Abstract
With regards to oil-based diesel fuel, the adoption of bio-derived diesel fuel was estimated to reduce CO2 emissions by approximately 75%, considering the whole life cycle. In this paper, we present a novel continuous-flow process able to transfer an equimolar amount of CO2 (through urea) to glycerol, producing glycerol carbonate. This represents a convenient tool, able to both improve the efficiency of the biodiesel production through the conversion of waste streams into added-value chemicals and to beneficially contribute to the whole carbon cycle. By means of a Design of Experiments approach, the influence of key operating variables on the product yield was studied and statistically modeled.
1. Introduction
In addition to different natural dynamics outside of our control, such as solar activity, developments over the last hundred years have been accompanied by a general and progressive increase in the mean global temperature, collectively known as ‘global warming’. This variation is strictly related to several climate alterations such as melting glaciers and rising sea levels, but also to extreme weather events, a change in wildlife habitats, and an array of other impacts []. Beyond the heating due to direct irradiation from the sun, the atmosphere plays a determinant role in retaining part of the thermal energy close to the Earth’s surface []. This heat-trapping phenomenon, known as the “greenhouse effect”, comes from some naturally occurring gases, such as CO2, methane, and nitrous oxides, and helps our planet to maintain stable conditions for life.
The continuous and ever-increasing exploitation of fossil carbon resources to sustain our development brought a symmetrical increase in greenhouse gas (GHG) emissions, especially CO2, which have been linked to an increased greenhouse effect.
More sustainable strategies to drive our development without hampering the progress of future generations are urgently required and, in this context, a great deal of attention has recently been devoted to the study and implementation of biorefineries. Biorefineries are production facilities able to exploit renewable biomass instead of a fossil carbon resource and convert it into carbon-based products, such materials, fuels, or energy []. CO2 fixation during plant growth can considerably reduce the impact on carbon balance when a vegetable source is exploited. This provides biorefineries with a greatly improved GHG emissions profile compared to traditional refineries. Among the different biorefinery types, biodiesel biorefineries focus on the production of carboxylic acid esters that can be directly blended with regular diesel fuel, starting from vegetable or animal fats. The latter are subjected to acid- or base-catalyzed transesterification in the presence of an excess of a short chain alcohol, to give a mixture of fatty acid methyl (or ethyl) esters, which is biodiesel. Depending on the triglyceride source, it has been estimated that the adoption of bio-derived diesel fuel brings between a 63% (energy crops) and 86% (waste fat) reduction in GHG emission compared to oil-based diesel fuel []. Moreover, a common factor in all transesterification processes is the co-production of glycerol (Gly) that is usually considered a waste stream, especially in small-scale productions where further refining costs are not justified. The present crude Gly price is as low as between USD 0.04 and 0.09/lb and it is expected to decrease further according to the growing industrial availability. Current global production is approximately 42 billion liters per year [].
Gly shows some direct potential uses in the pharma, food, cosmetic, and polymer industries; however, its features (limited miscibility with organics, high boiling point, chemoselectivity of primary vs. secondary hydroxyl), as well as the presence of contaminants in the raw material, pose several barriers to its free applicability. As a result, the identification of effective processes able to economically convert Gly into value-added chemicals represents a key development needed to improve the sustainability of the whole biodiesel industry [].
Various Gly-derived upgraded structures with enhanced properties and extended application profiles are well-described. Solketal [] and other acetals, 1,3-propanediol [,] and dihydroxy acetone [], represent known examples, and among these, a leading position is occupied by glycerol carbonate (GC), which represents the main focus of this work. As a multiple-site electrophile, GC can efficiently and selectively interact with diverse nucleophiles, such as amines [,,], amino acids [,], phenols [], carboxylates [], and others []. The nucleophilic property of the free hydroxyl function can be directly exploited [,] or can be used to obtain a new electrophilic site capable of additional reactivity [,]. Valuable GC-derived oligomers or polymers, especially polyglycerols [,], polycarbonates, and polyesters [], find application in biomedical tissue engineering [] and in the controlled release of active pharmaceutical ingredients []. Polyglycerols are also applied in water-dispersible polymers, adhesives [,], and bio-based surfactants []. As a building block, GC has been included in diverse classes of polymeric materials, such as acrylates [,], polyesters [,,], isocyanate-free polyurethanes [,,,,], hybrid structures [,], and others [,]. Finally, GC can also be used as a carbonate carrier able to transfer the same function to other polyols, including sugars [,], through transcarbonylation processes. The carbonate function can also be considered as a precursor to highly electrophilic (and valuable) epoxy function [,].
Thanks to the excellent properties of GC, a number of methods able to convert Gly into GC were devised [,]. Based on the nature of the main interacting reagent involved, they can be divided into three groups, as follows:
- (i).
- processes using activated phosgene-sourced reagents, such as phosgene itself, chloroformates, or carbonyldiimidazole;
- (ii).
- processes using activated reagents not sourced from phosgene, such as dialkyl carbonates, diaryl carbonates, or CO + O2;
- (iii).
- processes using non-activated CO2-sourced reagents, such as CO2 itself or urea.
Class (i) processes are characterized by high Gly conversion and product selectivity, but also by health and environmental issues due to the toxicity of the involved reagent (or precursor). The reactivity of class (ii) reagents allows high efficiency as well and, thanks to the better sustainability profile, the related processes are the most investigated [,,]. Different implementations of these processes are described, extending through various catalysts and plant engineering, including continuous-flow techniques [,]. Organic carbonates can, in principle, be prepared from CO2, however, aside from promising research advancements [,], no industrial process is currently available, possibly due to known issues such as process reversibility and hydrolytic instability of the product [].
Dealing with class (iii) reagents, CO2 certainly represents the more atom economical choice, but the same hurdles described to form alkyl carbonates also hamper the direct carbonylation of glycerol to GC [,]. The option to use highly toxic glycidol as an activated Gly-sourced substrate [,] able to overcome the chemical inertness of CO2 presents critical toxicity features similar to phosgene or gaseous CO []. To pursue chemical upgrading routes characterized by a non-toxic and stable reactant as well as featuring high atom economy, our attention was devoted to urea. Urea is a white crystalline solid containing 46 wt% of nitrogen and, being non-toxic, is largely used as an animal feed additive and fertilizer []. It is even more resonance-stabilized [] than CO2, thus, its reactivity with polyols is expected to not be very manifest; in fact, thermally stable mixtures between urea and polyols are well-described [] and are used as deep eutectic solvents. What makes urea an attractive carbonylation agent is its multi-basic nature [], which makes the coordination of metal cations possible []. These urea–cation complexes are the key to converting urea into a highly electrophilic “masked” isocyanate species [,], where the resonance is strongly reduced by the interaction with the metal center. This allowed the easy nucleophilic attack by Gly, as shown in Figure 1. The process is reversible [] and proceeds stepwise, with the formation of a carbamic acid intermediate (I) (Figure 1) [], which is converted in the final product through the elimination of a second ammonia molecule [].
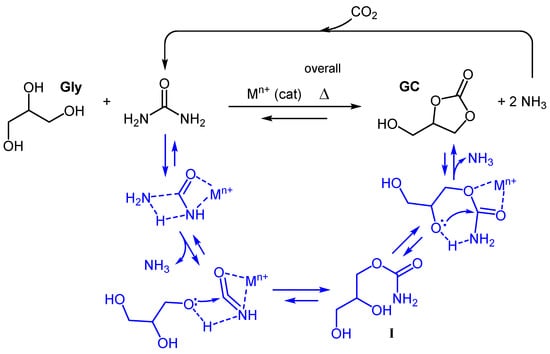
Figure 1.
Metal-catalyzed carbonylation of Gly with urea.
Urea is industrially prepared from ammonia and CO2 [], therefore, it can be considered a CO2 carrier [] and promises additional advantages regarding the overall carbon balance, especially when the proper recycling of ammonia is implemented. In other words, the upgrading of Gly to GC involves the fixation of one mole of CO2 per mole of vegetable oil (i.e., ~5 wt%, when 880 g/mol is taken as a mean molecular mass of a vegetable oil), a fact that, together with refined farming practices [], can give additional support to reducing GHG emissions within the whole biodiesel industry.
In this paper, we present a novel continuous-flow process able to convert Gly into GC. The influence of key operating variables, such as time, temperature, and urea/Gly molar ratio (MR), on the product yield and selectivity are studied and statistically modeled by means of a Design of Experiments (DoE) approach.
2. Materials and Methods
2.1. General Information
Solvents and reagents were commercial grade and used as received. Glycerol (from vegetable source) was purchased from Merck (Milano, Italy). ZnSO4·H2O was obtained by heating ZnSO4·7H2O overnight in an oven at 130 °C, which was then stored in a desiccator. Gly was vacuum dried for 4 h in the rotavapor (water bath 40 °C) and stored in a capped bottle. Urea was dried at 65 °C overnight and stored in a desiccator. 1H NMR spectra were acquired with a Bruker Avance 400 spectrometer (Billerica, MA, USA).
2.2. Procedure for Preliminary Batch Reactions
In a 10 mL Schlenk tube with a screw cap and equipped with a stirring bar, Gly (1.0 g, 10.9 mmol), solid catalyst (0.05 mol/mol Gly, see later), and urea (0.82 g, 13.65 mmol, 1.25 mol/mol Gly) were inserted. A membrane vacuum pump was connected through the side arm and the vacuum was set at 400 mmHg. The capped tube was inserted in an oil bath (pre-heated at 150 °C) and stirred for 4 h. The crude reaction mixture, once cooled to room temperature (r.t.), was extracted with EtOAc:Et2O (4:1 mixture, 3 × 5 mL), and the crude colorless product, obtained from the dried organic phase, was analyzed with 1H NMR using CDCl3 or D2O as the solvent.
2.3. Procedure for Continuous Flow Reactions
The plant assembly is depicted in Figure 2. The reacting mixture was continuously recirculated through a heated tubular reactor (R) by means of a Bellco (model: BL 758, Mirandola, Italy) peristaltic pump (P). A mixing chamber (M), composed of a 25 mL two-necked round bottom flask, was heated and stirred through a standard stirring plate and an oil bath. A three-arm distillation connector acted as an expanding chamber (E) and connected the reactor output to an air-cooled condenser (A, also connected to M) and a vacuum line, set at 400 mm Hg. The said reactor was composed of a metallic AISI 316 stainless steel tube (1/16 in od × 1.2 mm id, 3 m long, ~4 mL internal volume, sourced from Restek, Milano, Italy) coiled around a 4 cm diameter cylindrical aluminum block, featuring a slot for a heating resistor and temperature sensor. Both of these heating and measuring elements were connected to a PID controller (Rex-C100, sourced from RobotDigg, Shangai, China), which allowed for setting and maintaining the desired temperature. Some thermal insulation (not shown in figure) was wrapped around the external side of the reactor.
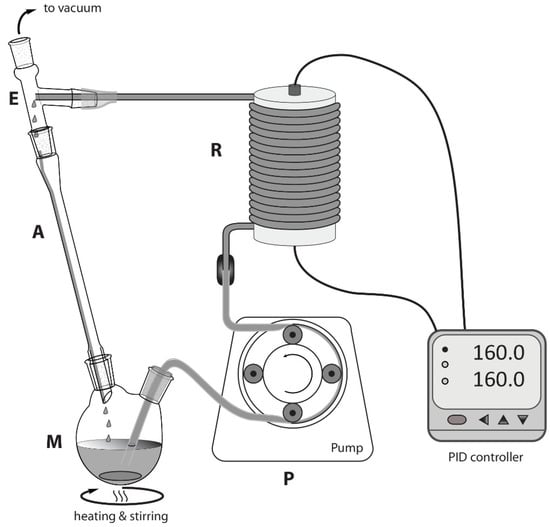
Figure 2.
Layout of the continuous flow reactor. M = mixing chamber; P = peristaltic pump; R = coiled tubular reactor; E = expansion chamber; A = air-cooled condenser.
Gly (11.0 g, 0.119 mol), ZnSO4·H2O (0.643 g; 3.58 mmol, 0.03 mol/mol Gly), and urea (e.g., 8.97 g, 0.149 mol for 1.25:1.00 urea:Gly molar ratio) were stirred at 65 °C within the mixing chamber (M, Figure 2) until a homogeneous solution was obtained. By that time, the temperature of the coiled reactor (R) was set at the desired value. Then, the peristaltic pump was started at constant flow (3.0 mL/min), and the mixture was recirculated for the desired time. To assess yield and conversion of each experiment, a 2.0 g portion of the crude reaction mixture was extracted with EtOAc:Et2O (4:1 mixture, 3 × 5 mL), and the raw colorless product, obtained from the organic phase, was carefully dried at the rotavapor. Purity was directly assessed with 1H NMR spectroscopy (see Supplementary Materials). GC gave the following signals (Figure S1): 1H NMR (400 MHz, CDCl3, 298 K): δH (ppm) = 4.81 (1H, CH, m), 4.49 (2H, CH2, m), 3.99 (1H, CH2, dd, J = 12.84 Hz; 3.1 Hz), 3.72 (1H, CH2, dd, J = 12.84; 3.5 Hz), 2.21 (1H, OH, br). The metal catalyst and some carbamic acid intermediates (currently not characterized) constituted the denser, polar phase insoluble in the extracting mixture. No residual Gly was detected.
2.4. Design of Experiments (DoE)
Nineteen experiments were suggested, and the relative output data were statistically analyzed by means of Design Expert® v.12 software (Stat-Ease Inc., 1300 Godward Street NE, Suite 6400, Minneapolis, MN 55413 USA). A three-level face-centered cubic design with five replicates of the central point was implemented. The investigated independent parameters were time (90–210 min), temperature (175–195 °C), and urea/Gly ratio (1.2–1.8), while output parameters were GC yield (%) and GC selectivity (%). Reagent flow was demonstrated to be not significant during the preliminary experiments, therefore, it was kept constant at 3.0 mL/min. Pressure was set to 400 mmHg because of hardware technical limits (squeezing of peristaltic pump tube) as well as urea losses through sublimation.
3. Results
Gly carbonylation with urea commonly involves solventless operation, temperatures ranging from 120 °C to 160 °C, reaction times from 3 to 24 h, and the presence of a catalyst (metal salt or metal oxide). Some degree of vacuum is also commonly applied in order to favor the removal of ammonia, obtaining a desired equilibrium shift [,] (Figure 1). A number of catalysts were described for this transformation [,,,] and, among these, zinc-containing species are by far the most active and popular. Irrespective of its initial form, soluble zinc-based catalysts are expected to give the same performance thanks to the formation of the metal glycerolate [,,], while insoluble forms [,,,] are involved in heterogeneous reactions.
It is interesting to note that most of the recent papers have focused on an accurate description of the catalyst but have not given enough attention to the separation and isolation of GC, which represent key steps for considering the industrial feasibility of the chemical process [].
Our investigations started with some preliminary experiments in which mixtures composed of Gly:urea:catalyst in a 1.00:1.25:0.05 molar ratio (MR) were heated in batch at 150 °C for 4 h, keeping the pressure at 400 mmHg (see materials and methods). After extraction with organic solvents, the amount of GC was evaluated with 1H NMR spectroscopy []. This screening, of which the results are collected in Table 1, let us draw some general observations, such as (i) yields are limited to 30%; (ii) longer reaction times do not improve the result (entry 3 vs. 2, Table 1); (iii) temperatures higher than 150 °C induce degradation (reaction mixture became brown and lower yields of GC were obtained); (iv) pressures lower than 400 mmHg induce urea losses through sublimation []; (v) catalyst anhydrification is beneficial (entry 2 vs. 1).

Table 1.
Screening of alternative catalysts in the batch conversion of Gly to GC a.
Cheap and easily available ZnSO4·H2O was chosen as the reference catalyst, especially because of its ability to give homogeneous mixtures that, being simpler reacting systems, let us focus on engineering approaches towards process improvement.
Some attempts to set up a reactive distillation [] were carried out, however, the high boiling point of GC (~140 °C at 0.5 mmHg) was a considerable hurdle to its isolation. Better success comes from the known implementation of microwave-heated batch reactors, giving improved isolated yields and reduced reaction times [,]. As this suggests, a main problem of the batch process featuring traditional heating is likely to be the supply of thermal energy to the reacting mixture that, being subjected to an endothermic event [], is affected by self-cooling, especially in the core (far from the heating bath). Therefore, to improve such heat transfer limitations, our focus was directed to the implementation of a high area-to-volume ratio reactor, such as a small diameter tubular reactor, together with a continuous flow of reactants inside it.
Solventless operation is a valuable feature of this process, but the high viscosity of Gly/urea mixtures poses serious troubles concerning their pumpability. Different HPLC pumps were not able to give a reliable flow, possibly due to the inability of check valves to promptly block the backflow. The addition of a high boiling point solvent such as DMSO was proposed as a solution [], but we judge this practice undesirable as it nullifies the “solventless advantage” and makes the isolation of reaction products troublesome. Fortunately, the use of a peristaltic pump in addition to a heated mixing chamber (M, Figure 2) allowed us to obtain a steady flow of reactants. In fact, by keeping the Gly/urea mixture at 65 °C, a useful reduction in viscosity was observed and, more importantly, the precipitation of urea within the tubes was avoided.
Using the flow chemistry technique in small volume reactors, thanks to strongly increased heat and mass transfer, typically benefits enhanced reaction control with respect to batch processes, especially in fast reactions []. Nevertheless, the process under study (Figure 1) features slow kinetics as evidenced by the long reaction times required for complete conversion, such as 2 h at 150 °C with microwave irradiation []; the meagre conversion obtained with reaction times of a few minutes, even with the implementation of a capillary reactor [], gives evidence of that. Using a coiled 3 m long tubular reactor, we first tried to get the best conversion through a single pass, keeping flow at a minimum (0.2 mL/min) and evaluating increased temperatures. Unluckily, unsatisfactory conversion was obtained even at 195 °C, while greater temperatures resulted in brown (degraded) mixtures, containing low amounts of GC. A possible solution was to consider multiple passes through the reactor, thus, a recirculating layout was set up, as shown in Figure 2.
Gly, urea, and the catalyst were stirred at 65 °C in the mixing chamber (M, Figure 2) to obtain a homogeneous mixture. This was continuously pumped through the heated coiled tubular reactor (R, Figure 2) and the outflow was recirculated through an air-cooled condenser (A) into the mixing chamber. The total volume of the reacting mixture was set to ~1.2 times of the total piping volume (reactor and interconnections) to maximize the number of passes through R. The entire system was maintained at reduced pressure by a membrane pump connected to E. At time intervals, a sample of reacting mixture was withdrawn from the mixing chamber to assess conversions and yields. Several trials let us delineate the general features of the system, as follows:
- temperatures greater than 175 °C resulted in better GC yields;
- a minimum time of 90 min was required to obtain complete conversion of Gly;
- pressures lower than 400 mmHg resulted in unreliable flow due to peristaltic pump malfunction (tube squeezing) and minor urea losses due to sublimation;
- yields were unaffected by changes in flow rate, within the range from 0.5–5.0 mL/min;
- yields were unaffected by changes in catalyst amount, within the range from 0.03–0.05 molZnSO4·H2O/molGly;
- diglycerol tricarbonate (DGTC) was identified as the main by-product [].
In order to get a better understanding of the influence of multiple process parameters, we decided to implement a multivariate statistical evaluation based on a DoE approach. In particular, a three-level face-centered central composite design (CCD) was chosen and was used to define an appropriate number of experiments within the variable domains arising from the above observations. The included independent variables were the reactor temperature (ranging from 175 to 195 °C), the recirculation time (from 90 to 210 min), and the urea:Gly MR (from 1.2 to 1.8); while the other process parameters such as pressure, flow, amount of catalyst, and mixing chamber temperature were fixed at 400 mmHg, 3.0 mL/min, 0.03 molZnSO4·H2O/molGly, and 65 °C, respectively. As suggested by the DoE CCD model, we planned nineteen experiments, with five replicates of the central point, as shown in Table 2. All the experiments were conducted by the same operator to minimize systematic errors, while the order of experiments was randomized. The two monitored responses were GC yield and GC purity. A quantitative evaluation of the reaction selectivity (100·mol GC/(mol GC + mol DGTC)) was obtained by the integration of isolated 1H NMR signals of the product and isolated 1H NMR signals of the main by-product (DGTC), as described in the Supplementary Materials (Figure S2). This was supported by the fact that the 1H NMR signals due to other substances always presented at a very low intensity.
Table 2.
Experiments suggested by the DoE model a.
4. Discussion
The ability to statistically evaluate the interrelation between process variables is one of the main advantages offered by the DoE approach. For instance, the influence of the three independent variables on the output responses are shown in Figure 3. GC-isolated yields (Figure 3a–c) strongly depends upon recirculation time and reactor temperature, while less marked is the influence of the urea:Gly MR. Best GC yields are obtained at temperatures between 180 and 190 °C, times between 110 and 160 min, and for urea:Gly MR in the range from 1.3–1.7. GC selectivity (Figure 3d–f) strongly depends upon urea:Gly MR and reactor temperature, while recirculation time is found to be the less influent parameter. Best selectivities are obtained at lower temperatures, lower urea:Gly MR, and times between 110 and 170 min.

Figure 3.
All factors graphs. (a) GC yield% vs. temperature, (b) GC yield% vs. urea/Gly MR, (c) GC yield% vs. recirculation time, (d) GC selectivity% vs. temperature, (e) GC selectivity% vs. urea/Gly ratio and (f) GC selectivity% vs. recirculation time. Dashed blue lines refers to minimum and maximum values of the data set. Continuous black lines refers to the mean values.
The simultaneous influence of two independent variables on each physical property is of particular value. Figure 4 shows some “two factors response surfaces”, which put into evidence the dependency of GC yield% (left of Figure 4) from time–temperature or urea:Gly MR–time couples. On the right of the same figure, the influence of time–temperature or ratio–temperature couples on GC selectivity% are shown.
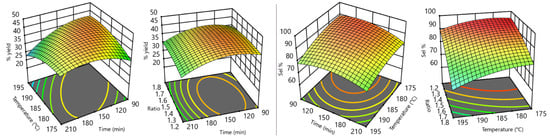
Figure 4.
Two factors response surfaces.
Finally, both GC yield% and GC selectivity% response parameters can be conveniently composed in a single factor, called “desirability”, which is able to describe the best process conditions in a simple and effective way. In the present case, desirability is obtained by the product of the said response parameters, normalized to unity. A desirability of 1.0 means that both parameters are maximized, while the lowest desirability (0.0) describes conditions where both responses are at minimum levels. In Figure 5, some contour heatmap plots of desirability as a function of independent variable couples are shown.
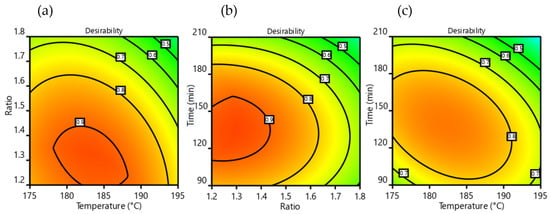
Figure 5.
Desirability contour maps, (a) vs. urea/Gly MR-temperature, (b) vs. time-urea/Gly MR and (c) vs. time-temperature.
The strong reduction in process performance at high urea:Gly MR (Figure 5a,b) comes from a loss of selectivity (see also Figure 3b,e), meaning an increased formation of DGTC. This behavior is not surprising, as DGTC is described as an overreaction product of GC when the carbonylation reagent is in high molar excess [,,]. This also suggests good potential for DGTC production as a possible future implementation of the process. The significant yield decrement at high times and temperatures (Figure 3 and Figure 5c) found good correlation with the experimental observation of the brownish color acquired by the reacting mixture in these circumstances, a sign of partial degradation. Overall, best operating conditions of the process were found to be a reactor temperature from 180 to 185 °C, a recirculation time from 120 to 150 min, and a urea:Gly MR of 1.25.
Regarding the potential risk of isocyanic acid emission coming from carbamate decomposition [,], the process conditions (low operating temperatures, low urea:Gly MR, presence of glycerol) make this event highly unlikely. As an additional safety measure, it might be considerable, for further developments, to add a “water trap” on the output of the vacuum pump.
5. Conclusions
The most challenging issue we faced during the development of the process was the pumping of the viscous reacting mixture. This was solved by means of a peristaltic pump (P, Figure 2) and by a heated mixing chamber (M) able to lower the viscosity and increase urea solubility. The technical limits of this implementation were evidenced when we attempted to operate at a pressure lower than 400 mmHg. This pressure constraint is thought to be one of the factors limiting the GC-isolated yield to ~42%; therefore, the employment of different pumping hardware could significatively improve performance. Moreover, to the best of our knowledge, this is the first example of a multiple pass tubular reactor applied to the conversion of Gly into GC. This plant layout (Figure 2) allowed for the management of a kinetically slow process within a confined, highly thermally controlled reactor (R). The same layout also features a closed loop with a single evacuation point (E), which has the particular advantage of collecting the co-produced ammonia and recycling it to urea (Figure 1). This feature, coupled with the modest molar excess of urea required for the transformation, demonstrate the opportunity to sustainably transfer an equimolar amount of CO2 to Gly, beneficially contributing to the entire carbon cycle of the biodiesel industry.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/bioengineering9120778/s1, Figure S1: Typical 1H NMR spectra (CDCl3, 400 MHz) of a GC-rich raw mixture; Figure S2: Typical 1H NMR spectra (CDCl3, 400 MHz) of a DGTC-rich raw mixture.
Author Contributions
Conceptualization, F.R. and B.A.; validation, A.U. and L.F.; investigation, B.A. and E.C.; data curation, B.A.; writing—original draft preparation, F.R.; writing—review and editing, L.R., B.A. and L.F.; visualization, A.U.; supervision, F.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
The data presented in this study are available in the article and supplementary material.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Pörtner, H.-O.; Roberts, D.C.; Tignor, M.; Poloczanska, E.S.; Mintenbeck, K.; Alegría, A.; Craig, M.; Langsdorf, S.; Löschke, S.; Möller, V.; et al. (Eds.) Contribution of Working Group II to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change. In IPCC 2022: Climate Change 2022: Impacts, Adaptation and Vulnerability; Cambridge University Press: Cambridge, UK; New York, NY, USA, 2022; p. 3056. [Google Scholar] [CrossRef]
- Ubando, A.T.; Felix, C.B.; Chen, W.-H. Biorefineries in circular bioeconomy: A comprehensive review. Bioresour. Technol. 2020, 299, 122585. [Google Scholar] [CrossRef]
- Xu, H.; Ou, L.; Li, Y.; Hawkins, T.R.; Wang, M. Life Cycle Greenhouse Gas Emissions of Biodiesel and Renewable Diesel Production in the United States. Environ. Sci. Technol. 2022, 56, 7512–7521. [Google Scholar] [CrossRef] [PubMed]
- Nanda, M.R.; Zhang, Y.; Yuan, Z.; Qin, W.; Ghaziaskar, H.S.; Xu, C. Catalytic conversion of glycerol for sustainable production of solketal as a fuel additive: A review. Renew. Sustain. Energy Rev. 2015, 56, 1022–1031. [Google Scholar] [CrossRef]
- Zandalinas, S.I.; Fritschi, F.B.; Mittler, R. Global Warming, Climate Change, and Environmental Pollution: Recipe for a Multifactorial Stress Combination Disaster. Trends Plant Sci. 2021, 26, 588–599. [Google Scholar] [CrossRef] [PubMed]
- Roncaglia, F.; Forti, L.; D’Anna, S.; Maletti, L. An Expedient Catalytic Process to Obtain Solketal from Biobased Glycerol. Processes 2021, 9, 141. [Google Scholar] [CrossRef]
- Wu, F.; Jiang, H.; Zhu, X.; Lu, R.; Shi, L.; Lu, F. Effect of Tungsten Species on Selective Hydrogenolysis of Glycerol to 1,3-Propanediol. ChemSusChem 2020, 14, 569–581. [Google Scholar] [CrossRef]
- Sittijunda, S.; Reungsang, A. Valorization of crude glycerol into hydrogen, 1,3-propanediol, and ethanol in an up-flow anaerobic sludge blanket (UASB) reactor under thermophilic conditions. Renew. Energy 2020, 161, 361–372. [Google Scholar] [CrossRef]
- Zheng, Z.; Luo, M.; Yu, J.; Wang, J.; Ji, J. Novel Process for 1,3-Dihydroxyacetone Production from Glycerol. 1. Technological Feasibility Study and Process Design. Ind. Eng. Chem. Res. 2012, 51, 3715–3721. [Google Scholar] [CrossRef]
- Selva, M.; Fabris, M. The reaction of glycerol carbonate with primary aromatic amines in the presence of Y- and X-faujasites: The synthesis of N-(2,3-dihydroxy)propyl anilines and the reaction mechanism. Green Chem. 2009, 11, 1161–1172. [Google Scholar] [CrossRef]
- Nohra, B.; Candy, L.; Blanco, J.; Raoul, Y.; Mouloungui, Z. Synthesis of five and six-membered cyclic glycerilic carbonates bearing exocyclic urethane functions. Eur. J. Lipid Sci. Technol. 2012, 115, 111–122. [Google Scholar] [CrossRef]
- Quienne, B.; Poli, R.; Pinaud, J.; Caillol, S. Enhanced aminolysis of cyclic carbonates by β-hydroxylamines for the production of fully biobased polyhydroxyurethanes. Green Chem. 2021, 23, 1678–1690. [Google Scholar] [CrossRef]
- Fricke, N.; Keul, H.; Möller, M. Carbonate Couplers and Functional Cyclic Carbonates from Amino Acids and Glucosamine. Macromol. Chem. Phys. 2009, 210, 242–255. [Google Scholar] [CrossRef]
- Bao, Y.-M.; Shen, G.-R.; He, J.; Li, Y.-S. Water-soluble hyperbranched poly(ester urethane)s based on d,l-alanine: Isocyanate-free synthesis, post-functionalization and application. Green Chem. 2012, 14, 2243–2250. [Google Scholar] [CrossRef]
- Galletti, G.; Prete, P.; Vanzini, S.; Cucciniello, R.; Fasolini, A.; De Maron, J.; Cavani, F.; Tabanelli, T. Glycerol Carbonate as a Versatile Alkylating Agent for the Synthesis of β-Aryloxy Alcohols. ACS Sustain. Chem. Eng. 2022, 10, 10922–10933. [Google Scholar] [CrossRef]
- Ghandi, M.; Mostashari, A.; Karegar, M.; Barzegar, M. Efficient Synthesis of α-Monoglycerides via Solventless Condensation of Fatty Acids with Glycerol Carbonate. J. Am. Oil Chem. Soc. 2007, 84, 681–685. [Google Scholar] [CrossRef]
- Simao, A.-C.; Lynikaite-Pukleviciene, B.; Rousseau, C.; Tatibouet, A.; Cassel, S.; Sackus, A.; Rauter, A.; Rollin, P. 1,2-Glycerol Carbonate: A Versatile Renewable Synthon. Lett. Org. Chem. 2006, 3, 744–748. [Google Scholar] [CrossRef]
- da Costa, P.L.F.; Melo, V.N.; Guimarães, B.M.; Schuler, M.; Pimenta, V.; Rollin, P.; Tatibouët, A.; de Oliveira, R.N. Glycerol carbonate in Ferrier reaction: Access to new enantiopure building blocks to develop glycoglycerolipid analogues. Carbohydr. Res. 2016, 436, 1–10. [Google Scholar] [CrossRef]
- Carré, C.; Zoccheddu, H.; Delalande, S.; Pichon, P.; Avérous, L. Synthesis and characterization of advanced biobased thermoplastic nonisocyanate polyurethanes, with controlled aromatic-aliphatic architectures. Eur. Polym. J. 2016, 84, 759–769. [Google Scholar] [CrossRef]
- Rousseau, J.; Rousseau, C.; Lynikaite, B.; Sackus, A.; de Leon, C.; Rollin, P.; Tatibouet, A. Tosylated glycerol carbonate, a versatile bis-electrophile to access new functionalized glycidol derivatives. Tetrahedron 2009, 65, 8571–8581. [Google Scholar] [CrossRef]
- Legros, V.; Taing, G.; Buisson, P.; Schuler, M.; Bostyn, S.; Rousseau, J.; Sinturel, C.; Tatibouët, A. Activated Glycerol Carbonates, Versatile Reagents with Aliphatic Amines: Formation and Reactivity of Glycidyl Carbamates and Trialkylamines. Eur. J. Org. Chem. 2017, 2017, 5032–5043. [Google Scholar] [CrossRef]
- Parzuchowski, P.G.; Świderska, A.; Roguszewska, M.; Frączkowski, T.; Tryznowski, M. Amine functionalized polyglycerols obtained by copolymerization of cyclic carbonate monomers. Polymer 2018, 151, 250–260. [Google Scholar] [CrossRef]
- Rokicki, G.; Rakoczy, P.; Parzuchowski, P.; Sobiecki, M. Hyperbranched aliphatic polyethers obtained from environmentally benign monomer: Glycerol carbonate. Green Chem. 2005, 7, 529–539. [Google Scholar] [CrossRef]
- Vogt, L.; Ruther, F.; Salehi, S.; Boccaccini, A.R. Poly(Glycerol Sebacate) in Biomedical Applications—A Review of the Recent Literature. Adv. Heal. Mater. 2021, 10, e2002026. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Grinstaff, M.W. Recent Advances in Glycerol Polymers: Chemistry and Biomedical Applications. Macromol. Rapid Commun. 2014, 35, 1906–1924. [Google Scholar] [CrossRef]
- Ekladious, I.; Liu, R.; Zhang, H.; Foil, D.H.; Todd, D.A.; Graf, T.N.; Padera, R.F.; Oberlies, N.H.; Colson, Y.L.; Grinstaff, M.W. Synthesis of poly(1,2-glycerol carbonate)–paclitaxel conjugates and their utility as a single high-dose replacement for multi-dose treatment regimens in peritoneal cancer. Chem. Sci. 2017, 8, 8443–8450. [Google Scholar] [CrossRef]
- Kundys, A.; Plichta, A.; Florjańczyk, Z.; Zychewicz, A.; Lisowska, P.; Parzuchowski, P.; Wawrzyńska, E. Multi-arm star polymers of lactide obtained in melt in the presence of hyperbranched oligoglycerols. Polym. Int. 2016, 65, 927–937. [Google Scholar] [CrossRef]
- Mamiński, M.; Czarzasta, M.; Parzuchowski, P. Wood adhesives derived from hyperbranched polyglycerol cross-linked with hexamethoxymethyl melamines. Int. J. Adhes. Adhes. 2011, 31, 704–707. [Google Scholar] [CrossRef]
- Jansen, J.F.G.A.; Dias, A.A.; Dorschu, M.; Coussens, B. Fast Monomers: Factors Affecting the Inherent Reactivity of Acrylate Monomers in Photoinitiated Acrylate Polymerization. Macromolecules 2003, 36, 3861–3873. [Google Scholar] [CrossRef]
- Mhanna, A.; Sadaka, F.; Boni, G.; Brachais, C.-H.; Brachais, L.; Couvercelle, J.-P.; Plasseraud, L.; Lecamp, L. Photopolymerizable Synthons from Glycerol Derivatives. J. Am. Oil Chem. Soc. 2013, 91, 337–348. [Google Scholar] [CrossRef]
- Sacripante, G.G.; Zhou, K.; Farooque, M. Sustainable Polyester Resins Derived from Rosins. Macromolecules 2015, 48, 6876–6881. [Google Scholar] [CrossRef]
- Ibn El Alami, M.S.; Suisse, I.; Fadlallah, S.; Sauthier, M.; Visseaux, M. Telomerization of 1,3-butadiene with glycerol carbonate and subsequent ring-opening lactone co-polymerization. Comptes Rendus. Chim. 2016, 19, 299–305. [Google Scholar] [CrossRef]
- Wu, Z.; Tang, L.; Dai, J.; Qu, J. Synthesis and properties of aqueous cyclic carbonate dispersion and non-isocyanate polyurethanes under atmospheric pressure. Prog. Org. Coatings 2019, 136, 5209. [Google Scholar] [CrossRef]
- Ke, J.; Li, X.; Jiang, S.; Liang, C.; Wang, J.; Kang, M.; Li, Q.; Zhao, Y. Promising approaches to improve the performances of hybrid non-isocyanate polyurethane. Polym. Int. 2018, 68, 651–660. [Google Scholar] [CrossRef]
- Annunziata, L.; Diallo, A.K.; Fouquay, S.; Michaud, G.; Simon, F.; Brusson, J.-M.; Carpentier, J.-F.; Guillaume, S.M. α,ω-Di(glycerol carbonate) telechelic polyesters and polyolefins as precursors to polyhydroxyurethanes: An isocyanate-free approach. Green Chem. 2013, 16, 1947–1956. [Google Scholar] [CrossRef]
- Duval, C.; Kébir, N.; Jauseau, R.; Burel, F. Organocatalytic synthesis of novel renewable non-isocyanate polyhydroxyurethanes. J. Polym. Sci. Part A: Polym. Chem. 2015, 54, 758–764. [Google Scholar] [CrossRef]
- Quienne, B.; Kasmi, N.; Dieden, R.; Caillol, S.; Habibi, Y. Isocyanate-Free Fully Biobased Star Polyester-Urethanes: Synthesis and Thermal Properties. Biomacromolecules 2020, 21, 1943–1951. [Google Scholar] [CrossRef] [PubMed]
- Ekin, A.; Webster, D.C. Synthesis and Characterization of Novel Hydroxyalkyl Carbamate and Dihydroxyalkyl Carbamate Terminated Poly(dimethylsiloxane) Oligomers and Their Block Copolymers with Poly(ε-caprolactone). Macromolecules 2006, 39, 8659–8668. [Google Scholar] [CrossRef]
- Tachibana, Y.; Shi, X.; Graiver, D.; Narayan, R. The Use of Glycerol Carbonate in the Preparation of Highly Branched Siloxy Polymers. Silicon 2014, 7, 5–13. [Google Scholar] [CrossRef]
- Nomanbhay, S.; Ong, M.Y.; Chew, K.W.; Show, P.-L.; Lam, M.K.; Chen, W.-H. Organic Carbonate Production Utilizing Crude Glycerol Derived as By-Product of Biodiesel Production: A Review. Energies 2020, 13, 1483. [Google Scholar] [CrossRef]
- Magniont, C.; Escadeillas, G.; Oms-Multon, C.; De Caro, P. The benefits of incorporating glycerol carbonate into an innovative pozzolanic matrix. Cem. Concr. Res. 2010, 40, 1072–1080. [Google Scholar] [CrossRef]
- Hough, L.; Priddle, J.E.; Theobald, R.S. 363. Carbohydrate carbonates. Part II. Their preparation by ester-exchange methods. J. Chem. Soc. 1962, 9, 1934–1938. [Google Scholar] [CrossRef]
- Shen, Y.; Yang, X.; Song, Y.; Tran, D.K.; Wang, H.; Wilson, J.; Dong, M.; Vazquez, M.; Sun, G.; Wooley, K.L. Complexities of Regioselective Ring-Opening vs. Transcarbonylation-Driven Structural Metamorphosis during Organocatalytic Polymerizations of Five-Membered Cyclic Carbonate Glucose Monomers. JACS Au 2022, 2, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, L.; Cao, Y.; Xu, S.; He, P.; Li, H.; Liu, H. Tris-imidazolinium-based porous poly(ionic liquid)s as an efficient catalyst for decarboxylation of cyclic carbonate to epoxide. RSC Adv. 2021, 11, 14193–14202. [Google Scholar] [CrossRef] [PubMed]
- Ochoa Gómez, J.R.; Gómez de Miranda Jiménez de Aberastul, O.; Blanco Pérez, N.; Maestro Madurga, B.; Prieto Fernández, S. Glycidol Synthesis Method. Patent WO2017017307A1, 30 July 2015. [Google Scholar]
- Teng, W.K.; Ngoh, G.C.; Yusoff, R.; Aroua, M.K. A review on the performance of glycerol carbonate production via catalytic transesterification: Effects of influencing parameters. Energy Convers. Manag. 2014, 88, 484–497. [Google Scholar] [CrossRef]
- Van Mileghem, S.; De Borggraeve, W.M.; Baxendale, I.R. A Robust and Scalable Continuous Flow Process for Glycerol Carbonate. Chem. Eng. Technol. 2018, 41, 12. [Google Scholar] [CrossRef]
- Singh, D.; Reddy, B.; Ganesh, A.; Mahajani, S. Zinc/Lanthanum Mixed-Oxide Catalyst for the Synthesis of Glycerol Carbonate by Transesterification of Glycerol. Ind. Eng. Chem. Res. 2014, 53, 18786–18795. [Google Scholar] [CrossRef]
- Okoye, P.; Abdullah, A.; Hameed, B. Glycerol carbonate synthesis from glycerol and dimethyl carbonate using trisodium phosphate. J. Taiwan Inst. Chem. Eng. 2016, 68, 51–58. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, P.; Cui, P.; Cheng, W.; Zhang, S. Glycerol carbonate synthesis from glycerol and dimethyl carbonate using guanidine ionic liquids. Chin. J. Chem. Eng. 2017, 25, 1182–1186. [Google Scholar] [CrossRef]
- Nascimento, M.A.D.; Gotardo, L.E.; Leão, R.A.C.; de Castro, A.M.; de Souza, R.O.M.A.; Itabaiana, J.I. Enhanced Productivity in Glycerol Carbonate Synthesis under Continuous Flow Conditions: Combination of Immobilized Lipases from Porcine Pancreas and Candida antarctica (CALB) on Epoxy Resins. ACS Omega 2019, 4, 860–869. [Google Scholar] [CrossRef]
- Gérardy, R.; Estager, J.; Luis, P.; Debecker, D.P.; Monbaliu, J.-C.M. Versatile and scalable synthesis of cyclic organic carbonates under organocatalytic continuous flow conditions. Catal. Sci. Technol. 2019, 9, 6841–6851. [Google Scholar] [CrossRef]
- Zheng, Q.; Nishimura, R.; Sato, Y.; Inomata, H.; Ota, M.; Watanabe, M.; Camy, S. Dimethyl carbonate (DMC) synthesis from methanol and carbon dioxide in the presence of ZrO2 solid solutions and yield improvement by applying a natural convection circulation system. Chem. Eng. J. 2021, 429, 132378. [Google Scholar] [CrossRef]
- Liu, H.; Zhu, D.; Jia, B.; Huang, Y.; Cheng, Y.; Luo, X.; Liang, Z. Study on catalytic performance and kinetics of high efficiency CeO2 catalyst prepared by freeze drying for the synthesis of dimethyl carbonate from CO2 and methanol. Chem. Eng. Sci. 2022, 254, 117614. [Google Scholar] [CrossRef]
- Huang, S.; Yan, B.; Wang, S.; Ma, X. Recent advances in dialkyl carbonates synthesis and applications. Chem. Soc. Rev. 2015, 44, 3079–3116. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; He, D. Transformation of CO2 with glycerol to glycerol carbonate by a novel ZnWO4-ZnO catalyst. J. CO2 Util. 2018, 26, 370–379. [Google Scholar] [CrossRef]
- Park, C.-Y.; Nguyen-Phu, H.; Shin, E.W. Glycerol carbonation with CO2 and La2O2CO3/ZnO catalysts prepared by two different methods: Preferred reaction route depending on crystalline structure. Mol. Catal. 2017, 435, 99–109. [Google Scholar] [CrossRef]
- Chaugule, A.A.; Tamboli, A.H.; Kim, H. Ionic liquid as a catalyst for utilization of carbon dioxide to production of linear and cyclic carbonate. Fuel 2017, 200, 316–332. [Google Scholar] [CrossRef]
- He, Y.; Lu, H.; Li, X.; Wu, J.; Pu, T.; Du, W.; Li, H.; Ding, J.; Wan, H.; Guan, G. Insight into the reversible behavior of Lewis–Brønsted basic poly(ionic liquid)s in one-pot two-step chemical fixation of CO2 to linear carbonates. Green Chem. 2021, 23, 8571–8580. [Google Scholar] [CrossRef]
- Giannoccaro, P.; Casiello, M.; Milella, A.; Monopoli, A.; Cotugno, P.; Nacci, A. Synthesis of 5-membered cyclic carbonates by oxidative carbonylation of 1,2-diols promoted by copper halides. J. Mol. Catal. A Chem. 2012, 365, 162–171. [Google Scholar] [CrossRef]
- Glibert, P.M.; Harrison, J.; Heil, C.; Seitzinger, S. Escalating Worldwide use of Urea—A Global Change Contributing to Coastal Eutrophication. Biogeochemistry 2006, 77, 441–463. [Google Scholar] [CrossRef]
- Estiu, G.; Merz, J.K.M. The Hydrolysis of Urea and the Proficiency of Urease. J. Am. Chem. Soc. 2004, 126, 6932–6944. [Google Scholar] [CrossRef] [PubMed]
- Zdanowicz, M. Deep eutectic solvents based on urea, polyols and sugars for starch treatment. Int. J. Biol. Macromol. 2021, 176, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Newman, M.S.; Lala, L.K. Urea as a base in organic reactions. Tetrahedron Lett. 1967, 8, 3267–3269. [Google Scholar] [CrossRef]
- Rodríguez-Santiago, L.; Noguera, M.; Sodupe, M.; Salpin, J.Y.; Tortajada, J. Gas Phase Reactivity of Ni+ with Urea. Mass Spectrometry and Theoretical Studies. J. Phys. Chem. A 2003, 107, 9865–9874. [Google Scholar] [CrossRef]
- Li, Q.; Zhao, N.; Wei, W.; Sun, Y. Catalytic performance of metal oxides for the synthesis of propylene carbonate from urea and 1,2-propanediol. J. Mol. Catal. A: Chem. 2007, 270, 44–49. [Google Scholar] [CrossRef]
- Nguyen-Phu, H.; Shin, E.W. Disordered structure of ZnAl2O4 phase and the formation of a Zn NCO complex in ZnAl mixed oxide catalysts for glycerol carbonylation with urea. J. Catal. 2019, 373, 147–160. [Google Scholar] [CrossRef]
- Elman, A.R.; Davydov, I.E.; Stepanov, A.A. Synthesis of Urea by Ammonolysis of Propylene Carbonate. J. Chem. Chem. Eng. 2018, 12, 26–30. [Google Scholar] [CrossRef]
- Calvino-Casilda, V.; Mul, G.; Fernández, J.; Rubio-Marcos, F.; Bañares, M. Monitoring the catalytic synthesis of glycerol carbonate by real-time attenuated total reflection FTIR spectroscopy. Appl. Catal. A Gen. 2011, 409, 106–112. [Google Scholar] [CrossRef]
- Park, J.-H.; Choi, J.S.; Woo, S.K.; Lee, S.D.; Cheong, M.; Kim, H.S.; Lee, H. Isolation and characterization of intermediate catalytic species in the Zn-catalyzed glycerolysis of urea. Appl. Catal. A Gen. 2012, 433, 35–40. [Google Scholar] [CrossRef]
- Wang, H.; Xin, Z.; Li, Y. Synthesis of Ureas from CO2. Top. Curr. Chem. 2017, 375, 49. [Google Scholar] [CrossRef]
- Ishaq, H.; Siddiqui, O.; Chehade, G.; Dincer, I. A solar and wind driven energy system for hydrogen and urea production with CO2 capturing. Int. J. Hydrogen Energy 2020, 46, 4749–4760. [Google Scholar] [CrossRef]
- Li, J.; Wang, T. Chemical equilibrium of glycerol carbonate synthesis from glycerol. J. Chem. Thermodyn. 2011, 43, 731–736. [Google Scholar] [CrossRef]
- Ochoa-Gómez, J.R.; Gómez-Jiménez-Aberasturi, O.; Ramírez-López, C.; Belsué, M. A Brief Review on Industrial Alternatives for the Manufacturing of Glycerol Carbonate, a Green Chemical. Org. Process. Res. Dev. 2012, 16, 389–399. [Google Scholar] [CrossRef]
- Indran, V.P.; Saud, A.S.H.; Maniam, G.P.; Yusoff, M.M.; Taufiq-Yap, Y.H.; Rahim, M.H.A. Versatile boiler ash containing potassium silicate for the synthesis of organic carbonates. RSC Adv. 2016, 6, 34877–34884. [Google Scholar] [CrossRef]
- Fernandes, G.P.; Yadav, G.D. Selective glycerolysis of urea to glycerol carbonate using combustion synthesized magnesium oxide as catalyst. Catal. Today 2018, 309, 153–160. [Google Scholar] [CrossRef]
- Chaves, D.M.; Da Silva, M.J. A selective synthesis of glycerol carbonate from glycerol and urea over Sn(OH)2: A solid and recyclable in situ generated catalyst. New J. Chem. 2019, 43, 3698–3706. [Google Scholar] [CrossRef]
- Mallesham, B.; Rangaswamy, A.; Rao, B.G.; Rao, T.V.; Reddy, B.M. Solvent-Free Production of Glycerol Carbonate from Bioglycerol with Urea Over Nanostructured Promoted SnO2 Catalysts. Catal. Lett. 2020, 150, 3626–3641. [Google Scholar] [CrossRef]
- Turney, T.W.; Patti, A.; Gates, W.; Shaheen, U.; Kulasegaram, S. Formation of glycerol carbonate from glycerol and urea catalysed by metal monoglycerolates. Green Chem. 2013, 15, 1925–1931. [Google Scholar] [CrossRef]
- Fujita, S.-I.; Yamanishi, Y.; Arai, M. Synthesis of glycerol carbonate from glycerol and urea using zinc-containing solid catalysts: A homogeneous reaction. J. Catal. 2013, 297, 137–141. [Google Scholar] [CrossRef]
- Kulasegaram, S.; Shaheen, U.; Turney, T.W.; Gates, W.P.; Patti, A.F. Zinc monoglycerolate as a catalyst for the conversion of 1,3- and higher diols to diurethanes. RSC Adv. 2015, 5, 47809–47812. [Google Scholar] [CrossRef]
- Nguyen-Phu, H.; Do, L.T.; Shin, E.W. Investigation of glycerolysis of urea over various ZnMeO (Me = Co, Cr, and Fe) mixed oxide catalysts. Catal. Today 2020, 352, 80–87. [Google Scholar] [CrossRef]
- Nguyen-Phu, H.; Park, C.-Y.; Shin, E.W. Dual catalysis over ZnAl mixed oxides in the glycerolysis of urea: Homogeneous and heterogeneous reaction routes. Appl. Catal. A Gen. 2018, 552, 1–10. [Google Scholar] [CrossRef]
- Endah, Y.K.; Kim, M.S.; Choi, J.; Jae, J.; Lee, S.D.; Lee, H. Consecutive carbonylation and decarboxylation of glycerol with urea for the synthesis of glycidol via glycerol carbonate. Catal. Today 2017, 293, 136–141. [Google Scholar] [CrossRef]
- Kaur, A.; Prakash, R.; Ali, A. 1H NMR assisted quantification of glycerol carbonate in the mixture of glycerol and glycerol carbonate. Talanta 2018, 178, 1001–1005. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.P.; Isa, K. Thermal Decomposition of Urea and Urea Derivatives by Simultaneous TG/(DTA)/MS. J. Mass Spectrom. Soc. Jpn. 1998, 46, 299–303. [Google Scholar] [CrossRef]
- Lertlukkanasuk, N.; Phiyanalinmat, S.; Kiatkittipong, W.; Arpornwichanop, A.; Aiouache, F.; Assabumrungrat, S. Reactive distillation for synthesis of glycerol carbonate via glycerolysis of urea. Chem. Eng. Process. Process Intensif. 2013, 70, 103–109. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, Z.; Wu, C.; Qian, Q.; Ma, J.; Jiang, L.; Han, B. Microwave assisted synthesis of glycerol carbonate from glycerol and urea. Pure Appl. Chem. 2017, 90, 1–6. [Google Scholar] [CrossRef]
- Romano, G.; Paradisi, E.; Rosa, R.; Leonelli, C.; Roncaglia, F. Synthesis of Glycerol Carbonate from Glycerol and Urea Using a Microwave Reactor. AMPERE Newsl. 2022, 111, 1–8. Available online: www.ampereeurope.org/issue-111 (accessed on 22 November 2022).
- She, Q.M.; Liu, J.H.; Aymonier, C.; Zhou, C.H. In situ fabrication of layered double hydroxide film immobilizing gold nanoparticles in capillary microreactor for efficient catalytic carbonylation of glycerol. Mol. Catal. 2021, 513, 111825. [Google Scholar] [CrossRef]
- Plutschack, M.B.; Pieber, B.; Gilmore, K.; Seeberger, P.H. The Hitchhiker’s Guide to Flow Chemistry II. Chem. Rev. 2017, 117, 11796–11893. [Google Scholar] [CrossRef]
- Tennant, A.J.; Krause, M.J.; Kujawski, M.P.; Sherren, B.T. Compositions de Polyuréthane. Patent WO2021222192A1, 29 April 2020. [Google Scholar]
- Aresta, M.; Dibenedetto, A.; di Bitonto, L. New efficient and recyclable catalysts for the synthesis of di- and tri-glycerol carbonates. RSC Adv. 2015, 5, 64433–64443. [Google Scholar] [CrossRef]
- Atlantic Richfield Company. Patent GB1 458 595 A, 15 December 1976.
- Arne Them Jensen. Subtilases. Patent EP0 611 243 A1, 21 March 2021. [Google Scholar]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).