
PCR-Based Detection and Genetic Characterization of Parainfluenza Virus 5 Detected in Pigs in Korea from 2016 to 2018
 , , , and
, , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Screening for PIV5 in Pigs
2.2. Complete Genome Sequencing of PIV5
2.3. Genetic Characterization and Phylogenetic Classification of PIV5
2.4. Evolutionary Analyses of PIV5
3. Results
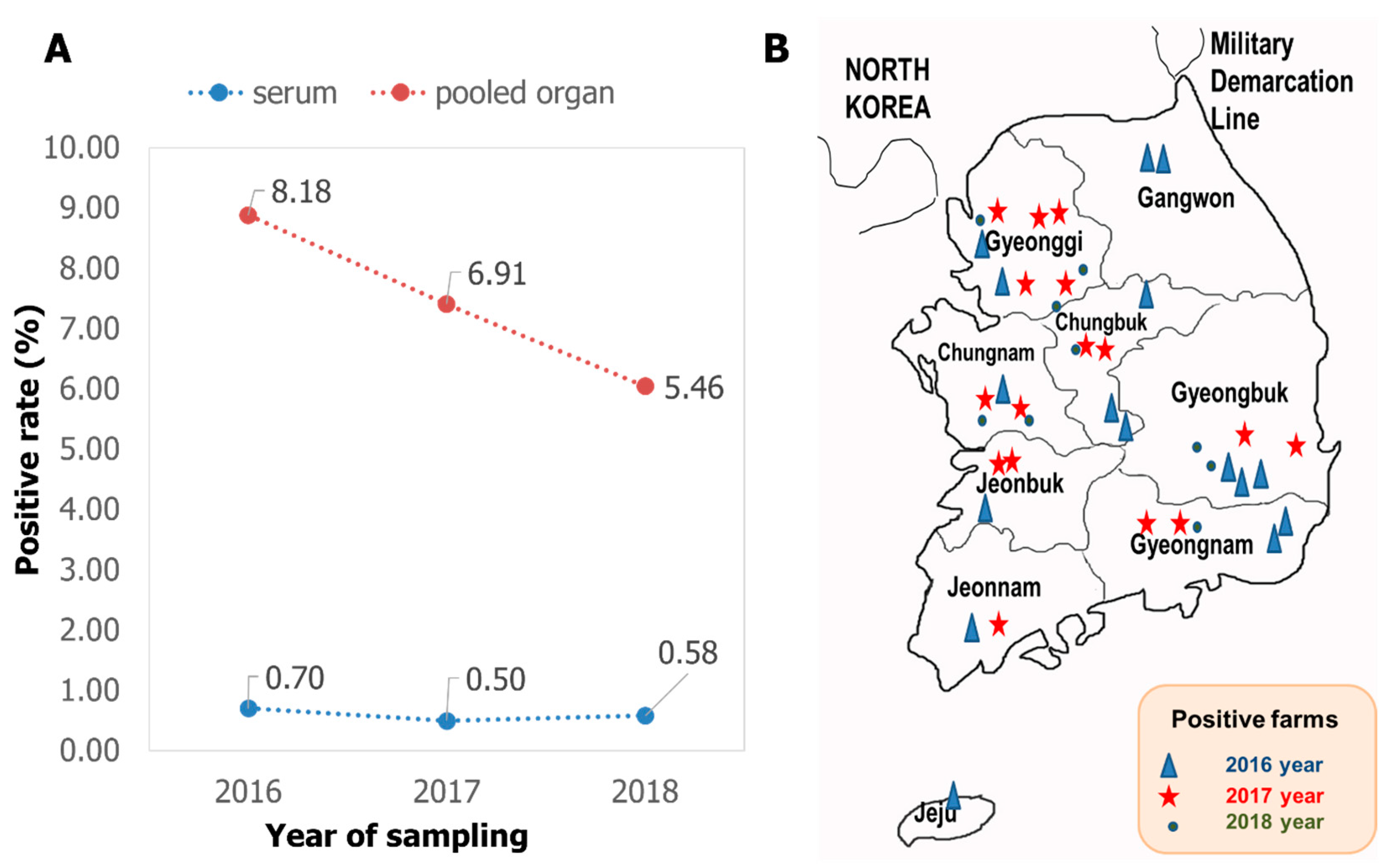
3.1. Detection of PIV5 in Pigs in Korea from 2016 to 2018
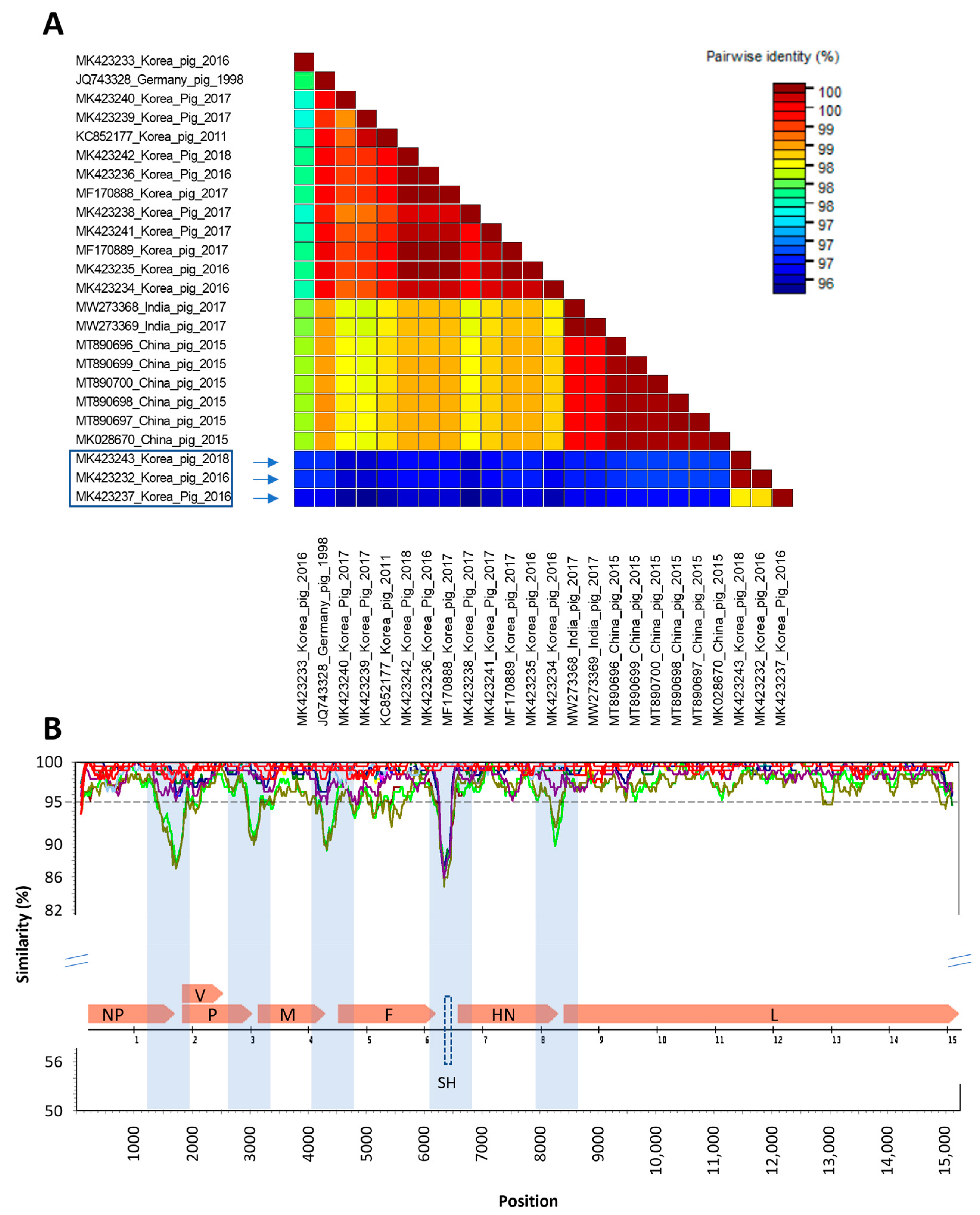
3.2. Genomic Comparison of PIV5 Detected in Pigs in Korea from 2016 to 2018
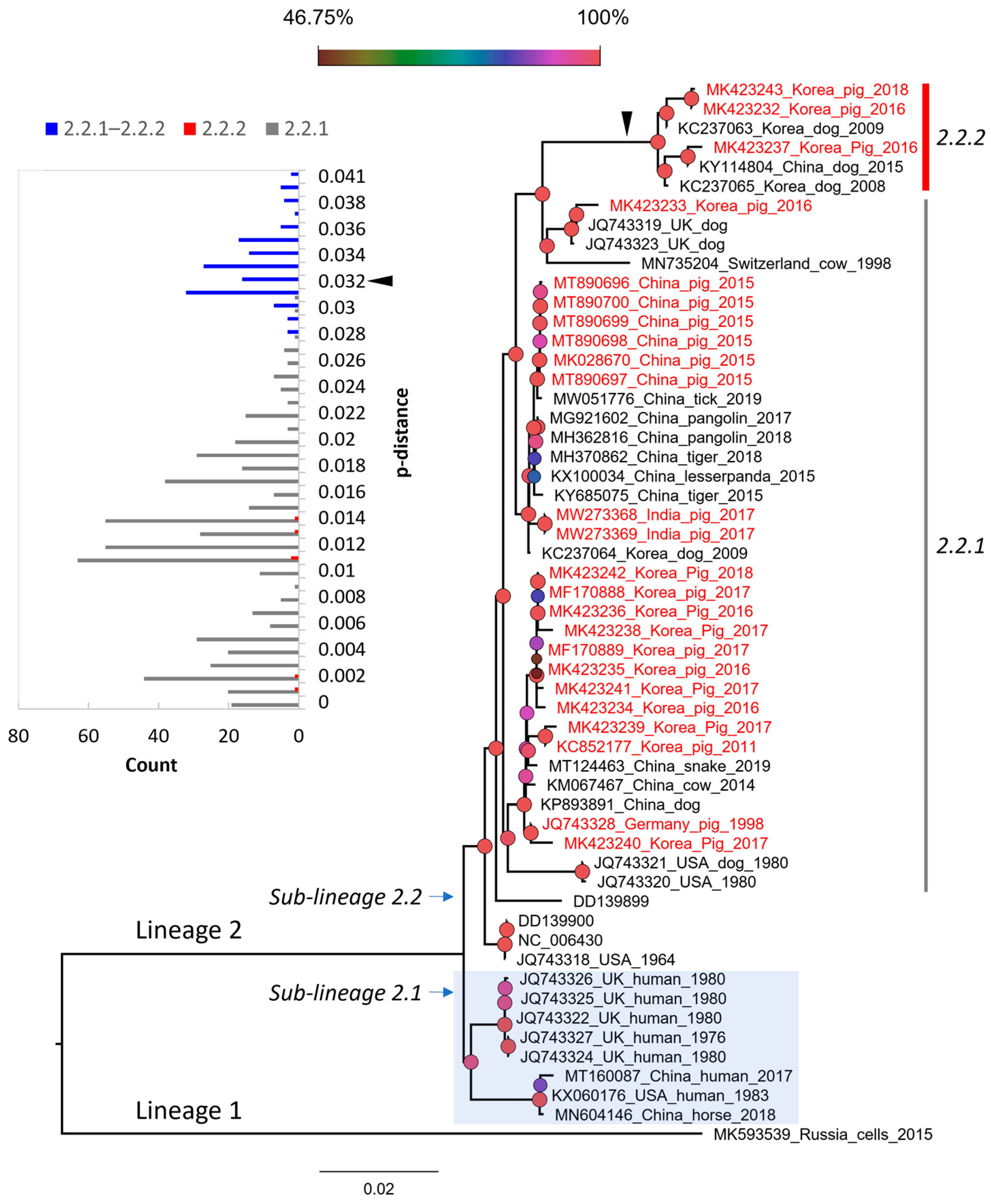
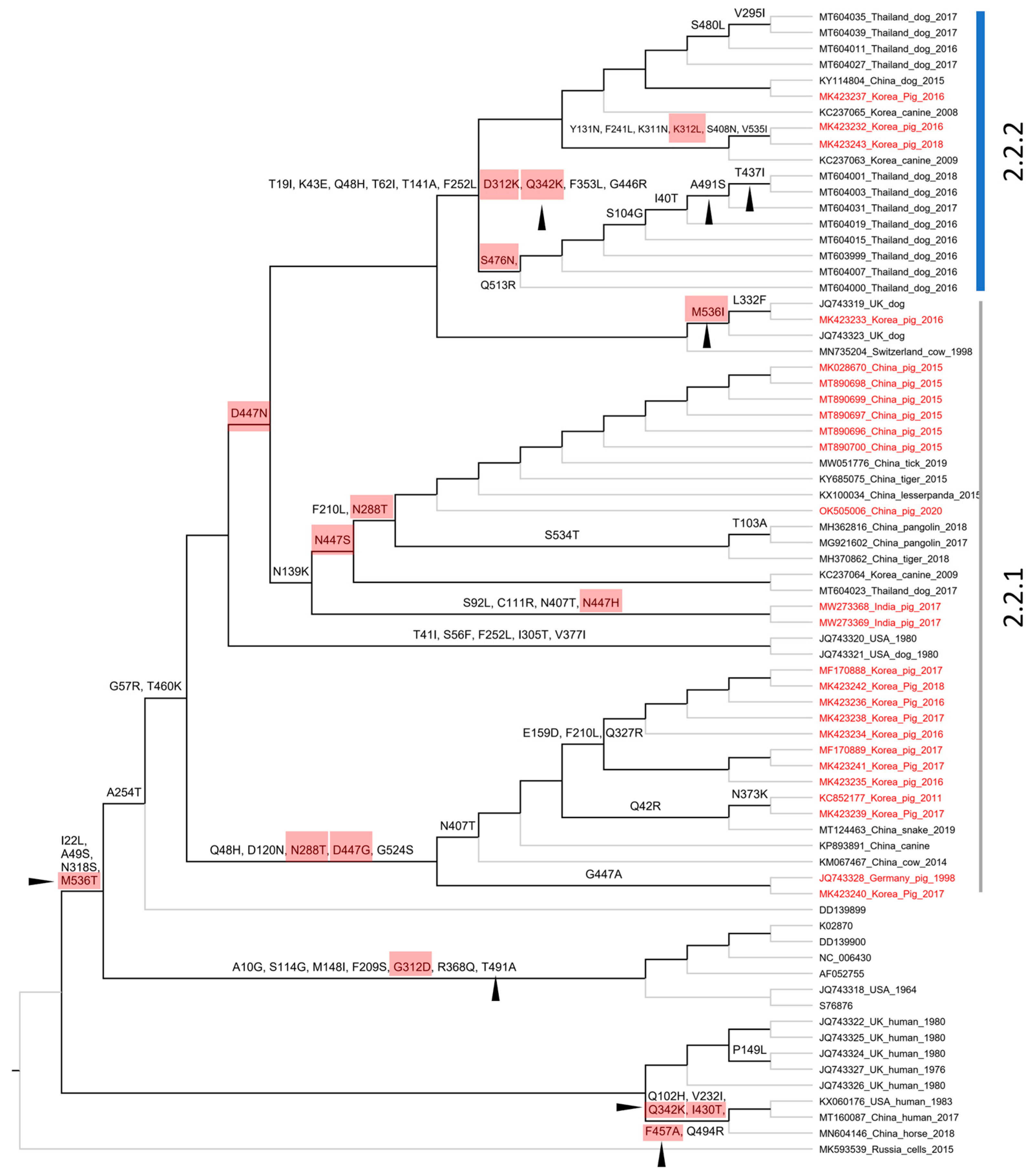
3.3. The Genetic Classification of PIV5
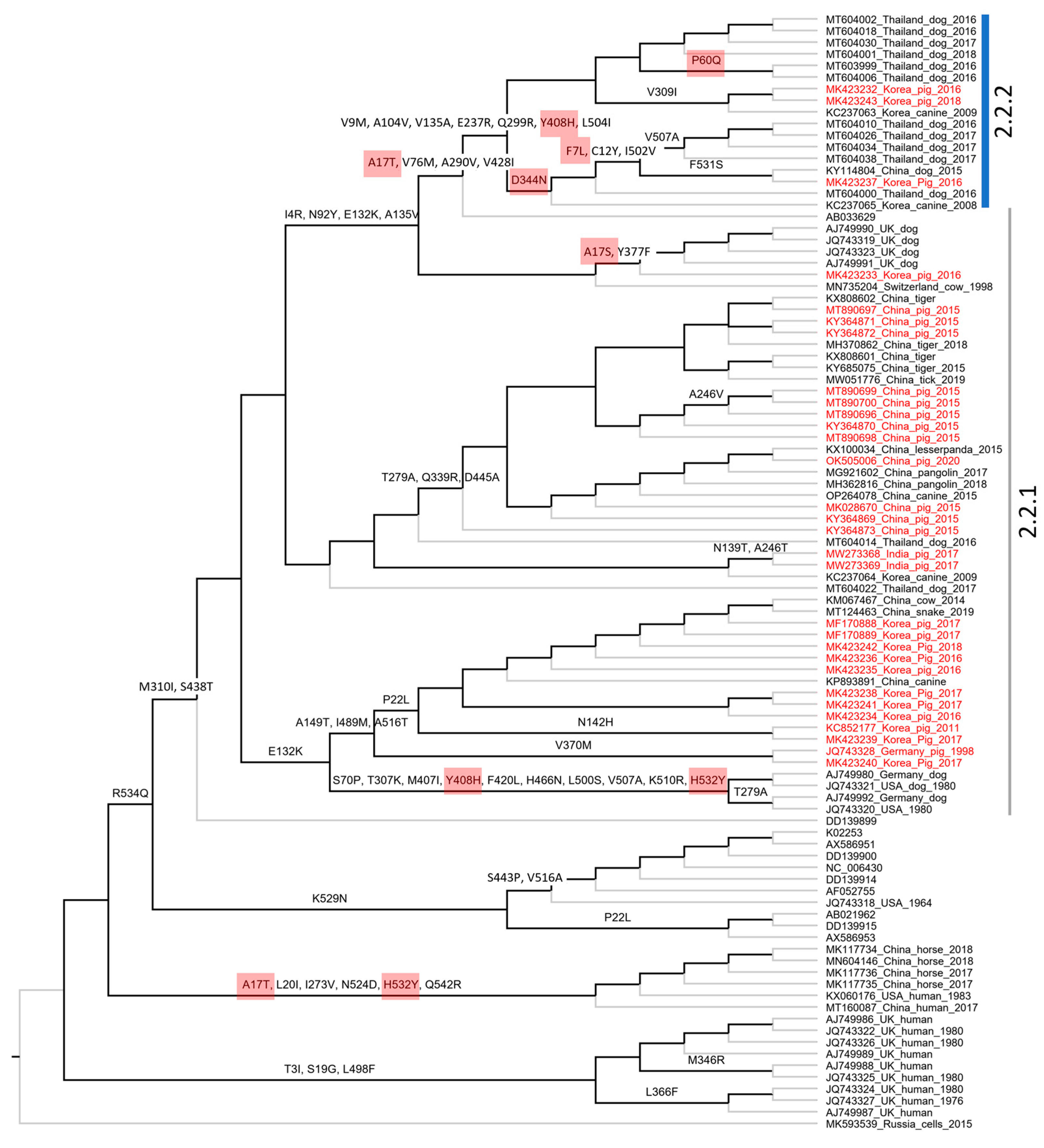
3.4. The Molecular Evolution of PIV5 Based on Structural Genes
4. Discussion
4.1. The Detection of PIV5 in Pigs in Korea
4.2. The Genetic Characterization and Classification of PIV5 Detected in Pigs
4.3. The Genetic Evolution of PIV5
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rima, B.; Balkema-Buschmann, A.; Dundon, W.G.; Duprex, P.; Easton, A.; Fouchier, R.; Kurath, G.; Lamb, R.; Lee, B.; Rota, P.; et al. ICTV Virus Taxonomy Profile: Paramyxoviridae. J. Gen. Virol. 2019, 100, 1593–1594. [Google Scholar] [CrossRef]
- Henrickson, K.J. Parainfluenza viruses. Clin. Microbiol. Rev. 2003, 16, 242–264. [Google Scholar] [CrossRef] [Green Version]
- Thomas, S.M.; Lamb, R.A.; Paterson, R.G. Two mRNAs that differ by two nontemplated nucleotides encode the amino coterminal proteins P and V of the paramyxovirus SV5. Cell 1988, 54, 891–902. [Google Scholar] [CrossRef]
- Lamb, R.A.; Parks, G.D. Paramyxoviridae: The viruses and their replication. In Fields Virology, 6th; Fields, B.N., Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; Volume 1, pp. 957–995. [Google Scholar]
- Lee, Y.N.; Lee, C. Complete genome sequence of a novel porcine parainfluenza virus 5 isolate in Korea. Arch. Virol. 2013, 158, 1765–1772. [Google Scholar] [CrossRef] [PubMed]
- Rima, B.K.; Gatherer, D.; Young, D.F.; Norsted, H.; Randall, R.E.; Davison, A.J. Stability of the parainfluenza virus 5 genome revealed by deep sequencing of strains isolated from different hosts and following passage in cell culture. J. Virol. 2014, 88, 3826–3836. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Singh, M.; Malashkevich, V.N.; Kim, P.S. Structural characterization of the human respiratory syncytial virus fusion protein core. Proc. Natl. Acad. Sci. USA 2000, 97, 14172–14177. [Google Scholar] [CrossRef] [Green Version]
- Dortmans, J.C.; Koch, G.; Rottier, P.J.; Peeters, B.P. Virulence of Newcastle disease virus: What is known so far? Vet. Res. 2011, 42, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baty, D.U.; Randall, R.E. Multiple amino acid substitutions in the HN protein of the paramyxovirus, SV5, are selected for in monoclonal antibody resistant mutants. Arch. Virol. 1993, 131, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.N.; Park, C.K.; Kim, S.H.; Lee, D.S.; Shin, J.H.; Lee, C. Characterization in vitro and in vivo of a novel porcine parainfluenza virus 5 isolate in Korea. Virus Res. 2013, 178, 423–429. [Google Scholar] [CrossRef]
- Liu, Y.; Li, N.; Zhang, S.; Zhang, F.; Lian, H.; Hu, R. Parainfluenza virus 5 as possible cause of severe respiratory disease in calves, China. Emerg. Infect. Dis. 2015, 21, 2242. [Google Scholar] [CrossRef]
- Binn, L.; Eddy, G.; Lazar, E.; Helms, J.; Murnane, T. Viruses recovered from laboratory dogs with respiratory disease. Proc. Soc. Exp. Biol. Med. 1967, 126, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Tribe, G. An investigation of the incidence, epidemiology and control of Simian virus 5. Br. J. Exp. Pathol. 1966, 47, 472. [Google Scholar]
- Chatziandreou, N.; Stock, N.; Young, D.; Andrejeva, J.; Hagmaier, K.; McGeoch, D.; Randall, R. Relationships and host range of human, canine, simian and porcine isolates of simian virus 5 (parainfluenza virus 5). J. Gen. Virol. 2004, 85, 3007–3016. [Google Scholar] [CrossRef]
- Zhai, J.Q.; Zhai, S.L.; Lin, T.; Liu, J.K.; Wang, H.X.; Li, B.; Zhang, H.; Zou, S.Z.; Zhou, X.; Wu, M.F.; et al. First complete genome sequence of parainfluenza virus 5 isolated from lesser panda. Arch. Virol. 2017, 162, 1413–1418. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, Y.M.; Zhang, W.; Werid, G.M.; Zhang, H.; Pan, Y.; Zhang, L.; Xu, Y.; Li, C.; Chen, H.; Wang, Y. Characterization of parainfluenza virus 5 from diarrheic piglet highlights its zoonotic potential. Transbound. Emerg. Dis. 2022, 69, e1510–e1525. [Google Scholar] [CrossRef]
- Welch, M.; Krueger, K.; Zhang, J.; Neveau, M.; Piñeyro, P.; Magstadt, D.; Main, R.; Gauger, P. Characterization of Two Porcine Parainfluenza Virus 1 Isolates and Human Parainfluenza Virus 1 Infection in Weaned Nursery Pigs. Vet. Sci. 2022, 10, 18. [Google Scholar] [CrossRef]
- Feehan, B.J.; Penin, A.A.; Mukhin, A.N.; Kumar, D.; Moskvina, A.S.; Khametova, K.M.; Yuzhakov, A.G.; Musienko, M.I.; Zaberezhny, A.D.; Aliper, T.I.; et al. Novel Mammalian orthorubulavirus 5 Discovered as Accidental Cell Culture Contaminant. Viruses 2019, 11, 777. [Google Scholar] [CrossRef] [Green Version]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A virus classification tool based on pairwise sequence alignment and identity calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 1999, 73, 152–160. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Lemoine, F.; Domelevo Entfellner, J.B.; Wilkinson, E.; Correia, D.; Davila Felipe, M.; De Oliveira, T.; Gascuel, O. Renewing Felsenstein’s phylogenetic bootstrap in the era of big data. Nature 2018, 556, 452–456. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemey, P.; Kosakovsky Pond, S.L.; Drummond, A.J.; Pybus, O.G.; Shapiro, B.; Barroso, H.; Taveira, N.; Rambaut, A. Synonymous Substitution Rates Predict HIV Disease Progression as a Result of Underlying Replication Dynamics. PLoS Comput. Biol. 2007, 3, e29. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.C.; Smith, S.E.; Carter, J.R.; Webb, S.R.; Gibson, K.M.; Hellman, L.M.; Fried, M.G.; Dutch, R.E. Trimeric transmembrane domain interactions in paramyxovirus fusion proteins: Roles in protein folding, stability, and function. J. Biol. Chem. 2013, 288, 35726–35735. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.S.; Wen, X.; Paterson, R.G.; Lamb, R.A.; Jardetzky, T.S. Structure of the parainfluenza virus 5 F protein in its metastable, prefusion conformation. Nature 2006, 439, 38–44. [Google Scholar] [CrossRef]
- Bose, S.; Zokarkar, A.; Welch, B.D.; Leser, G.P.; Jardetzky, T.S.; Lamb, R.A. Fusion activation by a headless parainfluenza virus 5 hemagglutinin-neuraminidase stalk suggests a modular mechanism for triggering. Proc. Natl. Acad. Sci. USA 2012, 109, E2625–E2634. [Google Scholar] [CrossRef] [Green Version]
- Welch, B.D.; Yuan, P.; Bose, S.; Kors, C.A.; Lamb, R.A.; Jardetzky, T.S. Structure of the parainfluenza virus 5 (PIV5) hemagglutinin-neuraminidase (HN) ectodomain. PLoS Pathog. 2013, 9, e1003534. [Google Scholar] [CrossRef] [Green Version]
- Oem, J.K.; Kim, S.H.; Kim, Y.H.; Lee, M.H.; Lee, K.K. Molecular characteristics of canine parainfluenza viruses type 5 (CPIV-5) isolated in Korea. Can. J. Vet. Res. Rev. Can. Rech. Vet. 2015, 79, 64–67. [Google Scholar]
- Yang, D.K.; Kim, H.H.; Choi, S.S.; Park, J.W. Isolation of novel bovine parainfluenza virus type 5 (bPIV5) and its incidence in Korean cattle. Korean J. Vet. Res. 2014, 54, 107–112. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.M.; Kim, H.R.; Jeon, G.T.; Baek, J.S.; Kwon, O.D.; Park, C.K. Molecular Detection of Porcine Parainfluenza Viruses 1 and 5 Using a Newly Developed Duplex Real-Time RT-PCR in South Korea. Animals 2023, 13, 598. [Google Scholar] [CrossRef] [PubMed]
- Brunborg, I.M.; Moldal, T.; Jonassen, C.M. Quantitation of porcine circovirus type 2 isolated from serum/plasma and tissue samples of healthy pigs and pigs with postweaning multisystemic wasting syndrome using a TaqMan-based real-time PCR. J. Virol. Methods 2004, 122, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, A.; Milek, D.; Stadejek, T. Wide Range of the Prevalence and Viral Loads of Porcine Circovirus Type 3 (PCV3) in Different Clinical Materials from 21 Polish Pig Farms. Pathogens 2020, 9, 411. [Google Scholar] [CrossRef]
- Hierweger, M.M.; Werder, S.; Seuberlich, T. Parainfluenza Virus 5 Infection in Neurological Disease and Encephalitis of Cattle. Int. J. Mol. Sci. 2020, 21, 498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, N.; Wang, E.; Guo, D.; Wang, X.; Su, M.; Kong, F.; Yuan, D.; Zhai, J.; Sun, D. Isolation and molecular characterization of parainfluenza virus 5 in diarrhea-affected piglets in China. J. Vet. Med. Sci. 2018, 80, 590–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, P.W.; Hsiung, G.D. Experimental Infection of Parainfluenza Virus Type 5 in Mice, Hamsters and Monkeys. J. Immunol. 1965, 95, 591–601. [Google Scholar] [CrossRef]
- Kryazhimskiy, S.; Bazykin, G.A.; Dushoff, J. Natural selection for nucleotide usage at synonymous and nonsynonymous sites in influenza A virus genes. J. Virol. 2008, 82, 4938–4945. [Google Scholar] [CrossRef] [Green Version]
- Bose, S.; Heath, C.M.; Shah, P.A.; Alayyoubi, M.; Jardetzky, T.S.; Lamb, R.A. Mutations in the parainfluenza virus 5 fusion protein reveal domains important for fusion triggering and metastability. J. Virol. 2013, 87, 13520–13531. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Samples | Number of Samples According to Year of Collection | ||
|---|---|---|---|
| 2016 | 2017 | 2018 | |
| Serum | 2142 | 1413 | 1035 |
| Pooled organs | 489 | 304 | 183 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Truong, H.-T.; Nguyen, V.-G.; Pham, L.-B.-H.; Huynh, T.-M.-L.; Lee, J.; Hwang, S.-J.; Lee, J.-M.; Chung, H.-C. PCR-Based Detection and Genetic Characterization of Parainfluenza Virus 5 Detected in Pigs in Korea from 2016 to 2018. Vet. Sci. 2023, 10, 414. https://doi.org/10.3390/vetsci10070414
Truong H-T, Nguyen V-G, Pham L-B-H, Huynh T-M-L, Lee J, Hwang S-J, Lee J-M, Chung H-C. PCR-Based Detection and Genetic Characterization of Parainfluenza Virus 5 Detected in Pigs in Korea from 2016 to 2018. Veterinary Sciences. 2023; 10(7):414. https://doi.org/10.3390/vetsci10070414
Chicago/Turabian StyleTruong, Ha-Thai, Van-Giap Nguyen, Le-Bich-Hang Pham, Thi-My-Le Huynh, Jasper Lee, Su-Jin Hwang, Jae-Myun Lee, and Hee-Chun Chung. 2023. "PCR-Based Detection and Genetic Characterization of Parainfluenza Virus 5 Detected in Pigs in Korea from 2016 to 2018" Veterinary Sciences 10, no. 7: 414. https://doi.org/10.3390/vetsci10070414