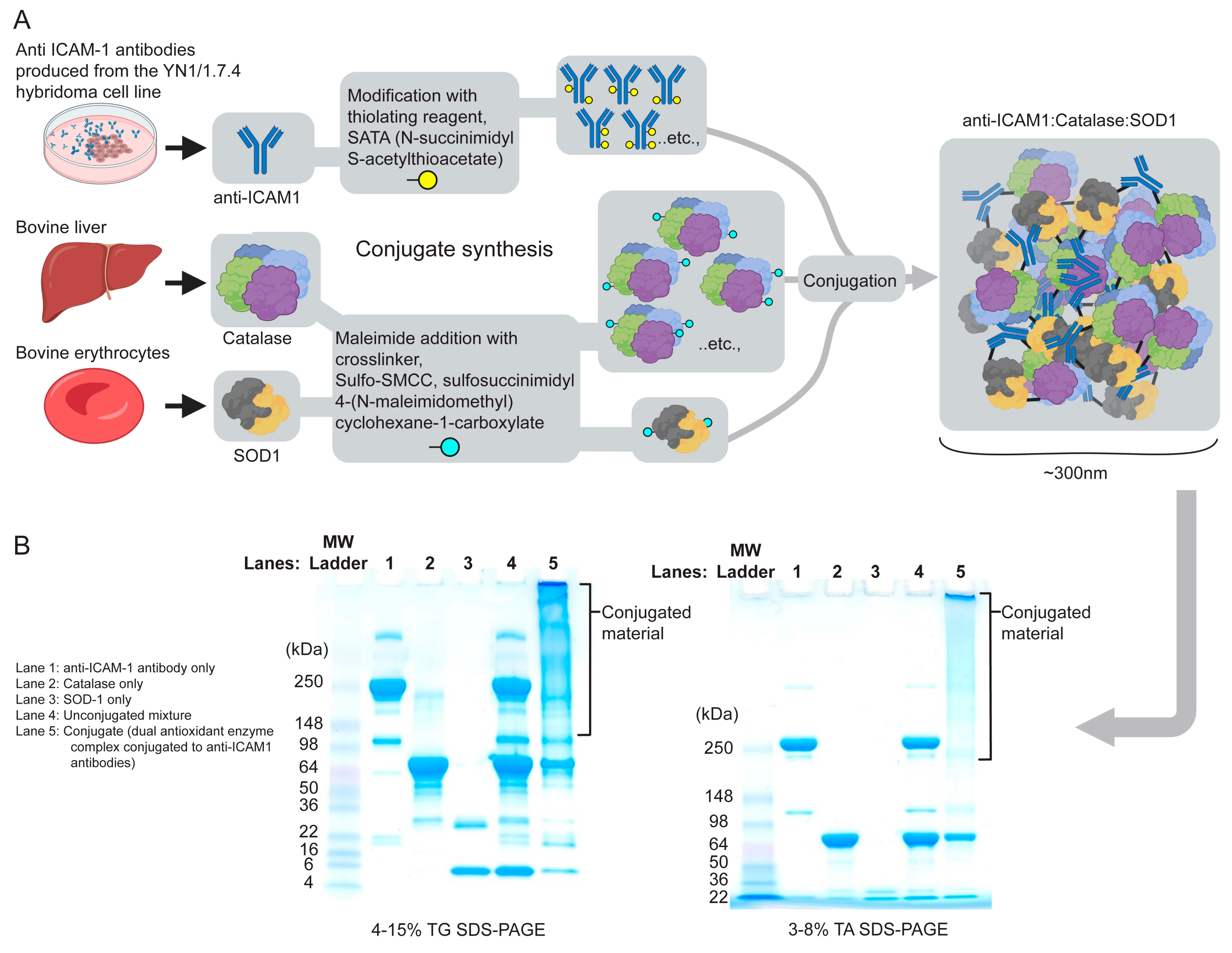
2.1. Dual-Conjugate Preparation
2.2. Cell Culture and Reagents
2.3. Paracellular Permeability Assays
2.4. Metabolic Flux Assays
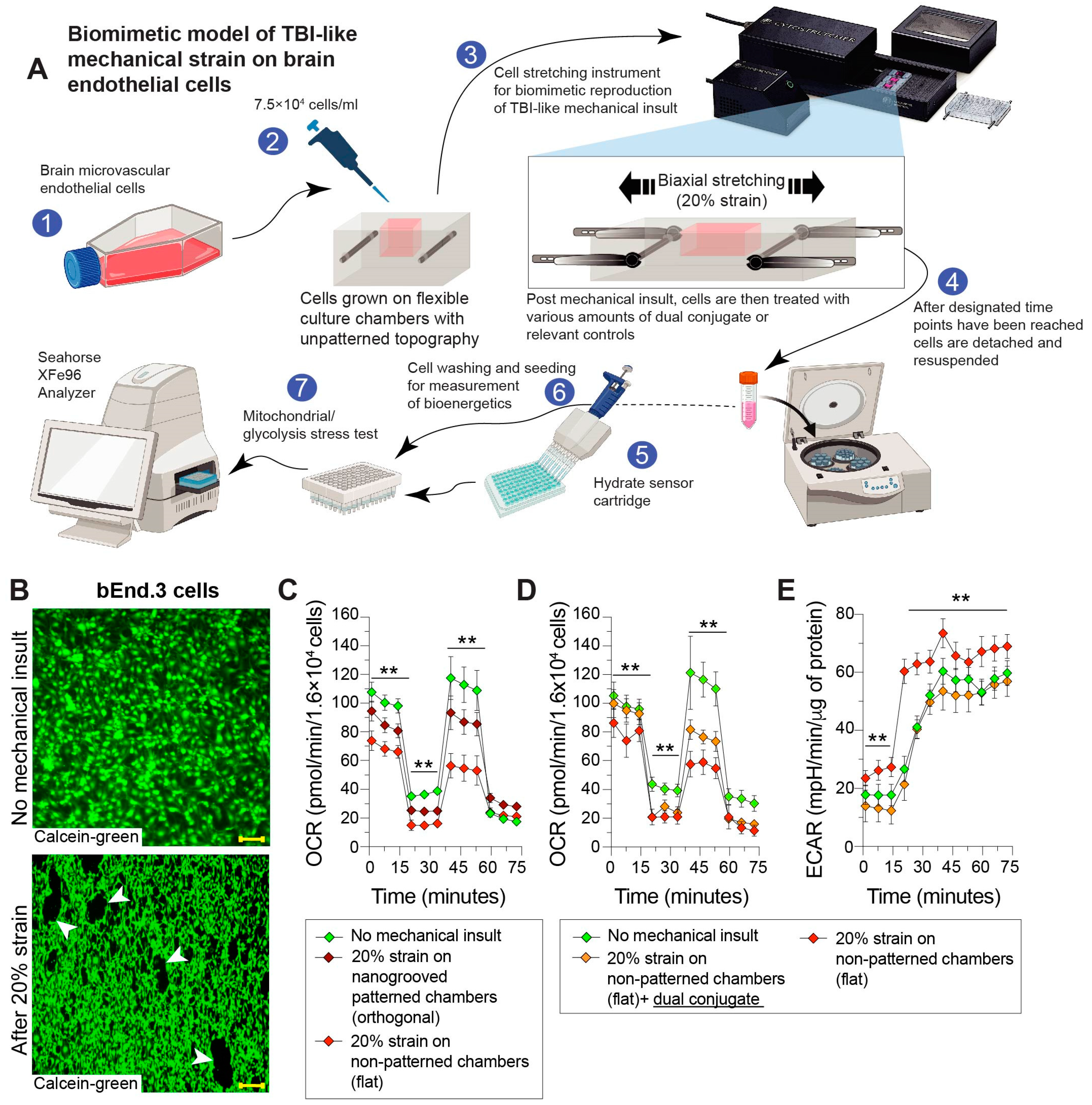
2.5. Mechanical Stretch Injury to Cells (In Vitro TBI Model)
2.6. Vertebrate Animals and Controlled Cortical Impact (CCI) Traumatic Brain Injury Model
2.7. Immunohistochemistry
2.8. Reactive Oxygen Species (ROS) Detection Assay
2.9. Immunofluorescence Assay
2.10. Imaging and Image Analysis
2.11. Statistical Analysis
3. Results
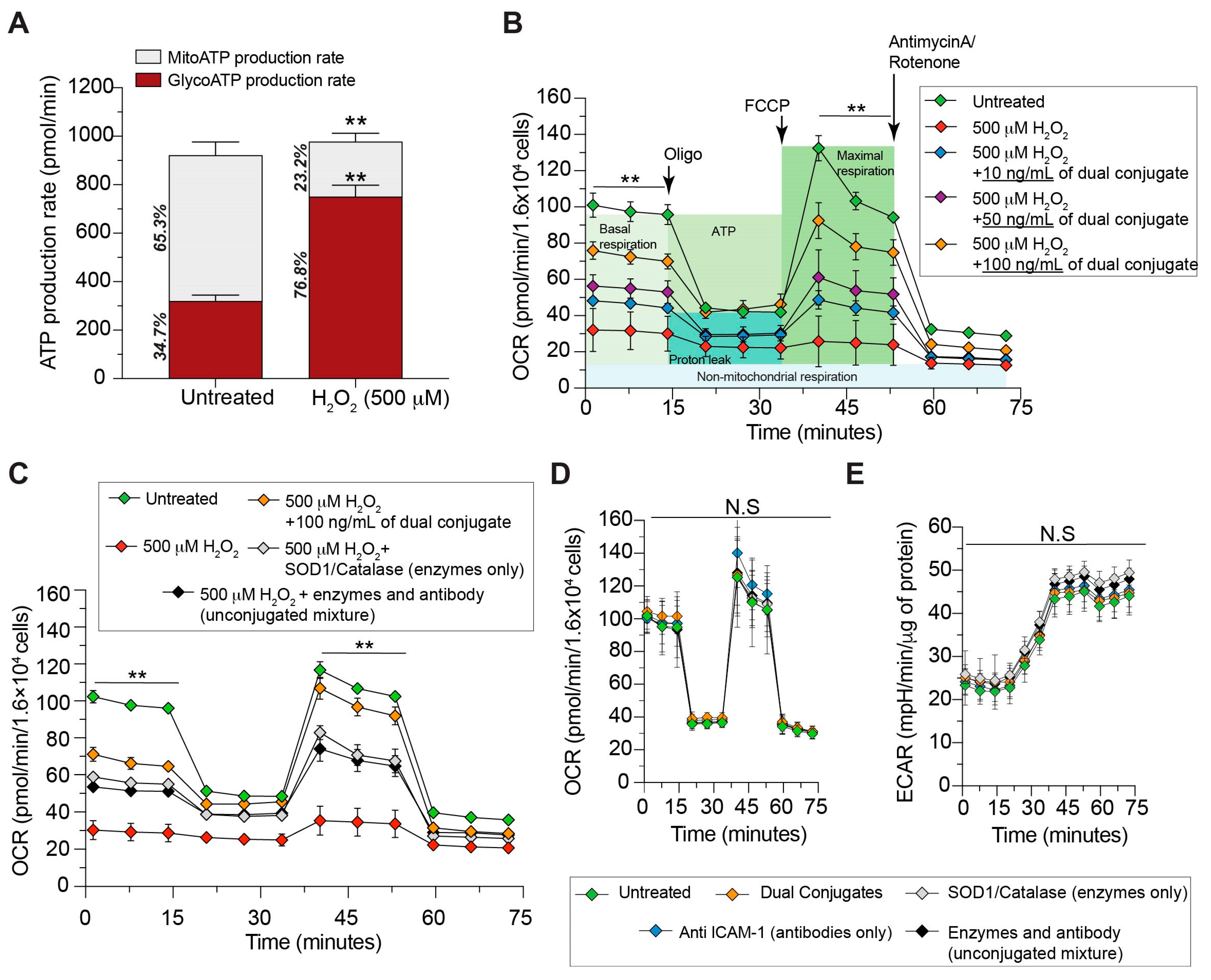
3.1. Dual Antioxidant Enzymes and Targeting Anti-ICAM-1 Ab Mitigate Mitochondrial Stress Due to H2O2
3.2. Anti-ICAM-1–SOD-1–Catalase Conjugate Preserves Brain Endothelial Barrier Integrity Following Exposure to H2O2
3.3. Mechanical Stretch of Brain Endothelial Monolayer Induces Strain-Associated Injury That Is Rescued by Anti-ICAM-1–SOD-1–Catalase Dual Conjugates
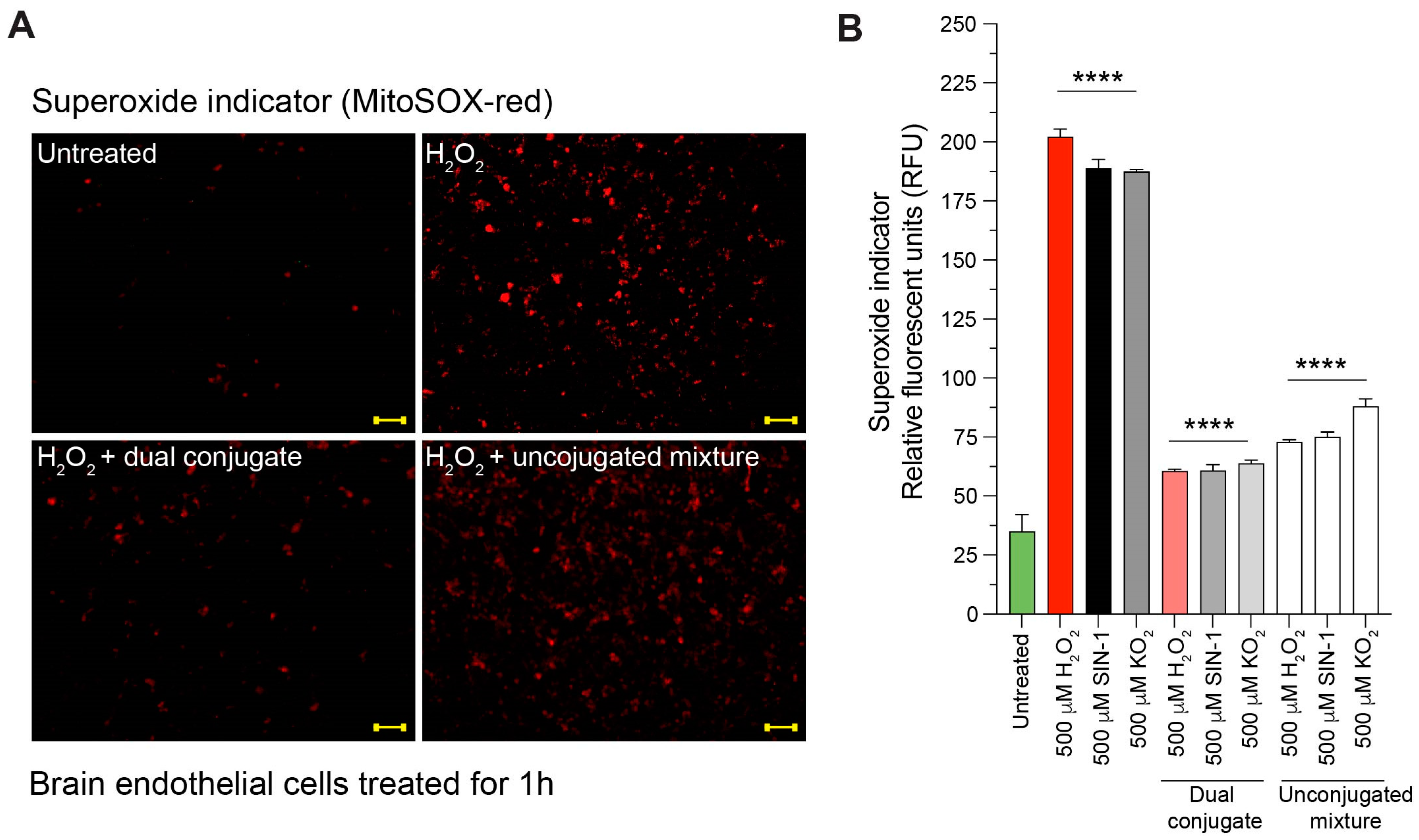
3.4. Anti-ICAM-1–SOD-1–Catalase Reduces Intracellular Superoxide Content in Cells Treated with H2O2, SIN-1, and KO2
3.5. Detection of Dual Conjugates Internalized in Stimulated Brain Endothelial Cells
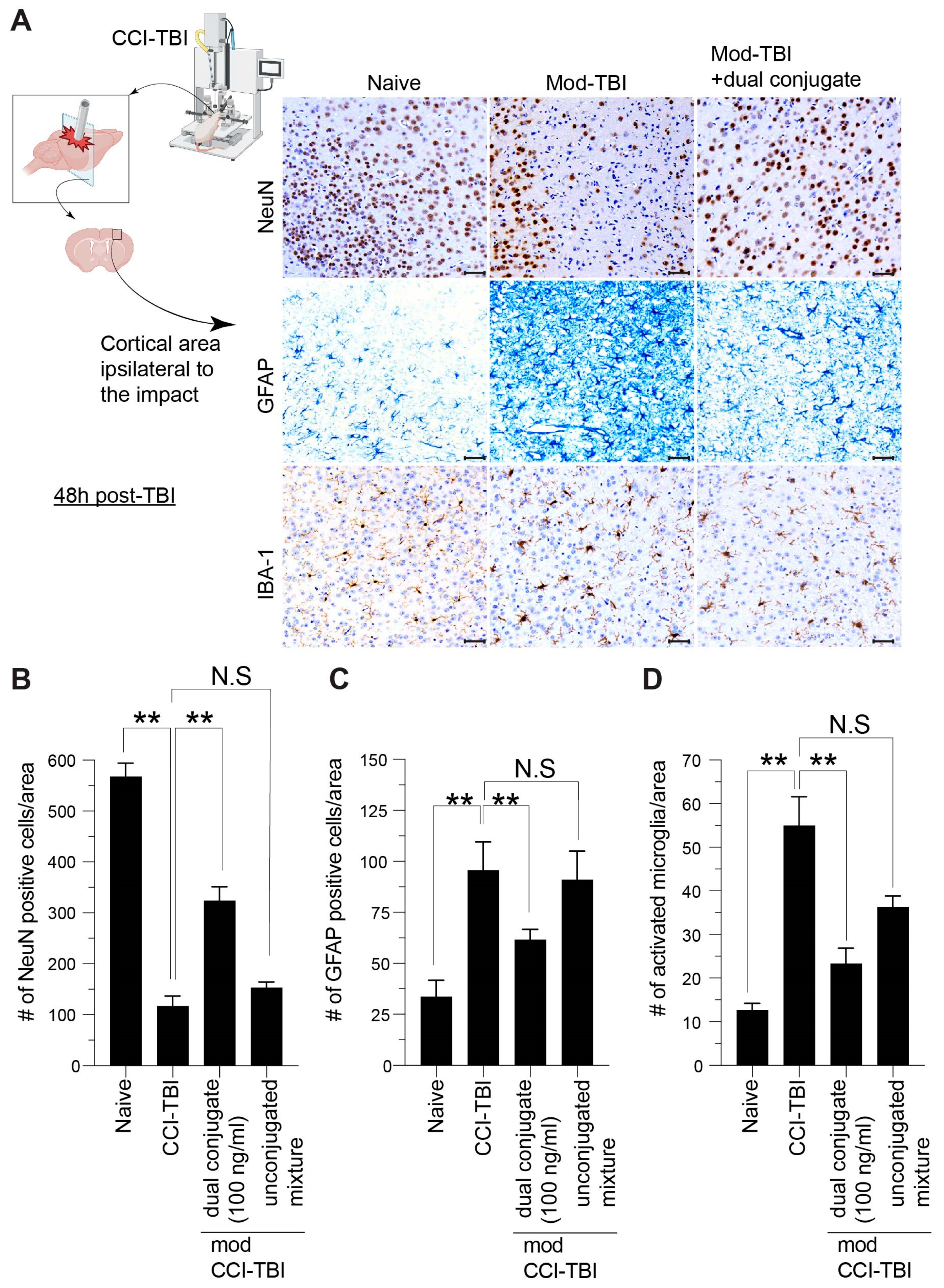
3.6. Histological Indices of CCI-TBI Neuropathology Are Attenuated by Administration of Dual-Conjugate (Anti-ICAM-1–SOD-1–Catalase) Intervention
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Coronado, V.G.; Xu, L.; Basavaraju, S.V.; McGuire, L.C.; Wald, M.M.; Faul, M.D.; Hemphill, J.D. Surveillance for traumatic brain injury-related deaths—United States, 1997–2007. MMWR Surveill Summ. 2011, 60, 1–32. [Google Scholar] [PubMed]
- Satz, P.; Zaucha, K.; McCleary, C.; Light, R.; Asarnow, R.; Becker, D. Mild head injury in children and adolescents: A review of studies (1970–1995). Psychol. Bull. 1997, 122, 107–131. [Google Scholar] [CrossRef] [PubMed]
- Langlois, J.A.; Rutland-Brown, W.; Wald, M.M. The epidemiology and impact of traumatic brain injury: A brief overview. J. Head Trauma Rehabil. 2006, 21, 375–378. [Google Scholar] [CrossRef] [PubMed]
- Prevention CfDCa. Surveillance Report of Traumatic Brain Injury-Related Deaths by Age Group, Sex, and Mechanism of Injury—United States, 2018 and 2019; Centers for Disease Control and Prevention, US Department of Health and Human Services: Atlanta, GA, USA, 2022.
- Rabinowitz, A.R.; Levin, H.S. Cognitive sequelae of traumatic brain injury. Psychiatr. Clin. N. Am. 2014, 37, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Habgood, M.D.; Bye, N.; Dziegielewska, K.M.; Ek, C.J.; Lane, M.A.; Potter, A.; Morganti-Kossmann, C.; Saunders, N.R. Changes in blood-brain barrier permeability to large and small molecules following traumatic brain injury in mice. Eur. J. Neurosci. 2007, 25, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Kawata, K.; Liu, C.Y.; Merkel, S.F.; Ramirez, S.H.; Tierney, R.T.; Langford, D. Blood biomarkers for brain injury: What are we measuring? Neurosci. Biobehav. Rev. 2016, 68, 460–473. [Google Scholar] [CrossRef]
- Algattas, H.; Huang, J.H. Traumatic Brain Injury pathophysiology and treatments: Early, intermediate, and late phases post-injury. Int. J. Mol. Sci. 2013, 15, 309–341. [Google Scholar] [CrossRef] [PubMed]
- McKee, A.C.; Daneshvar, D.H. The neuropathology of traumatic brain injury. Handb. Clin. Neurol. 2015, 127, 45–66. [Google Scholar]
- Ng, S.Y.; Lee, A.Y.W. Traumatic Brain Injuries: Pathophysiology and Potential Therapeutic Targets. Front. Cell Neurosci. 2019, 13, 528. [Google Scholar] [CrossRef]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Carvey, P.M.; Hendey, B.; Monahan, A.J. The blood-brain barrier in neurodegenerative disease: A rhetorical perspective. J. Neurochem. 2009, 111, 291–314. [Google Scholar] [CrossRef]
- Bazarian, J.J.; Wong, T.; Harris, M.; Leahey, N.; Mookerjee, S.; Dombovy, M. Epidemiology and predictors of post-concussive syndrome after minor head injury in an emergency population. Brain Inj. 1999, 13, 173–189. [Google Scholar] [CrossRef]
- Rodriguez-Rodriguez, A.; Egea-Guerrero, J.J.; Murillo-Cabezas, F.; Carrillo-Vico, A. Oxidative stress in traumatic brain injury. Curr. Med. Chem. 2014, 21, 1201–1211. [Google Scholar] [CrossRef]
- Kontos, H.A.; Wei, E.P. Superoxide production in experimental brain injury. J. Neurosurg. 1986, 64, 803–807. [Google Scholar] [CrossRef]
- Lewen, A.; Matz, P.; Chan, P.H. Free radical pathways in CNS injury. J. Neurotrauma 2000, 17, 871–890. [Google Scholar] [CrossRef]
- Shao, C.; Roberts, K.N.; Markesbery, W.R.; Scheff, S.W.; Lovell, M.A. Oxidative stress in head trauma in aging. Free Radic. Biol. Med. 2006, 41, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Kaur, P.; Sharma, S. Recent Advances in Pathophysiology of Traumatic Brain Injury. Curr. Neuropharmacol. 2018, 16, 1224–1238. [Google Scholar] [CrossRef]
- Ichimura, H.; Parthasarathi, K.; Quadri, S.; Issekutz, A.C.; Bhattacharya, J. Mechano-oxidative coupling by mitochondria induces proinflammatory responses in lung venular capillaries. J. Clin. Investig. 2003, 111, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, M.C.; Dunlay, R.P.; Lazartigues, E.; Zhang, Y.; Sharma, R.V.; Engelhardt, J.F.; Davisson, R.L. Requirement for Rac1-dependent NADPH oxidase in the cardiovascular and dipsogenic actions of angiotensin II in the brain. Circ. Res. 2004, 95, 532–539. [Google Scholar] [CrossRef] [PubMed]
- McCord, J.M.; Roy, R.S.; Schaffer, S.W. Free radicals and myocardial ischemia. The role of xanthine oxidase. Adv. Myocardiol. 1985, 5, 183–189. [Google Scholar]
- Lutton, E.M.; Farney, S.K.; Andrews, A.M.; Shuvaev, V.V.; Chuang, G.-Y.; Muzykantov, V.R.; Ramirez, S.H. Endothelial Targeted Strategies to Combat Oxidative Stress: Improving Outcomes in Traumatic Brain Injury. Front. Neurol. 2019, 10, 582. [Google Scholar] [CrossRef]
- Lutton, E.M.; Razmpour, R.; Andrews, A.M.; Cannella, L.A.; Son, Y.-J.; Shuvaev, V.V.; Muzykantov, V.R.; Ramirez, S.H. Acute administration of catalase targeted to ICAM-1 attenuates neuropathology in experimental traumatic brain injury. Sci. Rep. 2017, 7, 3846. [Google Scholar] [CrossRef] [PubMed]
- Takei, F. Inhibition of mixed lymphocyte response by a rat monoclonal antibody to a novel murine lymphocyte activation antigen (MALA-2). J. Immunol. 1985, 134, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Divakaruni, A.S.; Paradyse, A.; Ferrick, D.A.; Murphy, A.N.; Jastroch, M. Analysis and interpretation of microplate-based oxygen consumption and pH data. Methods Enzymol. 2014, 547, 309–354. [Google Scholar] [PubMed]
- Valentine, J.S.; Miksztal, A.R.; Sawyer, D.T. Methods for the study of superoxide chemistry in nonaqueous solutions. Methods Enzymol. 1984, 105, 71–81. [Google Scholar] [PubMed]
- Kladna, A.; Berczynski, P.; Kruk, I.; Michalska, T.; Aboul-Enein, H.Y. Superoxide anion radical scavenging property of catecholamines. Luminescence 2013, 28, 450–455. [Google Scholar] [CrossRef] [PubMed]
- Andrews, A.M.; Lutton, E.M.; Cannella, L.A.; Reichenbach, N.; Razmpour, R.; Seasock, M.J.; Kaspin, S.J.; Merkel, S.F.; Langford, D.; Persidsky, Y.; et al. Characterization of human fetal brain endothelial cells reveals barrier properties suitable for in vitro modeling of the BBB with syngenic co-cultures. J. Cereb. Blood Flow Metab. 2018, 38, 888–903. [Google Scholar] [CrossRef]
- Weydert, C.J.; Cullen, J.J. Measurement of superoxide dismutase, catalase and glutathione peroxidase in cultured cells and tissue. Nat. Protoc. 2010, 5, 51–66. [Google Scholar] [CrossRef]
- Chandran, R.; Kim, T.; Mehta, S.L.; Udho, E.; Chanana, V.; Cengiz, P.; Kim, H.; Kim, C.; Vemuganti, R. A combination antioxidant therapy to inhibit NOX2 and activate Nrf2 decreases secondary brain damage and improves functional recovery after traumatic brain injury. J. Cereb. Blood Flow Metab. 2018, 38, 1818–1827. [Google Scholar] [CrossRef]
- Carpenter, K.L.; Jalloh, I.; Hutchinson, P.J. Glycolysis and the significance of lactate in traumatic brain injury. Front. Neurosci. 2015, 9, 112. [Google Scholar] [CrossRef]
- Jalloh, I.; Carpenter, K.L.; Helmy, A.; Carpenter, T.A.; Menon, D.K.; Hutchinson, P.J. Glucose metabolism following human traumatic brain injury: Methods of assessment and pathophysiological findings. Metab. Brain Dis. 2015, 30, 615–632. [Google Scholar] [CrossRef] [PubMed]
- Lozano, D.; Gonzales-Portillo, G.S.; Acosta, S.; de la Pena, I.; Tajiri, N.; Kaneko, Y.; Borlongan, C.V. Neuroinflammatory responses to traumatic brain injury: Etiology, clinical consequences, and therapeutic opportunities. Neuropsychiatr. Dis. Treat. 2015, 11, 97–106. [Google Scholar]
- Luissint, A.C.; Artus, C.; Glacial, F.; Ganeshamoorthy, K.; Couraud, P.O. Tight junctions at the blood brain barrier: Physiological architecture and disease-associated dysregulation. Fluids Barriers CNS 2012, 9, 23. [Google Scholar] [CrossRef]
- Vorbrodt, A.W.; Dobrogowska, D.H. Molecular anatomy of intercellular junctions in brain endothelial and epithelial barriers: Electron microscopist’s view. Brain Res. Brain Res. Rev. 2003, 42, 221–242. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 2008, 57, 178–201. [Google Scholar] [CrossRef]
- Lifshitz, J.; Friberg, H.; Neumar, R.W.; Raghupathi, R.; Welsh, F.A.; Janmey, P.; Saatman, K.E.; Wieloch, T.; Grady, M.S.; McIntosh, T.K. Structural and functional damage sustained by mitochondria after traumatic brain injury in the rat: Evidence for differentially sensitive populations in the cortex and hippocampus. J. Cereb. Blood Flow Metab. 2003, 23, 219–231. [Google Scholar] [CrossRef]
- Vink, R.; Head, V.A.; Rogers, P.J.; McIntosh, T.K.; Faden, A.I. Mitochondrial metabolism following traumatic brain injury in rats. J. Neurotrauma 1990, 7, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Watson, W.D.; Buonora, J.E.; Yarnell, A.M.; Lucky, J.J.; D’acchille, M.I.; McMullen, D.C.; Boston, A.G.; Kuczmarski, A.V.; Kean, W.S.; Verma, A.; et al. Impaired cortical mitochondrial function following TBI precedes behavioral changes. Front. Neuroenergetics 2013, 5, 12. [Google Scholar]
- Cornelius, C.; Crupi, R.; Calabrese, V.; Graziano, A.; Milone, P.; Pennisi, G.; Radak, Z.; Calabrese, E.J.; Cuzzocrea, S. Traumatic brain injury: Oxidative stress and neuroprotection. Antioxid. Redox Signal. 2013, 19, 836–853. [Google Scholar] [CrossRef]
- Bayir, H.; Kagan, V.E.; Borisenko, G.G.; Tyurina, Y.Y.; Janesko, K.L.; Vagni, V.A.; Billiar, T.R.; Williams, D.L.; Kochanek, P.M. Enhanced oxidative stress in iNOS-deficient mice after traumatic brain injury: Support for a neuroprotective role of iNOS. J. Cereb. Blood Flow Metab. 2005, 25, 673–684. [Google Scholar] [CrossRef]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [PubMed]
- Blackwell, L.S.; Grell, R. Pediatric Traumatic Brain Injury: Impact on the Developing Brain. Pediatr. Neurol. 2023, 148, 215–222. [Google Scholar] [CrossRef]
- Roozenbeek, B.; Maas, A.I.; Menon, D.K. Changing patterns in the epidemiology of traumatic brain injury. Nat. Rev. Neurol. 2013, 9, 231–236. [Google Scholar] [CrossRef]
- Chodobski, A.; Zink, B.J.; Szmydynger-Chodobska, J. Blood-brain barrier pathophysiology in traumatic brain injury. Transl. Stroke Res. 2011, 2, 492–516. [Google Scholar] [CrossRef]
- Smith, S.L.; Andrus, P.K.; Zhang, J.R.; Hall, E.D. Direct measurement of hydroxyl radicals, lipid peroxidation, and blood-brain barrier disruption following unilateral cortical impact head injury in the rat. J. Neurotrauma 1994, 11, 393–404. [Google Scholar] [CrossRef]
- Scherpereel, A.; Wiewrodt, R.; Christofidou-Solomidou, M.; Gervais, R.; Murciano, J.; Albelda, S.M.; Muzykantov, V.R. Cell-selective intracellular delivery of a foreign enzyme to endothelium in vivo using vascular immunotargeting. FASEB J. 2001, 15, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Scherpereel, A.; Rome, J.J.; Wiewrodt, R.; Watkins, S.C.; Harshaw, D.W.; Alder, S.; Christofidou-Solomidou, M.; Haut, E.; Murciano, J.-C.; Nakada, M.; et al. Platelet-endothelial cell adhesion molecule-1-directed immunotargeting to cardiopulmonary vasculature. J. Pharmacol. Exp. Ther. 2002, 300, 777–786. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Shuvaev, V.V.; Muzykantov, V.R. Catalase and superoxide dismutase conjugated with platelet-endothelial cell adhesion molecule antibody distinctly alleviate abnormal endothelial permeability caused by exogenous reactive oxygen species and vascular endothelial growth factor. J. Pharmacol. Exp. Ther. 2011, 338, 82–91. [Google Scholar] [CrossRef]
- Hood, E.D.; Greineder, C.F.; Dodia, C.; Han, J.; Mesaros, C.; Shuvaev, V.V.; Blair, I.A.; Fisher, A.B.; Muzykantov, V.R. Antioxidant protection by PECAM-targeted delivery of a novel NADPH-oxidase inhibitor to the endothelium in vitro and in vivo. J. Control. Release 2012, 163, 161–169. [Google Scholar] [CrossRef]
- Shuvaev, V.V.; Tliba, S.; Pick, J.; Arguiri, E.; Christofidou-Solomidou, M.; Albelda, S.M.; Muzykantov, V.R. Modulation of endothelial targeting by size of antibody-antioxidant enzyme conjugates. J. Control. Release 2011, 149, 236–241. [Google Scholar] [CrossRef]
- Calderon, A.J.; Muzykantov, V.; Muro, S.; Eckmann, D.M. Flow dynamics, binding and detachment of spherical carriers targeted to ICAM-1 on endothelial cells. Biorheology 2009, 46, 323–341. [Google Scholar] [CrossRef]
- Ferrer, M.C.; Shuvaev, V.V.; Zern, B.J.; Composto, R.J.; Muzykantov, V.R.; Eckmann, D.M. Icam-1 targeted nanogels loaded with dexamethasone alleviate pulmonary inflammation. PLoS ONE 2014, 9, e102329. [Google Scholar]
- Andrews, A.M.; Lutton, E.M.; Merkel, S.F.; Razmpour, R.; Ramirez, S.H. Mechanical Injury Induces Brain Endothelial-Derived Microvesicle Release: Implications for Cerebral Vascular Injury during Traumatic Brain Injury. Front. Cell Neurosci. 2016, 10, 43. [Google Scholar] [CrossRef] [PubMed]
- Mertsch, K.; Blasig, I.; Grune, T. 4-Hydroxynonenal impairs the permeability of an in vitro rat blood-brain barrier. Neurosci. Lett. 2001, 314, 135–138. [Google Scholar] [CrossRef] [PubMed]
- Hall, E.D.; Vaishnav, R.A.; Mustafa, A.G. Antioxidant therapies for traumatic brain injury. Neurotherapeutics. 2010, 7, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Neuwelt, E.; Abbott, N.J.; Abrey, L.; Banks, W.A.; Blakley, B.; Davis, T.; Engelhardt, B.; Grammas, P.; Nedergaard, M.; Nutt, J.; et al. Strategies to advance translational research into brain barriers. Lancet Neurol. 2008, 7, 84–96. [Google Scholar] [CrossRef]
- Fischer, S.; Wiesnet, M.; Renz, D.; Schaper, W. H2O2 induces paracellular permeability of porcine brain-derived microvascular endothelial cells by activation of the p44/42 MAP kinase pathway. Eur. J. Cell Biol. 2005, 84, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Schreibelt, G.; Kooij, G.; Reijerkerk, A.; van Doorn, R.; Gringhuis, S.I.; van der Pol, S.; Weksler, B.B.; Romero, I.A.; Couraud, P.O.; Piontek, J.; et al. Reactive oxygen species alter brain endothelial tight junction dynamics via RhoA, PI3 kinase, and PKB signaling. FASEB J. 2007, 21, 3666–3676. [Google Scholar] [CrossRef] [PubMed]
- Beckman, J.S.; Koppenol, W.H. Nitric oxide, superoxide, and peroxynitrite: The good, the bad, and ugly. Am. J. Physiol. 1996, 271 Pt 1, C1424–C1437. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Mahmood, A.; Chopp, M. Animal models of traumatic brain injury. Nat. Rev. Neurosci. 2013, 14, 128–142. [Google Scholar] [CrossRef]
- Olsen, C.M.; Corrigan, J.D. Does Traumatic Brain Injury Cause Risky Substance Use or Substance Use Disorder? Biol. Psychiatry 2022, 91, 421–437. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leonard, B.M.; Shuvaev, V.V.; Bullock, T.A.; Galpayage Dona, K.N.U.; Muzykantov, V.R.; Andrews, A.M.; Ramirez, S.H. Engineered Dual Antioxidant Enzyme Complexes Targeting ICAM-1 on Brain Endothelium Reduce Brain Injury-Associated Neuroinflammation. Bioengineering 2024, 11, 200. https://doi.org/10.3390/bioengineering11030200
Leonard BM, Shuvaev VV, Bullock TA, Galpayage Dona KNU, Muzykantov VR, Andrews AM, Ramirez SH. Engineered Dual Antioxidant Enzyme Complexes Targeting ICAM-1 on Brain Endothelium Reduce Brain Injury-Associated Neuroinflammation. Bioengineering. 2024; 11(3):200. https://doi.org/10.3390/bioengineering11030200
Chicago/Turabian StyleLeonard, Brian M., Vladimir V. Shuvaev, Trent A. Bullock, Kalpani N. Udeni Galpayage Dona, Vladimir R. Muzykantov, Allison M. Andrews, and Servio H. Ramirez. 2024. "Engineered Dual Antioxidant Enzyme Complexes Targeting ICAM-1 on Brain Endothelium Reduce Brain Injury-Associated Neuroinflammation" Bioengineering 11, no. 3: 200. https://doi.org/10.3390/bioengineering11030200
APA StyleLeonard, B. M., Shuvaev, V. V., Bullock, T. A., Galpayage Dona, K. N. U., Muzykantov, V. R., Andrews, A. M., & Ramirez, S. H. (2024). Engineered Dual Antioxidant Enzyme Complexes Targeting ICAM-1 on Brain Endothelium Reduce Brain Injury-Associated Neuroinflammation. Bioengineering, 11(3), 200. https://doi.org/10.3390/bioengineering11030200