Comparative Analysis of the Pre-Parturition and Post-Parturition Genital Tract Microbiota in Plateau Bangor Sewa Sheep

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling
2.2. DNA Extraction
2.3. The Genital Tract Microbiome Amplicon Sequencing
2.4. Bioinformatic Analysis
2.5. Statistical Analysis
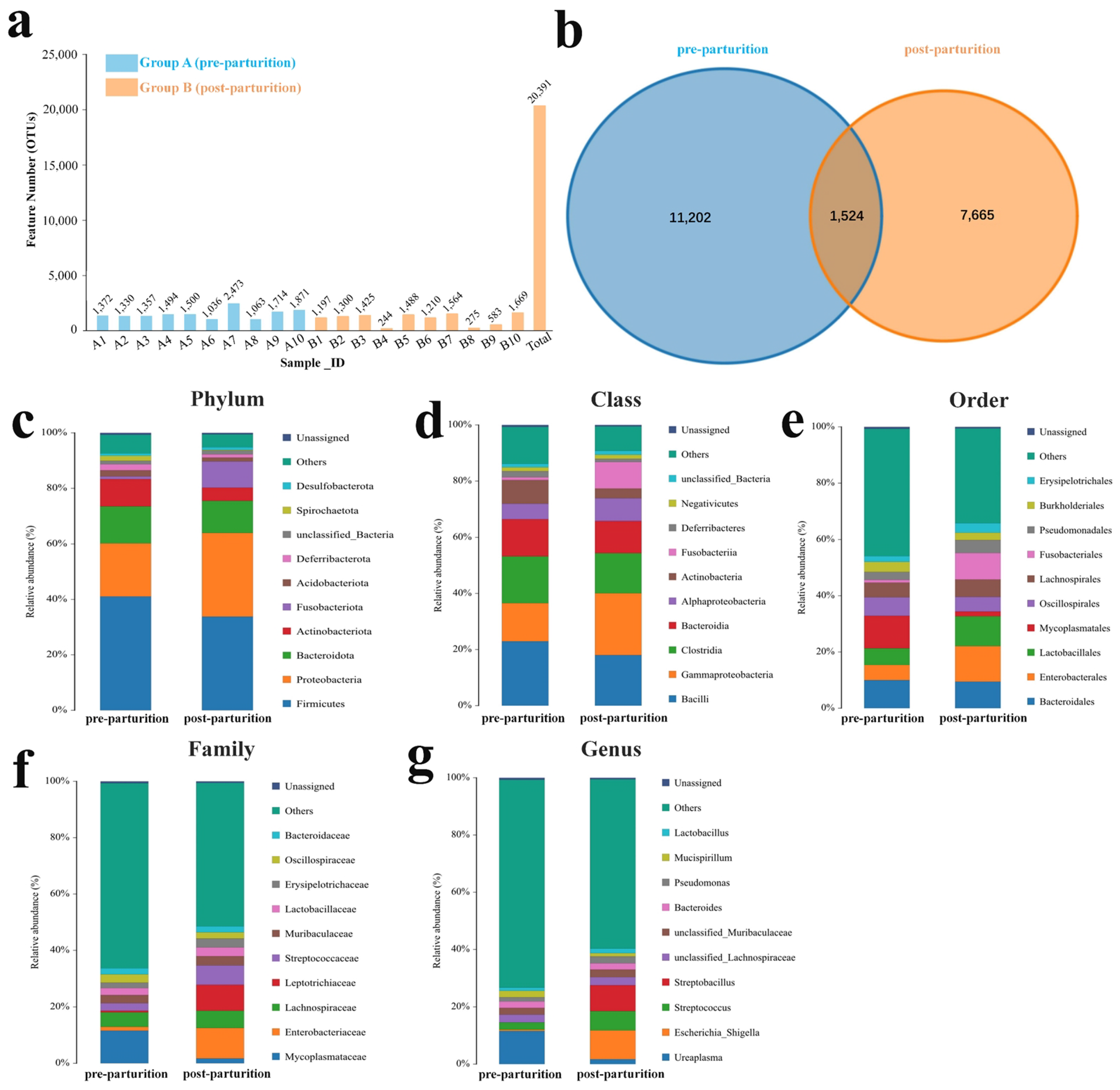
3. Results
3.1. Composition Analysis of Microbiota in the Birth Canal of Bangor Sewa Sheep
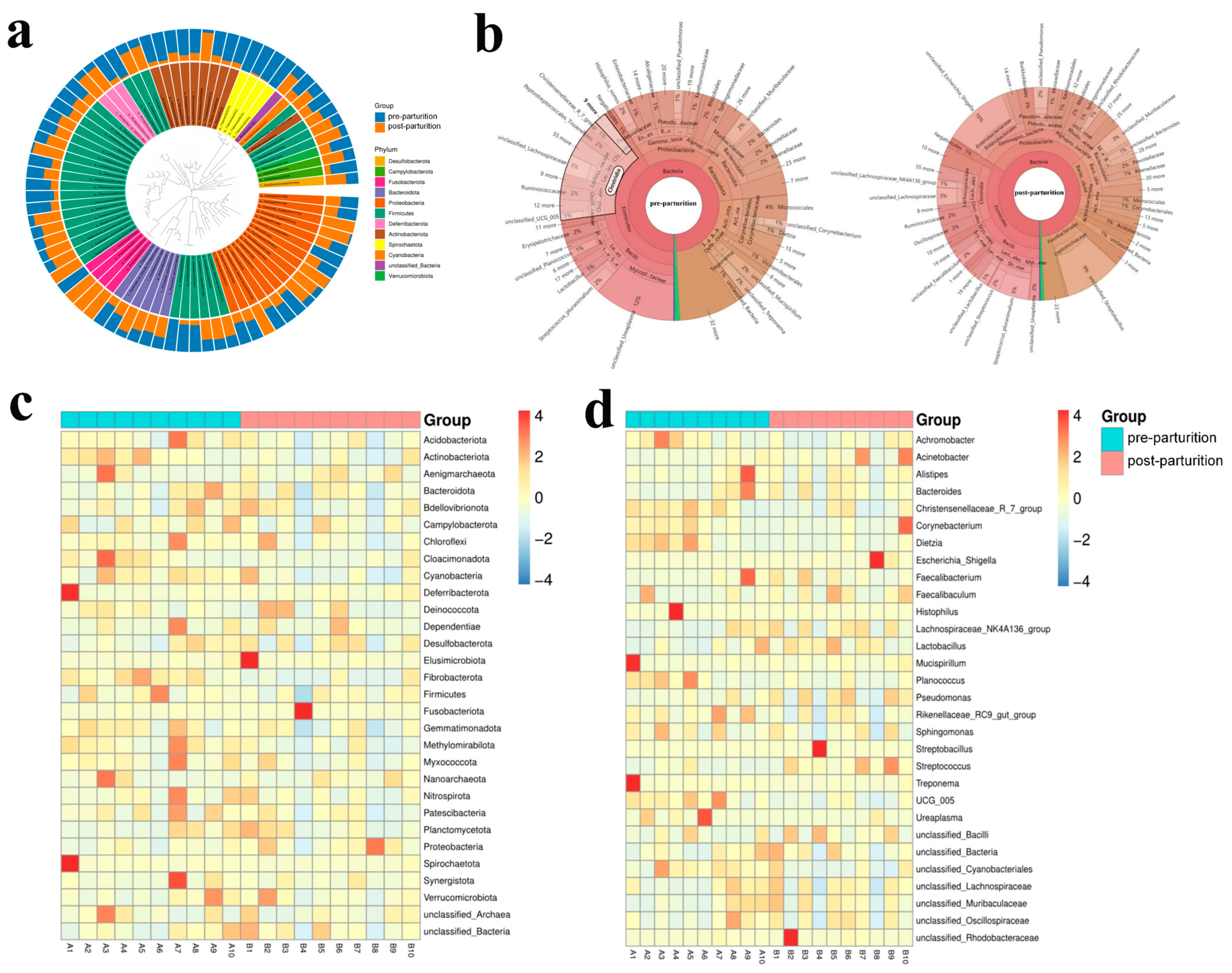
3.2. Comparing Microbiota Structure between the Pre- and Post-Parturition of Bangor Sewa Sheep Genital Tracts
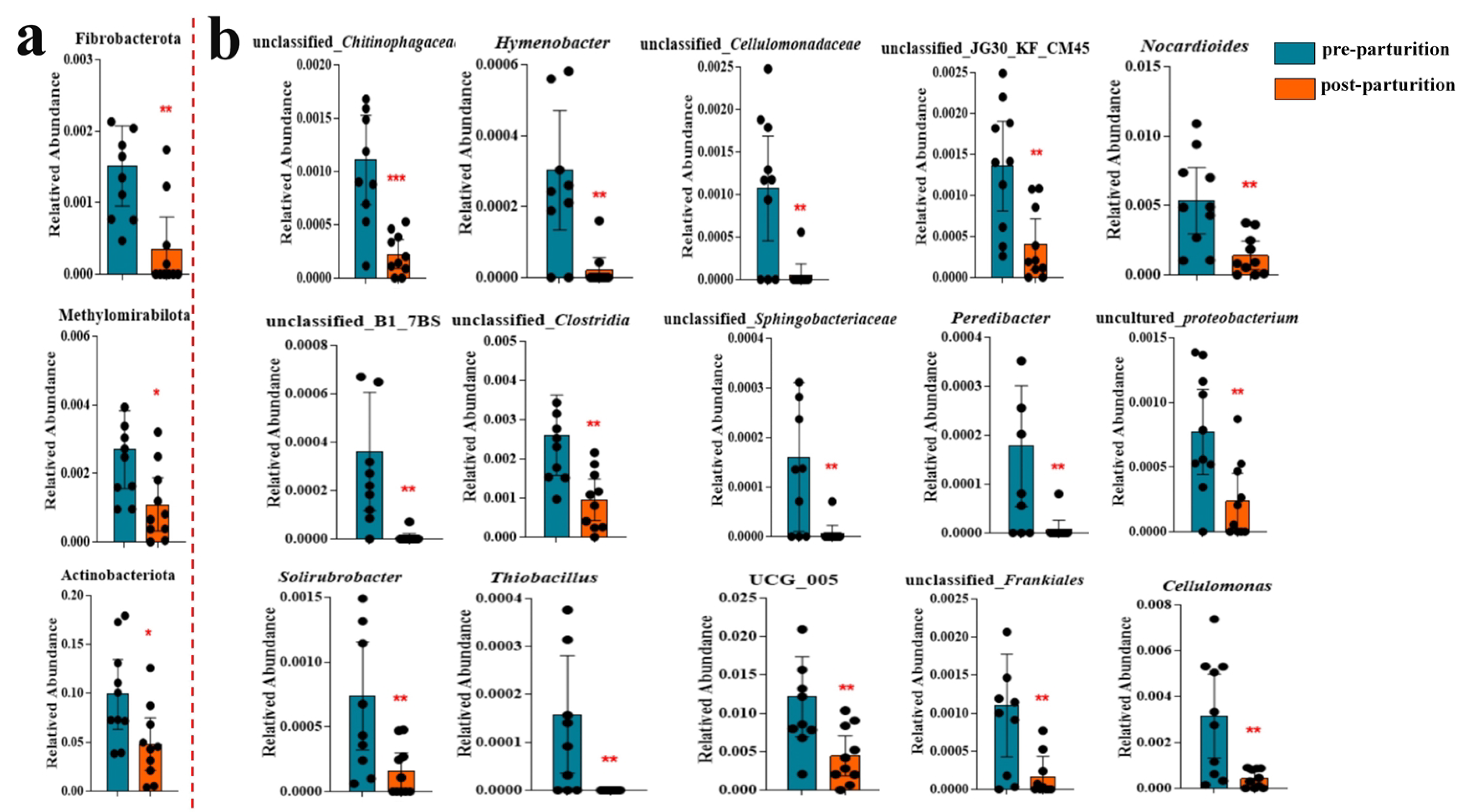
3.3. The Remarkable Species between Pre- and Post-Parturition of Bangor Sewa Sheep Genital Tract
3.4. Comparing the Microbiota Function between the Two Bangor Sewa Sheep Groups
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chikwanha, O.C.; Vahmani, P.; Muchenje, V.; Dugan, M.E.R.; Mapiye, C. Nutritional enhancement of sheep meat fatty acid profile for human health and wellbeing. Food Res. Int. 2018, 104, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Pethick, D.W.; Hocquette, J.F.; Scollan, N.D.; Dunshea, F.R. Review: Improving the nutritional, sensory and market value of meat products from sheep and cattle. Animal 2021, 15 (Suppl. 1), 100356. [Google Scholar] [PubMed]
- Dominguez-Bello, M.G.; Godoy-Vitorino, F.; Knight, R.; Blaser, M.J. Role of the microbiome in human development. Gut 2019, 68, 1108–1114. [Google Scholar] [CrossRef] [Green Version]
- Hussain, T.; Murtaza, G.; Kalhoro, D.H.; Kalhoro, M.S.; Metwally, E.; Chughtai, M.I.; Mazhar, M.U.; Khan, S.A. Relationship between gut microbiota and host-metabolism: Emphasis on hormones related to reproductive function. Anim. Nutr. 2021, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Mousa, W.K.; Chehadeh, F.; Husband, S. Recent Advances in Understanding the Structure and Function of the Human Microbiome. Front. Microbiol. 2022, 13, 825338. [Google Scholar] [CrossRef] [PubMed]
- Malard, F.; Dore, J.; Gaugler, B.; Mohty, M. Introduction to host microbiome symbiosis in health and disease. Mucosal Immunol. 2021, 14, 547–554. [Google Scholar] [CrossRef]
- Qi, X.; Yun, C.; Pang, Y.; Qiao, J. The impact of the gut microbiota on the reproductive and metabolic endocrine system. Gut Microbes 2021, 13, 1894070. [Google Scholar] [CrossRef]
- Chang, E.B.; Martinez-Guryn, K. Small intestinal microbiota: The neglected stepchild needed for fat digestion and absorption. Gut Microbes 2019, 10, 235–240. [Google Scholar] [CrossRef] [Green Version]
- D’Afflitto, M.; Upadhyaya, A.; Green, A.; Peiris, M. Association Between Sex Hormone Levels and Gut Microbiota Composition and Diversity-A Systematic Review. J. Clin. Gastroenterol. 2022, 56, 384–392. [Google Scholar] [CrossRef]
- Piccinni, M.P.; Raghupathy, R.; Saito, S.; Szekeres-Bartho, J. Cytokines, Hormones and Cellular Regulatory Mechanisms Favoring Successful Reproduction. Front. Immunol. 2021, 12, 717808. [Google Scholar] [CrossRef]
- Zhu, B.; Tao, Z.; Edupuganti, L.; Serrano, M.G.; Buck, G.A. Roles of the Microbiota of the Female Reproductive Tract in Gynecological and Reproductive Health. Microbiol. Mol. Biol. Rev. 2022, 86, e0018121. [Google Scholar] [CrossRef]
- Yoo, J.Y.; Groer, M.; Dutra, S.V.O.; Sarkar, A.; McSkimming, D.I. Gut Microbiota and Immune System Interactions. Microorganisms 2020, 8, 1587. [Google Scholar] [CrossRef] [PubMed]
- Libertucci, J.; Young, V.B. The role of the microbiota in infectious diseases. Nat. Microbiol. 2019, 4, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Moreno, I.; Simon, C. Deciphering the effect of reproductive tract microbiota on human reproduction. Reprod. Med. Biol. 2019, 18, 40–50. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Chen, L.; Yi, K.; Zhang, B.; Li, C.; Zhou, X. The effects of microbiota on reproductive health: A review. Crit. Rev. Food Sci. Nutr. 2022, 1–22. [Google Scholar] [CrossRef]
- Barba, M.; Martínez-Boví, R.; Quereda, J.J.; Mocé, M.L.; Plaza-Dávila, M.; Jiménez-Trigos, E.; Gómez-Martín, Á.; González-Torres, P.; Carbonetto, B.; García-Roselló, E. Vaginal Microbiota Is Stable throughout the Estrous Cycle in Arabian Maress. Animals 2020, 10, 2020. [Google Scholar] [CrossRef]
- Koester, L.R.; Petry, A.L.; Youngs, C.R.; Schmitz-Esser, S. Ewe Vaginal Microbiota: Associations With Pregnancy Outcome and Changes during Gestation. Front. Microbiol. 2021, 12, 745884. [Google Scholar] [CrossRef] [PubMed]
- Teshigawara, T.; Mouri, A.; Kubo, H.; Nakamura, Y.; Shiino, T.; Okada, T.; Morikawa, M.; Nabeshima, T.; Ozaki, N.; Yamamoto, Y.; et al. Changes in tryptophan metabolism during pregnancy and postpartum periods: Potential involvement in postpartum depressive symptoms. J. Affect Disord. 2019, 255, 168–176. [Google Scholar] [CrossRef]
- Siena, M.; Laterza, L.; Matteo, M.V.; Mignini, I.; Schepis, T.; Rizzatti, G.; Ianiro, G.; Rinninella, E.; Cintoni, M.; Gasbarrini, A. Gut and Reproductive Tract Microbiota Adaptation during Pregnancy: New Insights for Pregnancy-Related Complications and Therapy. Microorganisms 2021, 9, 473. [Google Scholar] [CrossRef]
- Nunn, K.L.; Witkin, S.S.; Schneider, G.M.; Boester, A.; Nasioudis, D.; Minis, E.; Gliniewicz, K.; Forney, L.J. Changes in the Vaginal Microbiome during the Pregnancy to Postpartum Transition. Reprod. Sci. 2021, 28, 1996–2005. [Google Scholar] [CrossRef]
- Al-Nasiry, S.; Ambrosino, E.; Schlaepfer, M.; Morré, S.A.; Wieten, L.; Voncken, J.W.; Spinelli, M.; Mueller, M.; Kramer, B.W. The Interplay Between Reproductive Tract Microbiota and Immunological System in Human Reproduction. Front. Immunol. 2020, 11, 378. [Google Scholar] [CrossRef] [PubMed]
- Ramos Bde, A.; Kanninen, T.T.; Sisti, G.; Witkin, S.S. Microorganisms in the female genital tract during pregnancy: Tolerance versus pathogenesis. Am. J. Reprod. Immunol. 2015, 73, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [Green Version]
- Ceccarani, C.; Foschi, C.; Parolin, C.; D’Antuono, A.; Gaspari, V.; Consolandi, C.; Laghi, L.; Camboni, T.; Vitali, B.; Severgnini, M.; et al. Diversity of vaginal microbiome and metabolome during genital infections. Sci. Rep. 2019, 9, 14095. [Google Scholar] [CrossRef] [Green Version]
- Anahtar, M.N.; Gootenberg, D.B.; Mitchell, C.M.; Kwon, D.S. Cervicovaginal Microbiota and Reproductive Health: The Virtue of Simplicity. Cell Host Microbe 2018, 23, 159–168. [Google Scholar] [CrossRef] [Green Version]
- Oliver, A.; LaMere, B.; Weihe, C.; Wandro, S.; Lindsay, K.L.; Wadhwa, P.D.; Mills, D.A.; Pride, D.T.; Fiehn, O.; Northen, T.; et al. Cervicovaginal Microbiome Composition Is Associated with Metabolic Profiles in Healthy Pregnancy. mBio 2020, 11, e01851-20. [Google Scholar] [CrossRef]
- Lewis, F.M.T.; Bernstein, K.T.; Aral, S.O. Vaginal Microbiome and Its Relationship to Behavior, Sexual Health, and Sexually Transmitted Diseases. Obstet. Gynecol. 2017, 129, 643–654. [Google Scholar] [CrossRef] [PubMed]
- Serrano, M.G.; Parikh, H.I.; Brooks, J.P.; Edwards, D.J.; Arodz, T.J.; Edupuganti, L.; Huang, B.; Girerd, P.H.; Bokhari, Y.A.; Bradley, S.P.; et al. Racioethnic diversity in the dynamics of the vaginal microbiome during pregnancy. Nat. Med. 2019, 25, 1001–1011. [Google Scholar] [CrossRef]
- Noyes, N.; Cho, K.C.; Ravel, J.; Forney, L.J.; Abdo, Z. Associations between sexual habits, menstrual hygiene practices, demographics and the vaginal microbiome as revealed by Bayesian network analysis. PLoS ONE 2018, 13, e0191625. [Google Scholar] [CrossRef] [Green Version]
- Dong, T.S.; Gupta, A. Influence of Early Life, Diet, and the Environment on the Microbiome. Clin. Gastroenterol. Hepatol. 2019, 17, 231–242. [Google Scholar] [CrossRef]
- Bradley, F.; Birse, K.; Hasselrot, K.; Noël-Romas, L.; Introini, A.; Wefer, H.; Seifert, M.; Engstrand, L.; Tjernlund, A.; Broliden, K.; et al. The vaginal microbiome amplifies sex hormone-associated cyclic changes in cervicovaginal inflammation and epithelial barrier disruption. Am. J. Reprod. Immunol. 2018, 80, e12863. [Google Scholar] [CrossRef]
- Mehta, S.D.; Zulaika, G.; Otieno, F.O.; Nyothach, E.; Agingu, W.; Bhaumik, R.; Green, S.J.; van Eijk, A.M.; Kwaro, D.; Phillips-Howard, P.A. High Prevalence of Lactobacillus crispatus Dominated Vaginal Microbiome Among Kenyan Secondary School Girls: Negative Effects of Poor Quality Menstrual Hygiene Management and Sexual Activity. Front. Cell. Infect. Microbiol. 2021, 11, 716537. [Google Scholar] [CrossRef] [PubMed]
- Chaban, B.; Links, M.G.; Jayaprakash, T.P.; Wagner, E.C.; Bourque, D.K.; Lohn, Z.; Albert, A.Y.; van Schalkwyk, J.; Reid, G.; Hemmingsen, S.M.; et al. Characterization of the vaginal microbiota of healthy Canadian women through the menstrual cycle. Microbiome 2014, 2, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holdcroft, A.M.; Ireland, D.J.; Payne, M.S. The Vaginal Microbiome in Health and Disease-What Role Do Common Intimate Hygiene Practices Play? Microorganisms 2023, 11, 298. [Google Scholar] [CrossRef]
- Wang, J.; Li, Z.; Ma, X.; Du, L.; Jia, Z.; Cui, X.; Yu, L.; Yang, J.; Xiao, L.; Zhang, B.; et al. Translocation of vaginal microbiota is involved in impairment and protection of uterine health. Nat. Commun. 2021, 12, 4191. [Google Scholar] [CrossRef] [PubMed]
- Bagga, R.; Arora, P. Genital Micro-Organisms in Pregnancy. Front. Public Health 2020, 8, 225. [Google Scholar] [CrossRef] [PubMed]
- Swartz, J.D.; Lachman, M.; Westveer, K.; O’Neill, T.; Geary, T.; Kott, R.W.; Berardinelli, J.G.; Hatfield, P.G.; Thomson, J.M.; Roberts, A.; et al. Characterization of the Vaginal Microbiota of Ewes and Cows Reveals a Unique Microbiota with Low Levels of Lactobacilli and Near-Neutral pH. Front. Vet. Sci. 2014, 1, 19. [Google Scholar] [CrossRef] [Green Version]
- Kuijper, E.A.; Ket, J.C.; Caanen, M.R.; Lambalk, C.B. Reproductive hormone concentrations in pregnancy and neonates: A systematic review. Reprod. Biomed. Online 2013, 27, 33–63. [Google Scholar] [CrossRef] [Green Version]
- Peterson, L.S.; Hedou, J.; Ganio, E.A.; Stelzer, I.A.; Feyaerts, D.; Harbert, E.; Adusumelli, Y.; Ando, K.; Tsai, E.S.; Tsai, A.S.; et al. Single-Cell Analysis of the Neonatal Immune System Across the Gestational Age Continuum. Front. Immunol. 2021, 12, 714090. [Google Scholar] [CrossRef] [PubMed]
- Zeng, B.; Zhao, J.; Guo, W.; Zhang, S.; Hua, Y.; Tang, J.; Kong, F.; Yang, X.; Fu, L.; Liao, K.; et al. High-Altitude Living Shapes the Skin Microbiome in Humans and Pigs. Front. Microbiol. 2017, 8, 1929. [Google Scholar] [CrossRef] [Green Version]
- Zhong, W.; Hu, L.; Zhao, Y.; Li, Z.; Zhuo, Y.; Jiang, X.; Li, J.; Zhao, X.; Che, L.; Feng, B.; et al. Effects of Dietary Choline Levels During Pregnancy on Reproductive Performance, Plasma Metabolome and Gut Microbiota of Sows. Front. Vet. Sci. 2021, 8, 771228. [Google Scholar] [CrossRef] [PubMed]
- Leclere, M.; Costa, M.C. Fecal microbiota in horses with asthma. J. Vet. Intern. Med. 2020, 34, 996–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Y.; Zhang, N.; Li, W.J.; Tan, K.; Zhou, Y.; She, C.; Chen, H.N. Correlations of changes in inflammatory factors, glucose and lipid metabolism indicators and adiponectin with alterations in intestinal flora in rats with coronary heart disease. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 10118–10125. [Google Scholar] [PubMed]
- Liu, B.; Ye, D.; Yang, H.; Song, J.; Sun, X.; Mao, Y.; He, Z. Two-Sample Mendelian Randomization Analysis Investigates Causal Associations Between Gut Microbial Genera and Inflammatory Bowel Disease, and Specificity Causal Associations in Ulcerative Colitis or Crohn’s Disease. Front. Immunol. 2022, 13, 921546. [Google Scholar] [CrossRef]
- Liu, Z.; Yin, B. Alterations in the Gut Microbial Composition and Diversity of Tibetan Sheep Infected With Echinococcus granulosus. Front. Vet. Sci. 2021, 8, 778789. [Google Scholar] [CrossRef] [PubMed]
- Click, R.E. A Potential ‘Curative’ Modality for Crohn’s Disease---Modeled after Prophylaxis of Bovine Johne’s Disease. Mycobact. Dis. 2012, 2, 117. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Nan, X.; Zhao, Y.; Wang, H.; Wang, M.; Jiang, L.; Zhang, F.; Xue, F.; Hua, D.; Li, K.; et al. Coupling 16S rDNA Sequencing and Untargeted Mass Spectrometry for Milk Microbial Composition and Metabolites from Dairy Cows with Clinical and Subclinical Mastitis. J. Agric. Food Chem. 2020, 68, 8496–8508. [Google Scholar] [CrossRef]
- Liu, X.L.; Zhao, Y.C.; Zhu, H.Y.; Wu, M.; Zheng, Y.N.; Yang, M.; Cheng, Z.Q.; Ding, C.B.; Liu, W.C. Taxifolin retards the D-galactose-induced aging process through inhibiting Nrf2-mediated oxidative stress and regulating the gut microbiota in mice. Food Funct. 2021, 12, 12142–12158. [Google Scholar] [CrossRef]
- Zhang, W.; Zhu, Y.H.; Yang, G.Y.; Liu, X.; Xia, B.; Hu, X.; Su, J.H.; Wang, J.F. Lactobacillus rhamnosus GG Affects Microbiota and Suppresses Autophagy in the Intestines of Pigs Challenged with Salmonella Infantis. Front. Microbiol. 2017, 8, 2705. [Google Scholar] [CrossRef] [Green Version]
- Rettedal, E.A.; Ilesanmi-Oyelere, B.L.; Roy, N.C.; Coad, J.; Kruger, M.C. The Gut Microbiome Is Altered in Postmenopausal Women With Osteoporosis and Osteopenia. JBMR Plus 2021, 5, e10452. [Google Scholar] [CrossRef]
- Palmas, V.; Pisanu, S.; Madau, V.; Casula, E.; Deledda, A.; Cusano, R.; Uva, P.; Vascellari, S.; Loviselli, A.; Manzin, A.; et al. Gut microbiota markers associated with obesity and overweight in Italian adults. Sci. Rep. 2021, 11, 5532. [Google Scholar] [CrossRef]
- Shi, Y.; Tang, L.; Bai, X.; Du, K.; Wang, H.; Jia, X.; Lai, S. Heat Stress Altered the Vaginal Microbiome and Metabolome in Rabbits. Front. Microbiol. 2022, 13, 813622. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Xu, Y.; Shen, L.; Huang, J.; Xu, S.; Li, J.; Sun, Z.; He, J.; Chen, M.; Pan, Y. GuanXinNing Tablet Attenuates Alzheimer’s Disease via Improving Gut Microbiota, Host Metabolites, and Neuronal Apoptosis in Rabbits. Evid. Based Complement Alternat. Med. 2021, 2021, 9253281. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Y.; Zhang, X.; Liu, J.; Zhang, Q.; Zhao, Y.; Peng, J.; Feng, Q.; Dai, J.; Sun, S.; et al. Gut Microbial Dysbiosis Is Associated with Altered Hepatic Functions and Serum Metabolites in Chronic Hepatitis B Patients. Front. Microbiol. 2017, 8, 2222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leach, D.A.; Chen, J.; Yang, L.; Chua, H.K.; Walther-António, M.R.S.; Occhino, J.A. Microbiome diversity predicts surgical success in patients with rectovaginal fistula. Int. Urogynecol. J. 2021, 32, 2491–2501. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Lu, J.; Sun, W.; Jia, G.; Zhao, H.; Chen, X.; Kim, I.H.; Zhang, R.; Wang, J. Tryptophan Supplementation Enhances Intestinal Health by Improving Gut Barrier Function, Alleviating Inflammation, and Modulating Intestinal Microbiome in Lipopolysaccharide-Challenged Piglets. Front. Microbiol. 2022, 13, 919431. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Ye, Y.; Ji, J.; Yang, X.; Xu, J.; Wang, J.S.; Han, X.; Zhang, T.; Sun, X. Diet composition affects long-term zearalenone exposure on the gut-blood-liver axis metabolic dysfunction in mice. Ecotoxicol. Environ. Saf. 2022, 236, 113466. [Google Scholar] [CrossRef]
- Yang, D.F.; Huang, W.C.; Wu, C.W.; Huang, C.Y.; Yang, Y.S.H.; Tung, Y.T. Acute sleep deprivation exacerbates systemic inflammation and psychiatry disorders through gut microbiota dysbiosis and disruption of circadian rhythms. Microbiol. Res. 2023, 268, 127292. [Google Scholar] [CrossRef]
- Kaiyrlykyzy, A.; Kozhakhmetov, S.; Babenko, D.; Zholdasbekova, G.; Alzhanova, D.; Olzhayev, F.; Baibulatova, A.; Kushugulova, A.R.; Askarova, S. Study of gut microbiota alterations in Alzheimer’s dementia patients from Kazakhstan. Sci. Rep. 2022, 12, 15115. [Google Scholar] [CrossRef]
- Sun, L.; Guo, L.; Xu, G.; Li, Z.; Appiah, M.O.; Yang, L.; Lu, W. Quercetin Reduces Inflammation and Protects Gut Microbiota in Broilers. Molecules 2022, 27, 3269. [Google Scholar] [CrossRef]
- Bamba, S.; Imai, T.; Sasaki, M.; Ohno, M.; Yoshida, S.; Nishida, A.; Takahashi, K.; Inatomi, O.; Andoh, A. Altered gut microbiota in patients with small intestinal bacterial overgrowth. J. Gastroenterol. Hepatol. 2023, 38, 61–69. [Google Scholar] [CrossRef]
- Rehman, A.U.; Siddiqui, N.Z.; Farooqui, N.A.; Alam, G.; Gul, A.; Ahmad, B.; Asim, M.; Khan, A.I.; Xin, Y.; Zexu, W.; et al. Morchella esculenta mushroom polysaccharide attenuates diabetes and modulates intestinal permeability and gut microbiota in a type 2 diabetic mice model. Front. Nutr. 2022, 9, 984695. [Google Scholar] [CrossRef] [PubMed]
- Zhuge, A.; Li, S.; Lou, P.; Wu, W.; Wang, K.; Yuan, Y.; Xia, J.; Li, B.; Li, L. Longitudinal 16S rRNA Sequencing Reveals Relationships among Alterations of Gut Microbiota and Nonalcoholic Fatty Liver Disease Progression in Mice. Microbiol. Spectr. 2022, 10, e0004722. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Guo, J.; Liu, R.; Zhang, F.; Wen, S.; Liu, Y.; Ren, W.; Zhang, X.; Shang, Y.; Gao, M.; et al. Predominance of Escherichia-Shigella in Gut Microbiome and Its Potential Correlation with Elevated Level of Plasma Tumor Necrosis Factor Alpha in Patients with Tuberculous Meningitis. Microbiol. Spectr. 2022, 10, e0192622. [Google Scholar] [CrossRef] [PubMed]
- Iebba, V.; Totino, V.; Gagliardi, A.; Santangelo, F.; Cacciotti, F.; Trancassini, M.; Mancini, C.; Cicerone, C.; Corazziari, E.; Pantanella, F.; et al. Eubiosis and dysbiosis: The two sides of the microbiota. New Microbiol. 2016, 39, 1–12. [Google Scholar]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, H.; Zhao, W.; Song, T.; Baijiu, Z.; Zhang, Z. Comparative Analysis of the Pre-Parturition and Post-Parturition Genital Tract Microbiota in Plateau Bangor Sewa Sheep. Vet. Sci. 2023, 10, 523. https://doi.org/10.3390/vetsci10080523
Ma H, Zhao W, Song T, Baijiu Z, Zhang Z. Comparative Analysis of the Pre-Parturition and Post-Parturition Genital Tract Microbiota in Plateau Bangor Sewa Sheep. Veterinary Sciences. 2023; 10(8):523. https://doi.org/10.3390/vetsci10080523
Chicago/Turabian StyleMa, Hongcai, Wangsheng Zhao, Tianzeng Song, Zhaxi Baijiu, and Zhenzhen Zhang. 2023. "Comparative Analysis of the Pre-Parturition and Post-Parturition Genital Tract Microbiota in Plateau Bangor Sewa Sheep" Veterinary Sciences 10, no. 8: 523. https://doi.org/10.3390/vetsci10080523
APA StyleMa, H., Zhao, W., Song, T., Baijiu, Z., & Zhang, Z. (2023). Comparative Analysis of the Pre-Parturition and Post-Parturition Genital Tract Microbiota in Plateau Bangor Sewa Sheep. Veterinary Sciences, 10(8), 523. https://doi.org/10.3390/vetsci10080523