
Preparation and Identification of a Monoclonal Antibody against the Pseudorabies Virus gE Glycoprotein through a Novel Strategy



 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Viruses, Cells, Experimental Animals, and Main Reagents
2.2. Immunization of Mice
2.3. Preparation of the mAB
2.4. Indirect ELISA
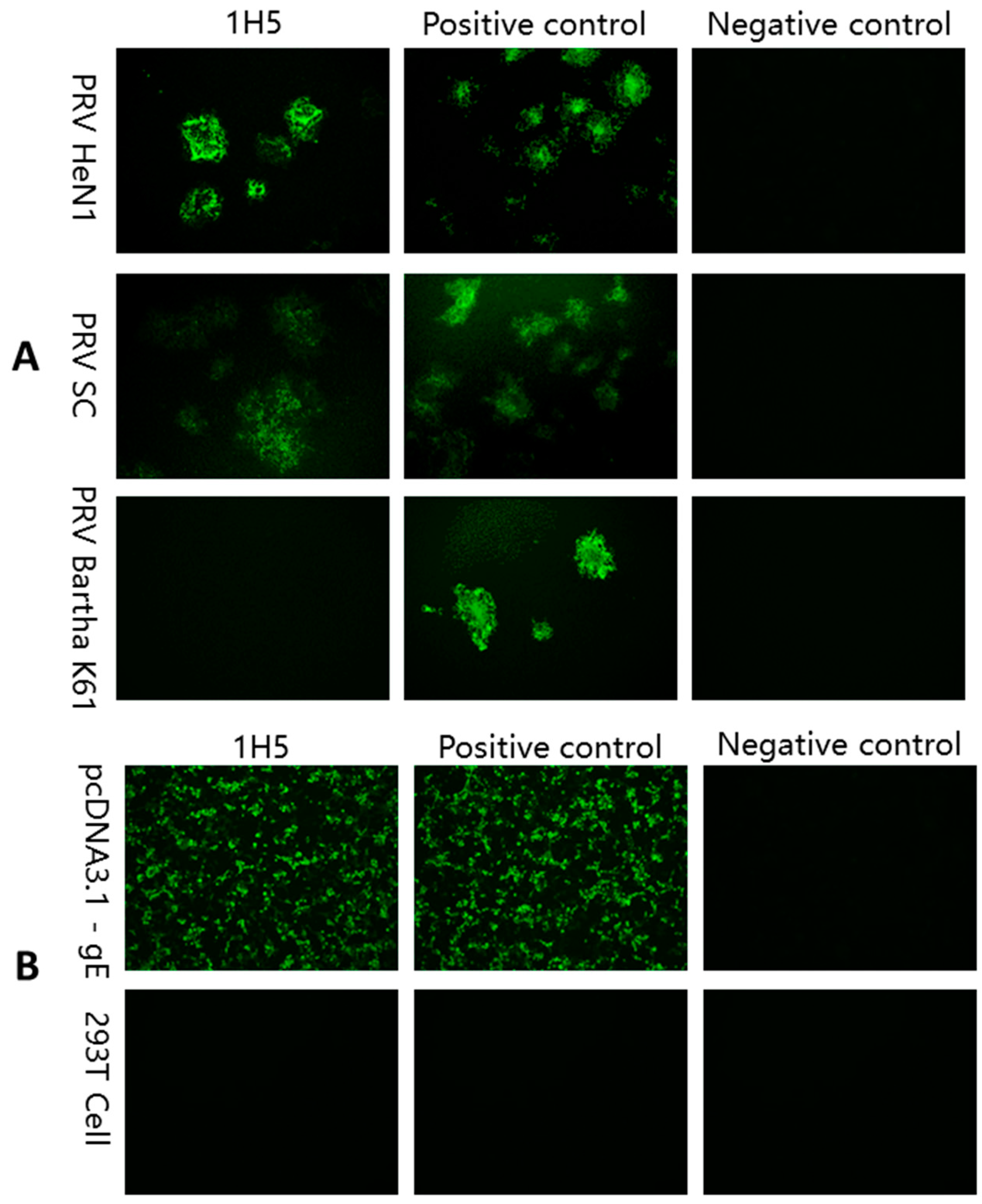
2.5. IFA
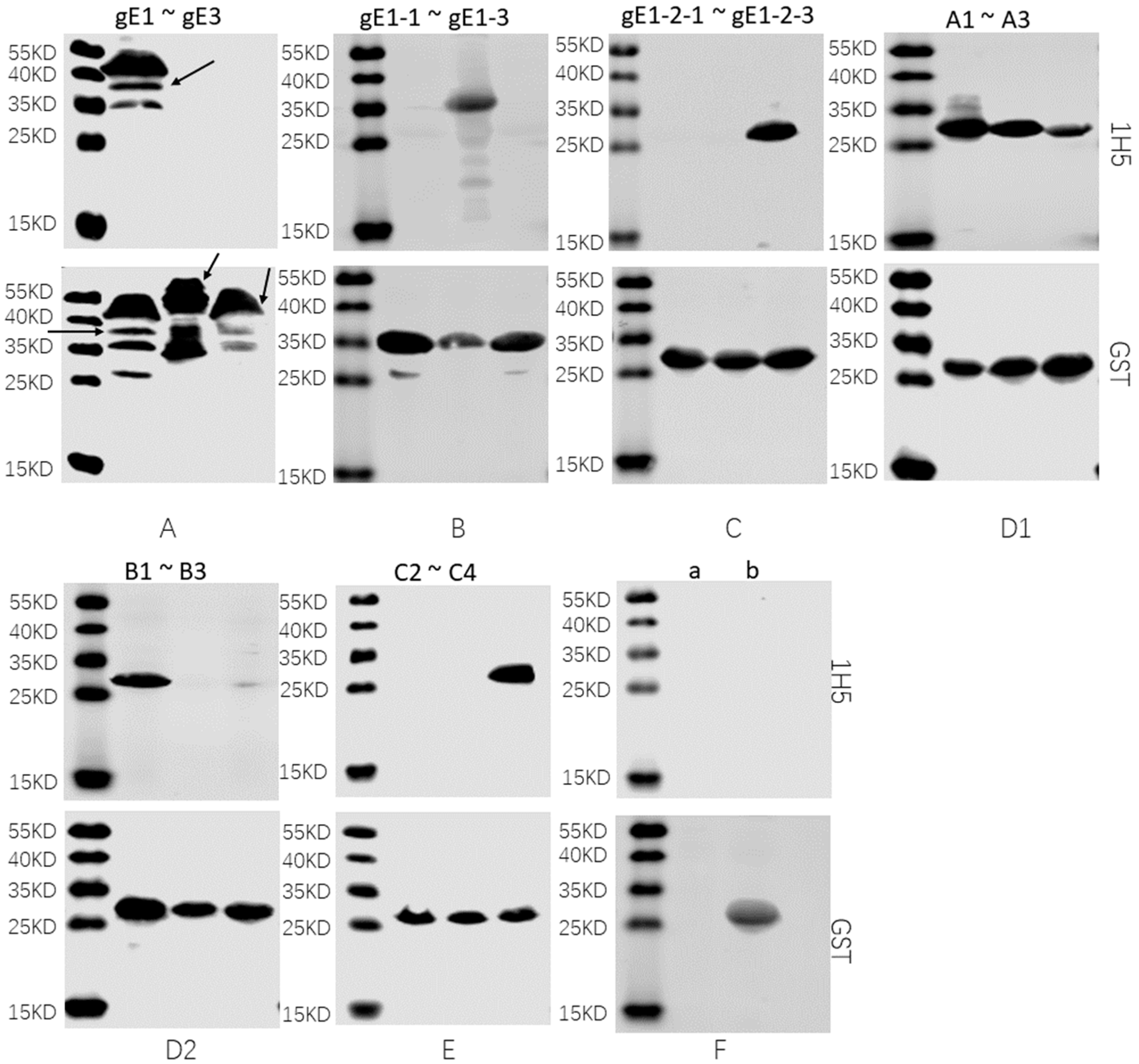
2.6. Western Blotting
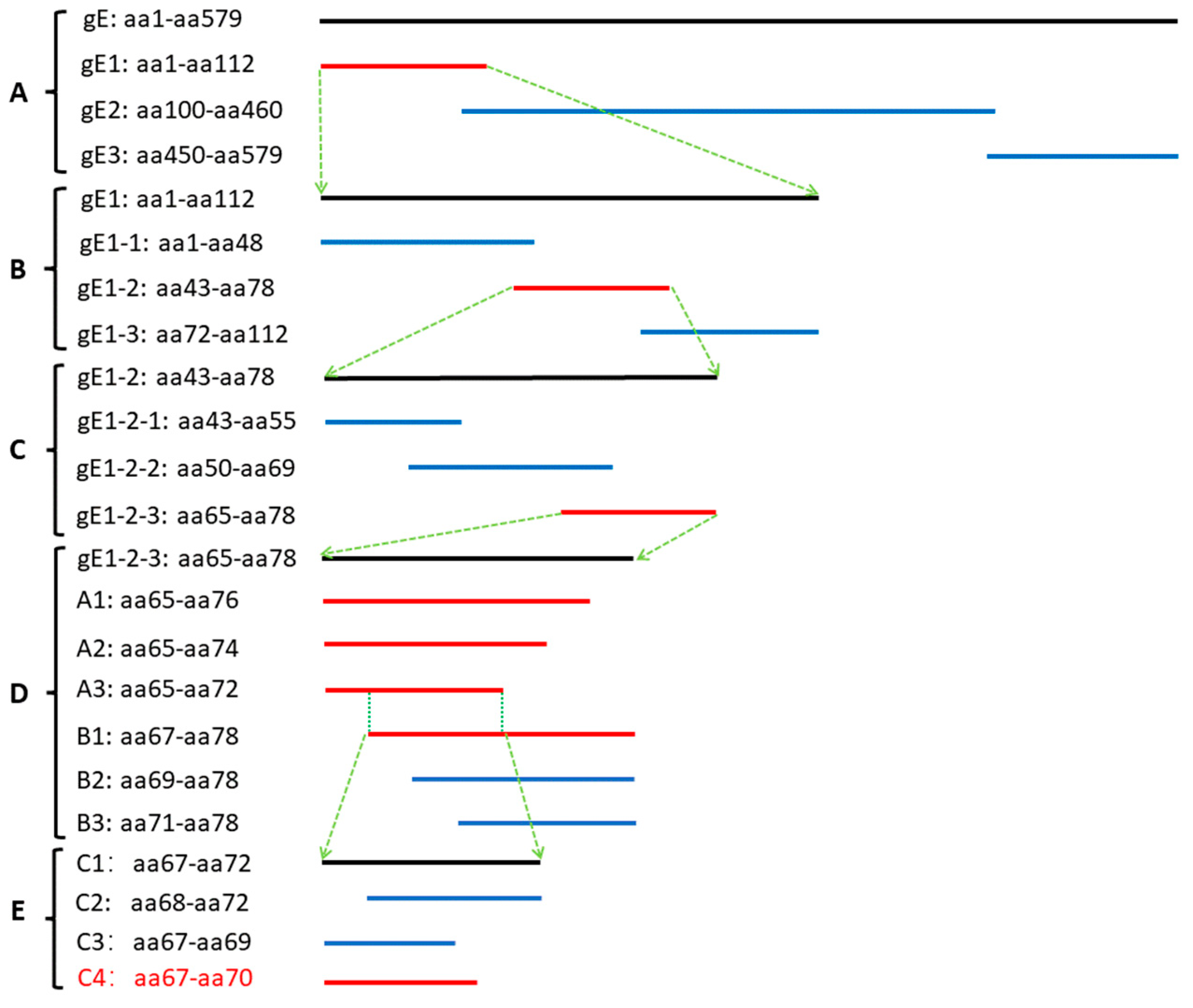
2.7. Preliminary Analysis of Antigenic Epitopes
3. Results
3.1. Immunization of Mice
3.2. Preparation of mAB
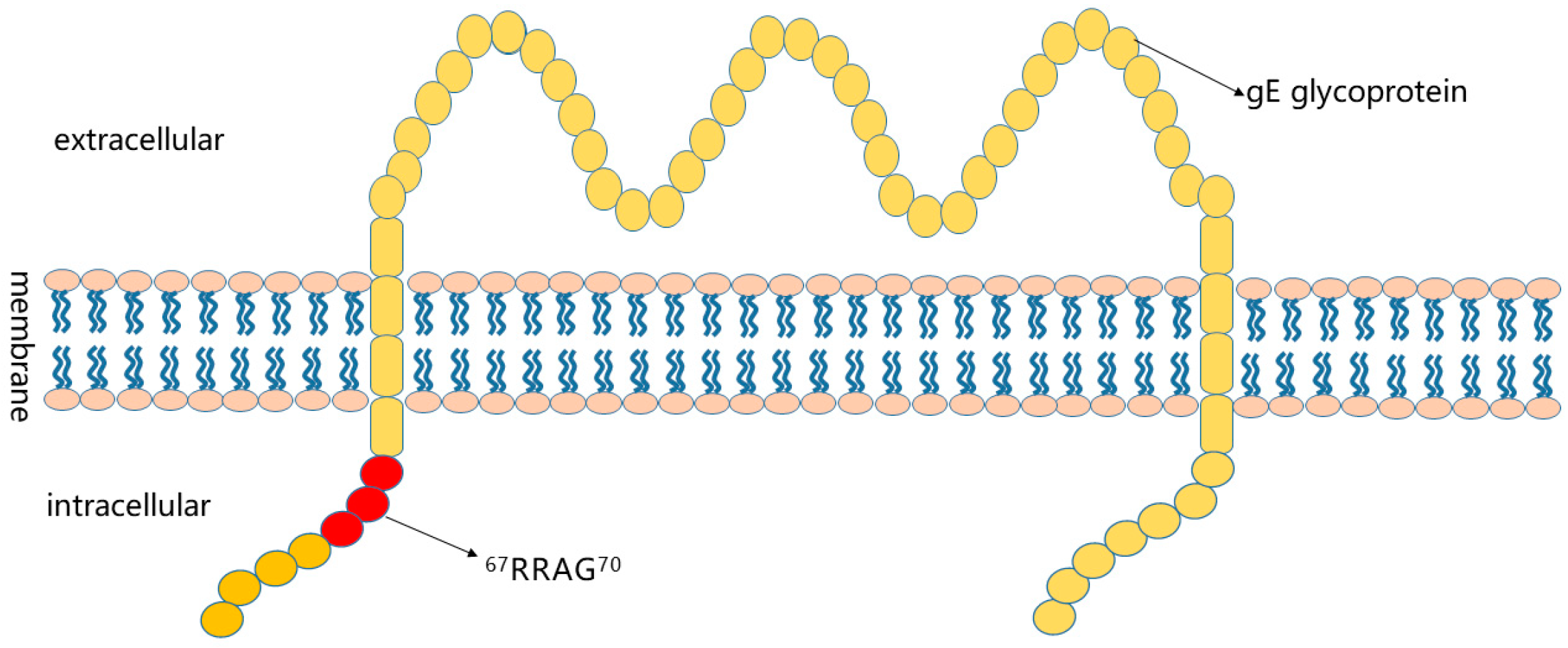
3.3. Preliminary Analysis of Antigenic Epitopes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bitsch, V.; Knox, B. On pseudorabies in carnivores in Denmark. II. The blue fox (Alopex lagopus). Acta Vet. Scand. 1971, 12, 285–292. [Google Scholar]
- Glass, C.M.; McLean, R.G.; Katz, J.B.; Maehr, D.S.; Cropp, C.B.; Kirk, L.J.; McKeirnan, A.J.; Evermann, J.F. Isolation of pseudorabies (Aujeszky’s disease) virus from a Florida panther. J. Wildl. Dis. 1994, 30, 180–184. [Google Scholar] [PubMed]
- Delva, J.L.; Nauwynck, H.J.; Mettenleiter, T.C.; Favoreel, H.W. The Attenuated Pseudorabies Virus Vaccine Strain Bartha K61: A Brief Review on the Knowledge Gathered During 60 Years of Research. Pathogens 2020, 9, 897. [Google Scholar]
- Ketusing, N.; Reeves, A.; Portacci, K.; Yano, T.; Olea-Popelka, F.; Keefe, T.; Salman, M. Evaluation of strategies for the eradication of Pseudorabies virus (Aujeszky’s disease) in commercial swine farms in Chiang-Mai and Lampoon Provinces, Thailand, using a simulation disease spread model. Transbound. Emerg. Dis. 2014, 61, 169–176. [Google Scholar]
- McFerran, J.B.; Dow, C. Studies on immunisation of pigs with the Bartha strain of Aujeszky’s disease virus. Res. Vet. Sci. 1975, 19, 17–22. [Google Scholar] [CrossRef]
- An, T.Q.; Peng, J.M.; Tian, Z.J.; Zhao, H.Y.; Li, N.; Liu, Y.M.; Chen, J.Z.; Leng, C.L.; Sun, Y.; Chang, D.; et al. Pseudorabies virus variant in Bartha-K61-vaccinated pigs, China, 2012. Emerg. Infect. Dis. 2013, 19, 1749–1755. [Google Scholar] [CrossRef]
- Peng, J.; An, T.; Zhao, H.; Liu, Y.; Chen, J.; Leng, C.; Sun, Y.; Chang, D.; Tian, Z.; Tong, G. Identification and antigenic variation of new epidemiology of pseudorabies virus from swine. Zhongguo Yufang Shouyi Xuebao/Chin. J. Prev. Vet. Med. 2013, 35, 1–4. [Google Scholar]
- Sun, Y.; Wang, X.; Liang, W.; Xie, S.; Peng, Z.; Chen, H.; Hua, L.; Song, W.; Tang, X.; Chen, H.; et al. Epidemiological and evolutionary characteristics of pseudorabies virus in China in 2018. Acta Vet. Zootech. Sin. 2020, 51, 584–593. [Google Scholar]
- Yao, J.; Li, J.; Gao, L.; He, Y.; Xie, J.; Zhu, P.; Zhang, Y.; Zhang, X.; Duan, L.; Yang, S.; et al. Epidemiological Investigation and Genetic Analysis of Pseudorabies Virus in Yunnan Province of China from 2017 to 2021. Viruses 2022, 14, 895. [Google Scholar]
- Hampl, H.; Ben-Porat, T.; Ehrlicher, L.; Habermehl, K.O.; Kaplan, A.S. Characterization of the envelope proteins of pseudorabies virus. J. Virol. 1984, 52, 583–590. [Google Scholar] [CrossRef]
- Lukacs, N.; Thiel, H.J.; Mettenleiter, T.C.; Rziha, H.J. Demonstration of three major species of pseudorabies virus glycoproteins and identification of a disulfide-linked glycoprotein complex. J. Virol. 1985, 53, 166–173. [Google Scholar]
- Gerdts, V.; Beyer, J.; Lomniczi, B.; Mettenleiter, T.C. Pseudorabies virus expressing bovine herpesvirus 1 glycoprotein B exhibits altered neurotropism and increased neurovirulence. J. Virol. 2000, 74, 817–827. [Google Scholar]
- Kit, S. Genetically engineered vaccines for control of Aujeszky’s disease (pseudorabies). Vaccine 1990, 8, 420–424. [Google Scholar] [CrossRef]
- Wu, C.Y.; Liao, C.M.; Chi, J.N.; Chien, M.S.; Huang, C. Growth properties and vaccine efficacy of recombinant pseudorabies virus defective in glycoprotein E and thymidine kinase genes. J. Biotechnol. 2016, 229, 58–64. [Google Scholar]
- Yang, D.K.; Kim, H.H.; Choi, S.S.; Hyun, B.H.; Song, J.Y. An oral Aujeszky’s disease vaccine (YS-400) induces neutralizing antibody in pigs. Clin. Exp. Vaccine Res. 2016, 5, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Freuling, C.M.; Muller, T.F.; Mettenleiter, T.C. Vaccines against pseudorabies virus (PrV). Vet. Microbiol. 2017, 206, 3–9. [Google Scholar]
- van Oirschot, J.T.; Gielkens, A.L.; Moormann, R.J.; Berns, A.J. Marker vaccines, virus protein-specific antibody assays and the control of Aujeszky’s disease. Vet. Microbiol. 1990, 23, 85–101. [Google Scholar]
- Wu, T.; Peng, J.; Tian, Z.; Wang, Q.; Chen, Z.; Zhao, R. Preparation of the monoclonal antibody against the gE protein and identification of the B-cell epitope in gE of pseudorabies virus. Zhongguo Yufang Shouyi Xuebao/Chin. J. Prev. Vet. Med. 2017, 39, 143–147. [Google Scholar]
- Zhang, P.; Lv, L.; Sun, H.; Li, S.; Fan, H.; Wang, X.; Bai, J.; Jiang, P. Identification of linear B cell epitope on gB, gC, and gE proteins of porcine pseudorabies virus using monoclonal antibodies. Vet. Microbiol. 2019, 234, 83–91. [Google Scholar] [PubMed]
- Xu, J.J.; Wu, J.Q.; Cheng, X.F.; Tong, W.; Zheng, H.; Zhu, H.J.; Liu, Y.T.; Jiang, Y.F.; Gao, F.; Yu, H.; et al. Identification of two novel epitopes targeting glycoprotein E of pseudorabies virus using monoclonal antibodies. Biochem. Biophys. Res. Commun. 2019, 519, 330–336. [Google Scholar] [CrossRef]
- Plagemann, P.G.; Rowland, R.R.; Faaberg, K.S. The primary neutralization epitope of porcine respiratory and reproductive syndrome virus strain VR-2332 is located in the middle of the GP5 ectodomain. Arch. Virol. 2002, 147, 2327–2347. [Google Scholar]
- Chang, H.T.; Liu, H.M.; Guo, Z.D.; Du, J.M.; Zhao, J.; Chen, L.; Yang, X.; Wang, X.W.; Yao, H.X.; Wang, C.Q. Investigation of etiology of massive infection with porcine pseudorabies virus in Henan and neighboring Provinces. Bing Du Xue Bao 2014, 30, 441–449. [Google Scholar]
- Tong, W.; Liu, F.; Zheng, H.; Liang, C.; Zhou, Y.J.; Jiang, Y.F.; Shan, T.L.; Gao, F.; Li, G.X.; Tong, G.Z. Emergence of a Pseudorabies virus variant with increased virulence to piglets. Vet. Microbiol. 2015, 181, 236–240. [Google Scholar] [CrossRef]
- Fuchs, W.; Granzow, H.; Klupp, B.G.; Karger, A.; Michael, K.; Maresch, C.; Klopfleisch, R.; Mettenleiter, T.C. Relevance of the interaction between alphaherpesvirus UL3.5 and UL48 proteins for virion maturation and neuroinvasion. J. Virol. 2007, 81, 9307–9318. [Google Scholar]
- Kimman, T.G.; Pol, J.M.; de Wind, N.; Oei-Lie, N.; Berns, A.J.; Gielkens, A.L. Role of different genes in the virulence and pathogenesis of Aujeszky’s disease virus. Vet. Microbiol. 1992, 33, 45–52. [Google Scholar] [CrossRef]
- Sun, Y.; Luo, Y.; Wang, C.H.; Yuan, J.; Li, N.; Song, K.; Qiu, H.J. Control of swine pseudorabies in China: Opportunities and limitations. Vet. Microbiol. 2016, 183, 119–124. [Google Scholar]
- Porowinska, D.; Wujak, M.; Roszek, K.; Komoszynski, M. Prokaryotic expression systems. Postep. Hig. Med. Dosw. 2013, 67, 119–129. [Google Scholar]
- Nosaki, S.; Hoshikawa, K.; Ezura, H.; Miura, K. Transient protein expression systems in plants and their applications. Plant Biotechnol. 2021, 38, 297–304. [Google Scholar]
- Popa, C.M.; Tabuchi, M.; Valls, M. Modification of Bacterial Effector Proteins Inside Eukaryotic Host Cells. Front. Cell. Infect. Microbiol. 2016, 6, 73. [Google Scholar]
- Tusnady, G.E.; Simon, I. Principles governing amino acid composition of integral membrane proteins: Application to topology prediction. J. Mol. Biol. 1998, 283, 489–506. [Google Scholar]
- Tusnady, G.E.; Simon, I. The HMMTOP transmembrane topology prediction server. Bioinformatics 2001, 17, 849–850. [Google Scholar]
- Tirabassi, R.S.; Townley, R.A.; Eldridge, M.G.; Enquist, L.W. Characterization of pseudorabies virus mutants expressing carboxy-terminal truncations of gE: Evidence for envelope incorporation, virulence, and neurotropism domains. J. Virol. 1997, 71, 6455–6464. [Google Scholar]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, Z.; Zhang, S.; Xu, H.; Li, W.; Li, C.; Zhao, J.; Gong, B.; Sun, Q.; Xiang, L.; Zhao, H.; et al. Preparation and Identification of a Monoclonal Antibody against the Pseudorabies Virus gE Glycoprotein through a Novel Strategy. Vet. Sci. 2023, 10, 133. https://doi.org/10.3390/vetsci10020133
Guo Z, Zhang S, Xu H, Li W, Li C, Zhao J, Gong B, Sun Q, Xiang L, Zhao H, et al. Preparation and Identification of a Monoclonal Antibody against the Pseudorabies Virus gE Glycoprotein through a Novel Strategy. Veterinary Sciences. 2023; 10(2):133. https://doi.org/10.3390/vetsci10020133
Chicago/Turabian StyleGuo, Zhenyang, Siyu Zhang, Hu Xu, Wansheng Li, Chao Li, Jing Zhao, Bangjun Gong, Qi Sun, Lirun Xiang, Hongyuan Zhao, and et al. 2023. "Preparation and Identification of a Monoclonal Antibody against the Pseudorabies Virus gE Glycoprotein through a Novel Strategy" Veterinary Sciences 10, no. 2: 133. https://doi.org/10.3390/vetsci10020133
APA StyleGuo, Z., Zhang, S., Xu, H., Li, W., Li, C., Zhao, J., Gong, B., Sun, Q., Xiang, L., Zhao, H., Wang, Q., Zhou, G., Tang, Y., An, T., Cai, X., Tian, Z., Zhang, H., & Peng, J. (2023). Preparation and Identification of a Monoclonal Antibody against the Pseudorabies Virus gE Glycoprotein through a Novel Strategy. Veterinary Sciences, 10(2), 133. https://doi.org/10.3390/vetsci10020133