

Monoclonal Antibody against Porcine LAG3 Inhibits Porcine Reproductive and Respiratory Syndrome Virus Infection

Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Virus Strains, Cells, Vectors, Antibodies, Tissues, and Animals
2.2. Isolation of Porcine Peripheral Blood Mononuclear Cells (PBMCs)
2.3. Expression and Purification of pLAG3
2.4. Production of mAbs against pLAG3
2.5. pcDNA3.1/V5-HisB-flpLAG3 Recombinant Plasmid Construction
2.6. Indirect Immunofluorescence Assay (IFA)
2.7. Western Blotting Detection
2.8. ELISA Additivity Test
2.9. Identification of the Antigen Region Recognized by mAb 1C2
2.10. Detection of LAG3, SLA-DRα, and FGL1 Expression in Tissues of PRRSV-Infected Pigs
2.11. Quantitative Real-Time PCR (qPCR)
2.12. Induction of Monocyte-Derived Dendritic Cells (MoDCs)
2.13. Establishment of a Co-Culture System
2.14. Statistical Analysis
3. Results
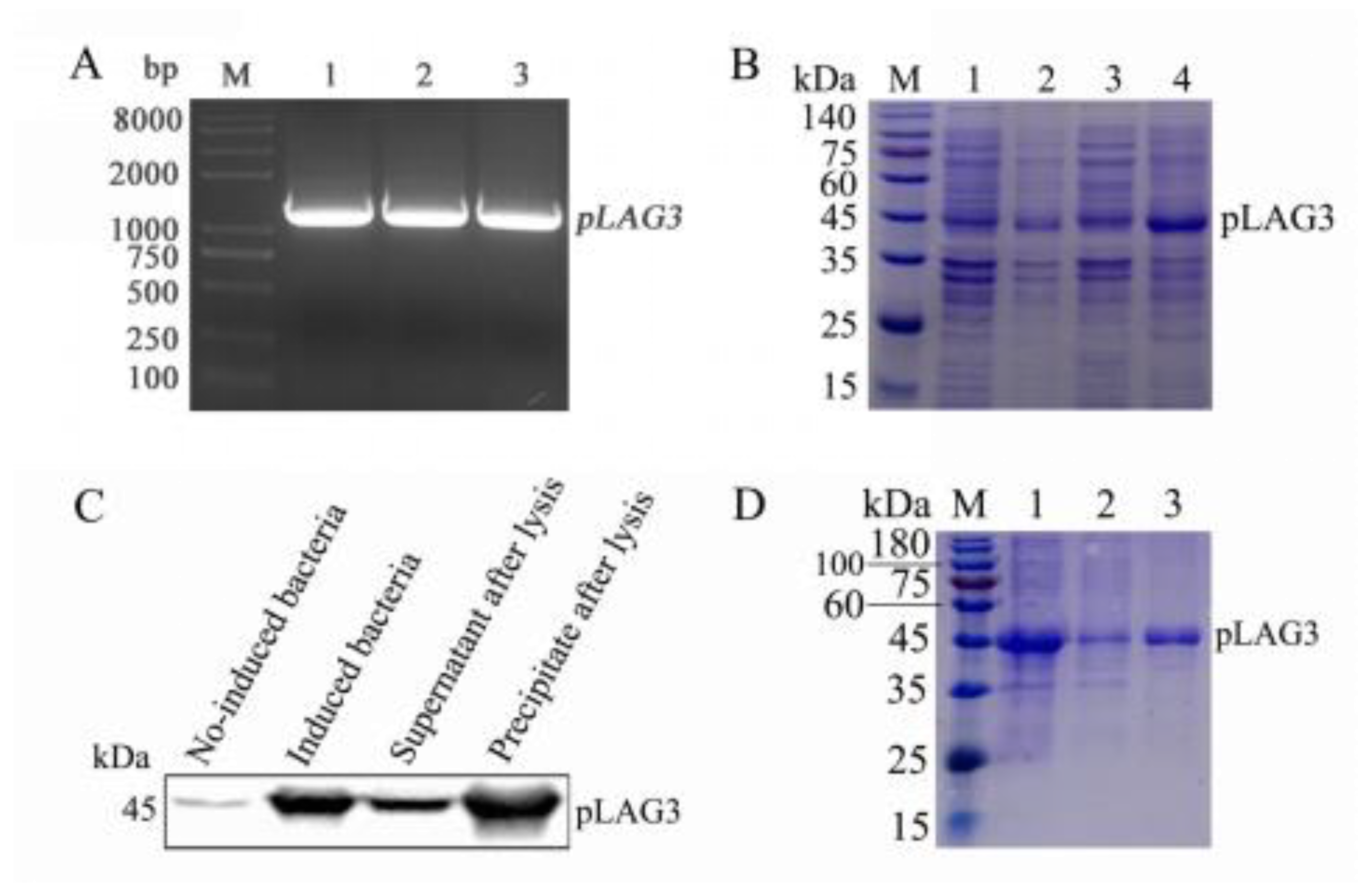
3.1. pLAG3 Gene Amplification and pLAG3 Protein Expression
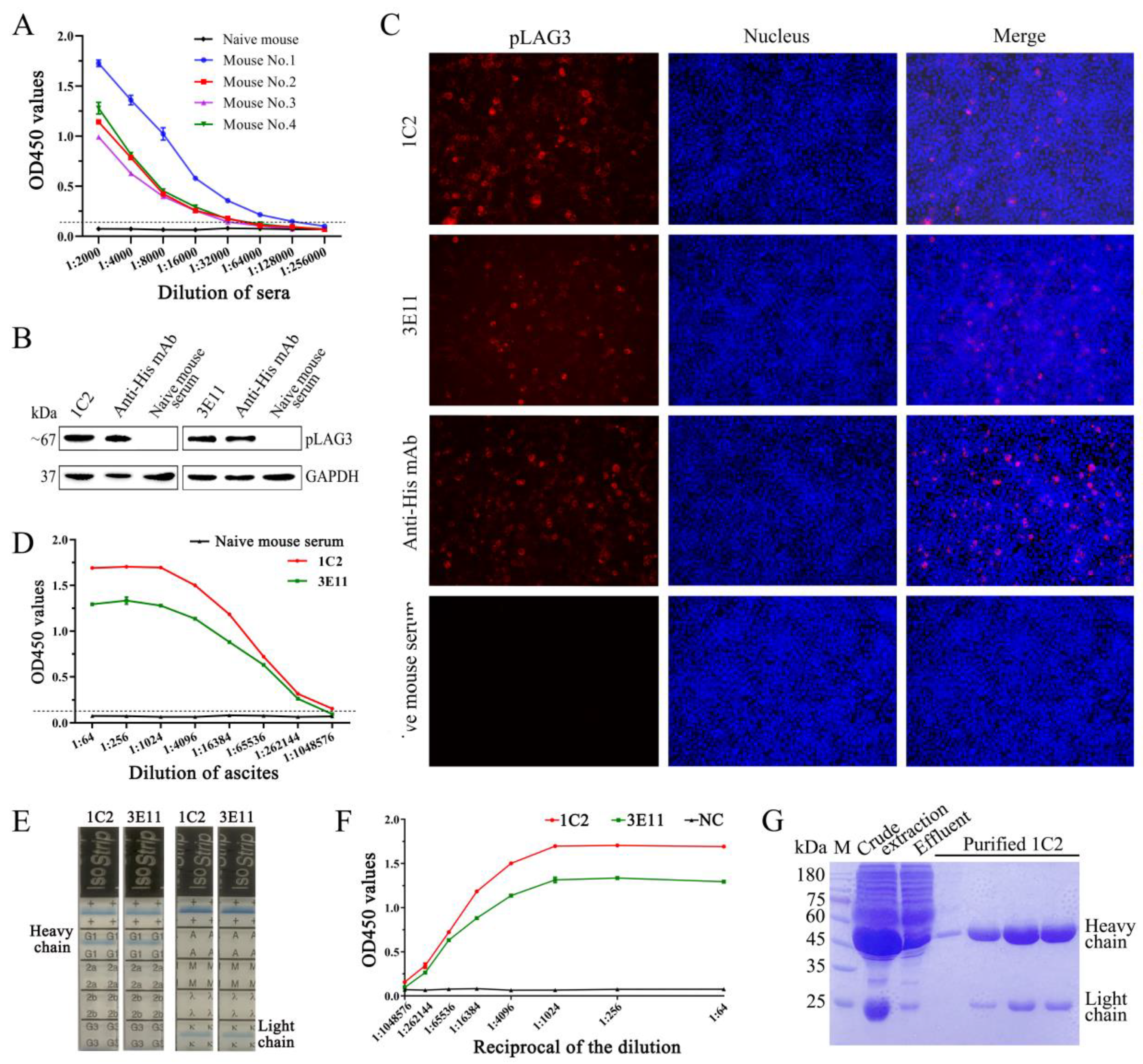
3.2. Generation and Characterization of pLAG3 mAbs
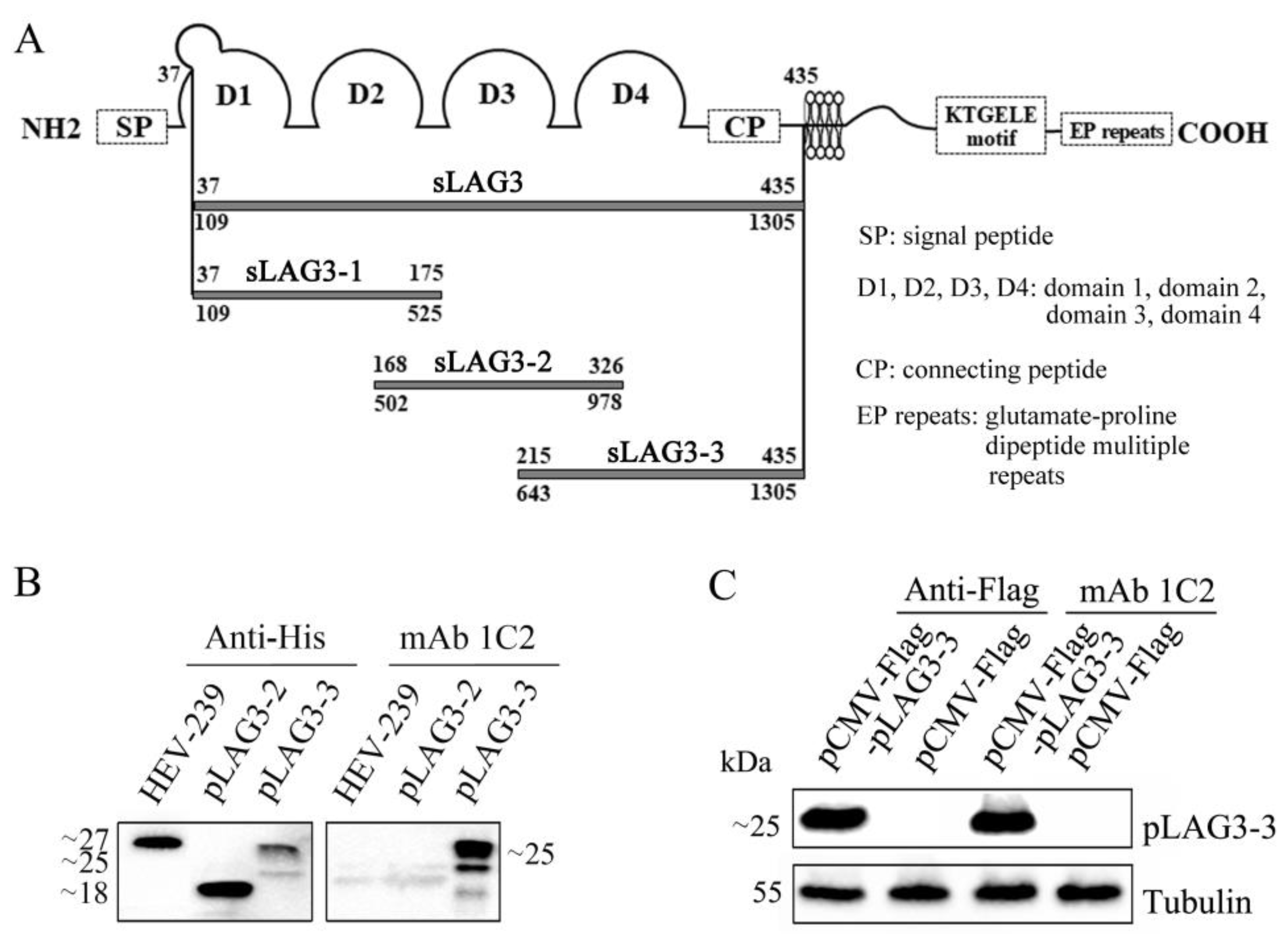
3.3. The Antigen Region Recognized by mAb 1C2 Located at 214–435 AA of pLAG3
3.4. PRRSV Infection Promotes LAG3, SLA-DRα, and FGL1 Expression In Vivo
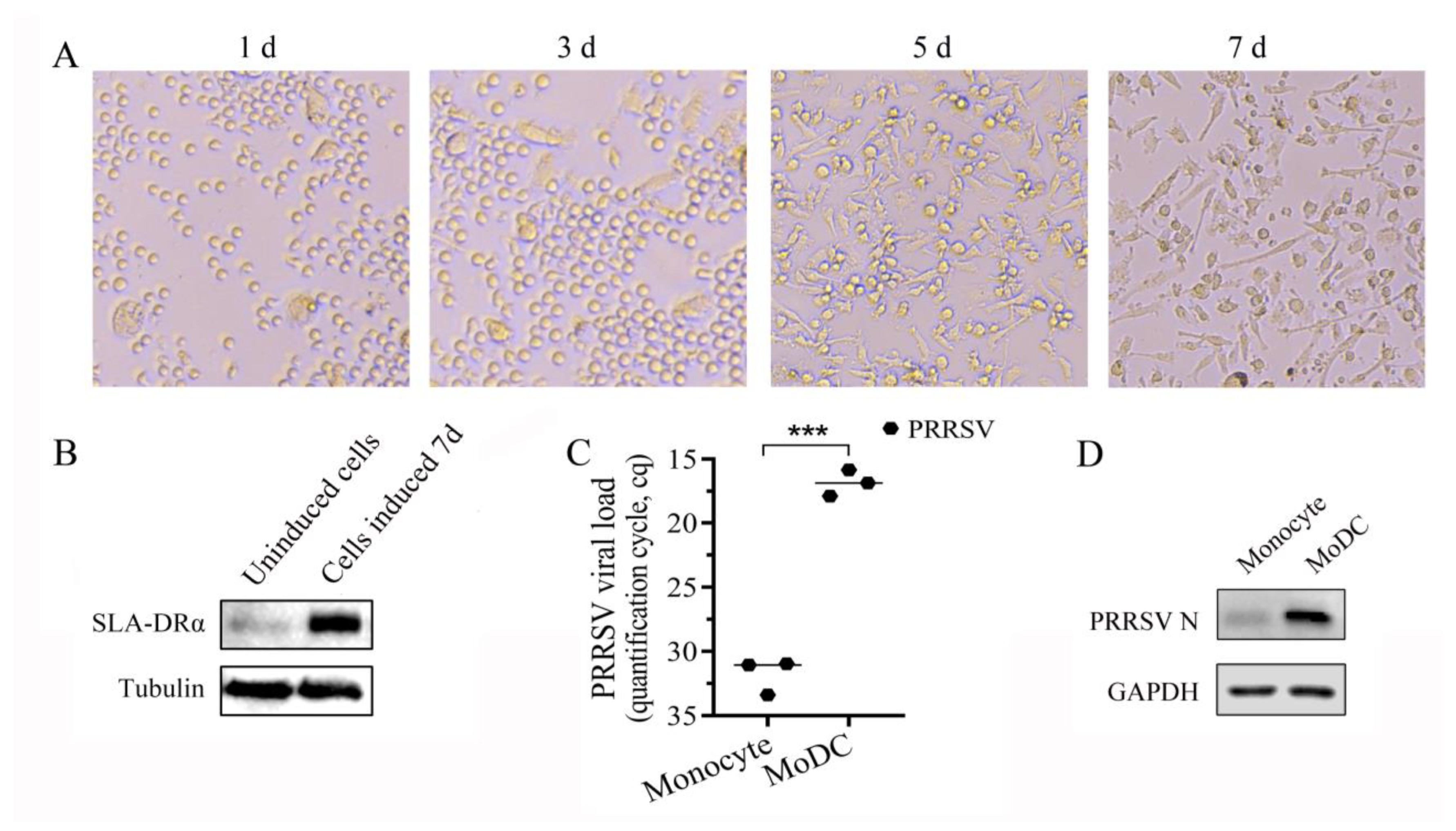
3.5. Monocytes Were Induced into MoDCs
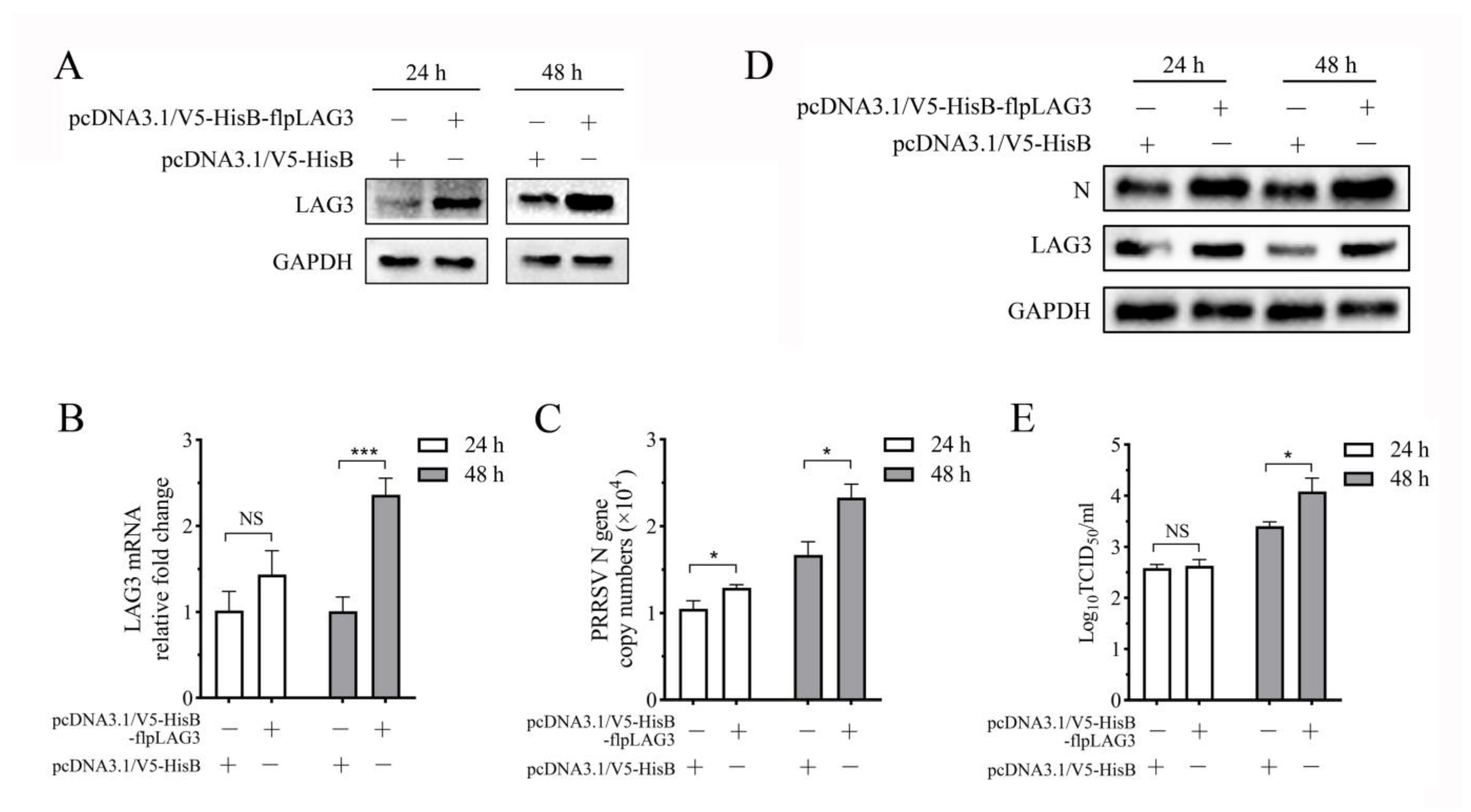
3.6. PRRSV Replication Increases in the Co-Culture System When Lymphocytes Overexpress LAG3
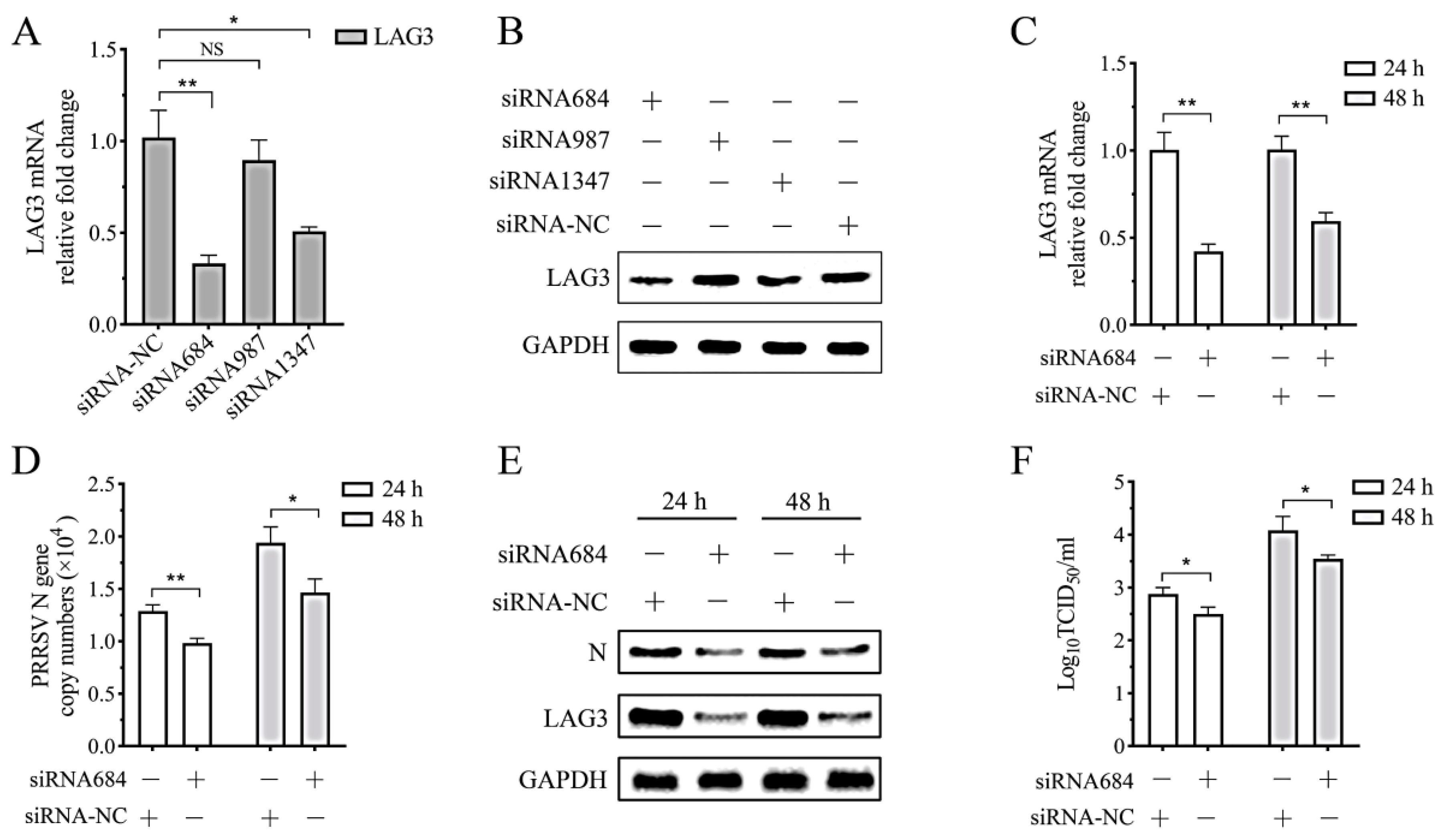
3.7. PRRSV Replication in the Co-Culture System Decreases with Interference of Lymphocyte LAG3 Expression by siRNA
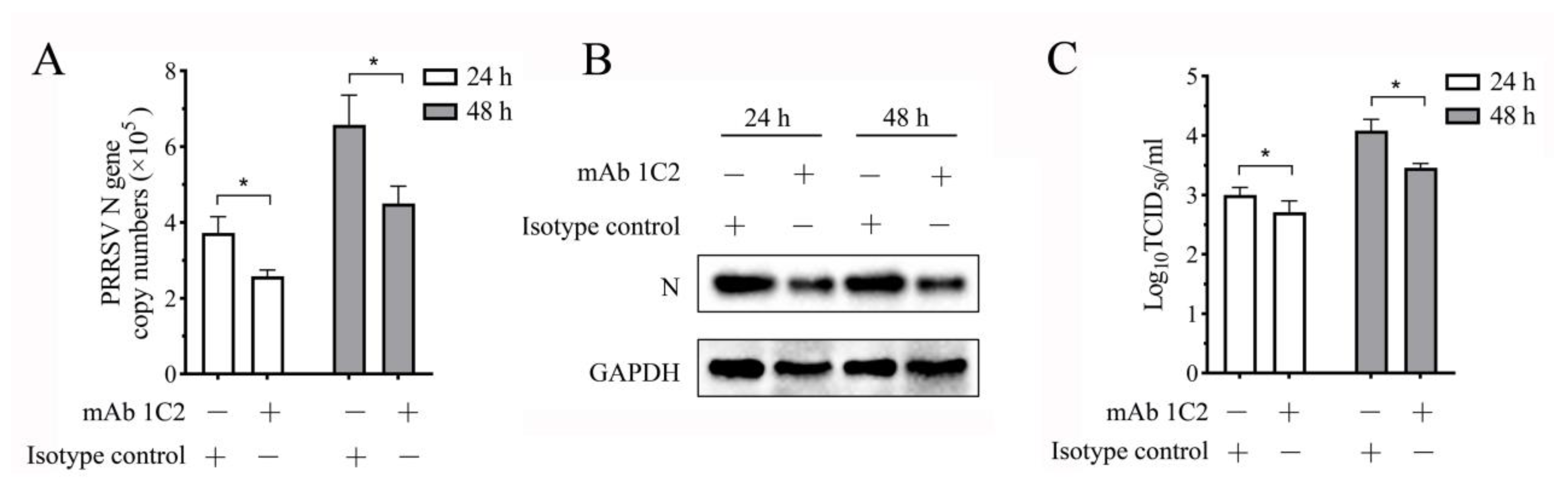
3.8. PRRSV Replication in the Co-Culture System Decreases When the LAG3 Signal Is Blocked by Specific mAb
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ruffo, E.; Wu, R.C.; Bruno, T.C.; Workman, C.J.; Vignali, D.A.A. Lymphocyte-activation gene 3 (LAG3): The next immune checkpoint receptor. Semin. Immunol. 2019, 42, 101305. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, S.D.; Shin, H.; Haining, W.N.; Zou, T.; Workman, C.J.; Polley, A.; Betts, M.R.; Freeman, G.J.; Vignali, D.A.; Wherry, E.J. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat. Immunol. 2009, 10, 29–37. [Google Scholar] [CrossRef]
- Graydon, C.G.; Mohideen, S.; Fowke, K.R. LAG3’s Enigmatic Mechanism of Action. Front. Immunol. 2020, 11, 615317. [Google Scholar] [CrossRef] [PubMed]
- Joller, N.; Kuchroo, V.K. Tim-3, Lag-3, and TIGIT. Curr. Top. Microbiol. Immunol. 2017, 410, 127–156. [Google Scholar] [PubMed]
- Wang, J.; Sanmamed, M.F.; Datar, I.; Su, T.T.; Ji, L.; Sun, J.; Chen, L.; Chen, Y.; Zhu, G.; Yin, W.; et al. Fibrinogen-like Protein 1 Is a Major Immune Inhibitory Ligand of LAG-3. Cell 2019, 176, 334–347.e312. [Google Scholar] [CrossRef]
- Huang, C.T.; Workman, C.J.; Flies, D.; Pan, X.; Marson, A.L.; Zhou, G.; Hipkiss, E.L.; Ravi, S.; Kowalski, J.; Levitsky, H.I.; et al. Role of LAG-3 in regulatory T cells. Immunity 2004, 21, 503–513. [Google Scholar] [CrossRef]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef]
- Guy, C.; Mitrea, D.M.; Chou, P.C.; Temirov, J.; Vignali, K.M.; Liu, X.; Zhang, H.; Kriwacki, R.; Bruchez, M.P.; Watkins, S.C.; et al. LAG3 associates with TCR-CD3 complexes and suppresses signaling by driving co-receptor-Lck dissociation. Nat. Immunol. 2022, 23, 757–767. [Google Scholar] [CrossRef]
- Garcia Cruz, D.; Giri, R.R.; Gamiotea Turro, D.; Balsbaugh, J.L.; Adler, A.J.; Rodriguez, A. Lymphocyte Activation Gene-3 Regulates Dendritic Cell Metabolic Programing and T Cell Priming Function. J. Immunol. 2021, 207, 2374–2384. [Google Scholar] [CrossRef]
- Durham, N.M.; Nirschl, C.J.; Jackson, C.M.; Elias, J.; Kochel, C.M.; Anders, R.A.; Drake, C.G. Lymphocyte Activation Gene 3 (LAG-3) modulates the ability of CD4 T-cells to be suppressed in vivo. PLoS ONE 2014, 9, e109080. [Google Scholar] [CrossRef]
- Aggarwal, V.; Workman, C.J.; Vignali, D.A.A. LAG-3 as the third checkpoint inhibitor. Nat. Immunol. 2023, 24, 1415–1422. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Chikina, M.; Szymczak-Workman, A.L.; Horne, W.; Kolls, J.K.; Vignali, K.M.; Normolle, D.; Bettini, M.; Workman, C.J.; Vignali, D.A.A. LAG3 limits regulatory T cell proliferation and function in autoimmune diabetes. Sci. Immunol. 2017, 2, eaah4569. [Google Scholar] [CrossRef] [PubMed]
- Yim-Im, W.; Anderson, T.K.; Paploski, I.A.D.; VanderWaal, K.; Gauger, P.; Krueger, K.; Shi, M.; Main, R.; Zhang, J. Refining PRRSV-2 genetic classification based on global ORF5 sequences and investigation of their geographic distributions and temporal changes. Microbiol. Spectr. 2023, 11, e0291623. [Google Scholar] [CrossRef] [PubMed]
- Ruedas-Torres, I.; Sánchez-Carvajal, J.M.; Carrasco, L.; Pallarés, F.J.; Larenas-Muñoz, F.; Rodríguez-Gómez, I.M.; Gómez-Laguna, J. PRRSV-1 induced lung lesion is associated with an imbalance between costimulatory and coinhibitory immune checkpoints. Front. Microbiol. 2022, 13, 1007523. [Google Scholar] [CrossRef]
- Ruedas-Torres, I.; Rodríguez-Gómez, I.M.; Sánchez-Carvajal, J.M.; Guil-Luna, S.; Larenas-Muñoz, F.; Pallarés, F.J.; Carrasco, L.; Gómez-Laguna, J. Up-Regulation of Immune Checkpoints in the Thymus of PRRSV-1-Infected Piglets in a Virulence-Dependent Fashion. Front. Immunol. 2021, 12, 671743. [Google Scholar] [CrossRef]
- Triebel, F.; Hacene, K.; Pichon, M.F. A soluble lymphocyte activation gene-3 (sLAG-3) protein as a prognostic factor in human breast cancer expressing estrogen or progesterone receptors. Cancer Lett. 2006, 235, 147–153. [Google Scholar] [CrossRef]
- Huard, B.; Prigent, P.; Tournier, M.; Bruniquel, D.; Triebel, F. CD4/major histocompatibility complex class II interaction analyzed with CD4- and lymphocyte activation gene-3 (LAG-3)-Ig fusion proteins. Eur. J. Immunol. 1995, 25, 2718–2721. [Google Scholar] [CrossRef]
- Maruhashi, T.; Okazaki, I.M.; Sugiura, D.; Takahashi, S.; Maeda, T.K.; Shimizu, K.; Okazaki, T. LAG-3 inhibits the activation of CD4(+) T cells that recognize stable pMHCII through its conformation-dependent recognition of pMHCII. Nat. Immunol. 2018, 19, 1415–1426. [Google Scholar] [CrossRef]
- Li, N.; Wang, Y.; Forbes, K.; Vignali, K.M.; Heale, B.S.; Saftig, P.; Hartmann, D.; Black, R.A.; Rossi, J.J.; Blobel, C.P.; et al. Metalloproteases regulate T-cell proliferation and effector function via LAG-3. EMBO J. 2007, 26, 494–504. [Google Scholar] [CrossRef]
- Woo, S.R.; Turnis, M.E.; Goldberg, M.V.; Bankoti, J.; Selby, M.; Nirschl, C.J.; Bettini, M.L.; Gravano, D.M.; Vogel, P.; Liu, C.L.; et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012, 72, 917–927. [Google Scholar] [CrossRef]
- Kim, S.S.; Kim, S.H.; Kang, H.S.; Chung, H.Y.; Choi, I.; Cheon, Y.P.; Lee, K.H.; Lee, D.M.; Park, J.; Lee, S.Y.; et al. Molecular cloning and expression analysis of pig lymphocyte activation gene-3 (LAG-3; CD223). Vet. Immunol. Immunopathol. 2010, 133, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Ma, L.; Yang, M.; Wu, W.; Feng, W.; Chen, Z. The Function of the PRRSV-Host Interactions and Their Effects on Viral Replication and Propagation in Antiviral Strategies. Vaccines 2021, 9, 364. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhu, H.; Zheng, X.; Jiao, Y.; Ning, L.; Zhou, E.M.; Mu, Y. A Double-Antibody Sandwich ELISA for Sensitive and Specific Detection of Swine Fibrinogen-like Protein 1. Front. Immunol. 2021, 12, 670626. [Google Scholar] [CrossRef] [PubMed]
- Zhou, E.M.; Xiao, Y.; Shi, Y.; Li, X.; Ma, L.; Jing, S.; Peng, J. Generation of internal image monoclonal anti-idiotypic antibodies against idiotypic antibodies to GP5 antigen of porcine reproductive and respiratory syndrome virus. J. Virol. Methods 2008, 149, 300–308. [Google Scholar] [CrossRef]
- Wu, C.; Shi, B.; Yang, D.; Zhang, K.; Li, J.; Wang, J.; Liu, H.; Zhao, Q.; Zhou, E.M.; Nan, Y. Porcine Reproductive and Respiratory Syndrome Virus Promotes SLA-DR-Mediated Antigen Presentation of Nonstructural Proteins To Evoke a Nonneutralizing Antibody Response In Vivo. J. Virol. 2020, 94, e01423. [Google Scholar] [CrossRef]
- Zhai, T.; Wu, C.; Wang, N.; Shi, B.; Li, J.; Chen, R.; Dong, J.; Zhang, Y.; Zhou, E.M.; Nan, Y. Development of a monoclonal antibody against swine leukocyte antigen (SLA)-DR α chain and evaluation of SLA-DR expression in bone marrow-derived dendritic cells after PRRSV infection. Vet. Immunol. Immunopathol. 2019, 211, 19–24. [Google Scholar] [CrossRef]
- Friguet, B.; Djavadi-Ohaniance, L.; Pages, J.; Bussard, A.; Goldberg, M. A convenient enzyme-linked immunosorbent assay for testing whether monoclonal antibodies recognize the same antigenic site. Application to hybridomas specific for the beta 2-subunit of Escherichia coli tryptophan synthase. J. Immunol. Methods 1983, 60, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wen, Y.; An, T.; Duan, G.; Sun, M.; Ge, J.; Li, X.; Yang, K.; Cai, X. Development of an Immunochromatographic Strip for Rapid Detection of Canine Adenovirus. Front. Microbiol. 2019, 10, 2882. [Google Scholar] [CrossRef]
- Chen, R.; Liu, B.; Zhang, X.; Qin, M.; Dong, J.; Gu, G.; Wu, C.; Wang, J.; Nan, Y. A porcine reproductive and respiratory syndrome virus (PRRSV)-specific IgM as a novel adjuvant for an inactivated PRRSV vaccine improves protection efficiency and enhances cell-mediated immunity against heterologous PRRSV challenge. Vet. Res. 2022, 53, 65. [Google Scholar] [CrossRef]
- Mu, Y.; Li, L.L.; Zhang, B.B.; Huang, B.C.; Gao, J.M.; Wang, X.P.; Wang, C.B.; Xiao, S.Q.; Zhao, Q.; Sun, Y.N.; et al. Glycoprotein 5 of porcine reproductive and respiratory syndrome virus strain SD16 inhibits viral replication and causes G2/M cell cycle arrest, but does not induce cellular apoptosis in Marc-145 cells. Virology 2015, 484, 136–145. [Google Scholar] [CrossRef]
- Kuzemtseva, L.; de la Torre, E.; Martín, G.; Soldevila, F.; Ait-Ali, T.; Mateu, E.; Darwich, L. Regulation of toll-like receptors 3, 7 and 9 in porcine alveolar macrophages by different genotype 1 strains of porcine reproductive and respiratory syndrome virus. Vet. Immunol. Immunopathol. 2014, 158, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Nedumpun, T.; Ritprajak, P.; Suradhat, S. Generation of potent porcine monocyte-derived dendritic cells (MoDCs) by modified culture protocol. Vet. Immunol. Immunopathol. 2016, 182, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Solinas, C.; Migliori, E.; De Silva, P.; Willard-Gallo, K. LAG3: The Biological Processes That Motivate Targeting This Immune Checkpoint Molecule in Human Cancer. Cancers 2019, 11, 1213. [Google Scholar] [CrossRef] [PubMed]
- Shan, C.; Li, X.; Zhang, J. Progress of immune checkpoint LAG-3 in immunotherapy. Oncol. Lett. 2020, 20, 207. [Google Scholar] [CrossRef]
- Workman, C.J.; Dugger, K.J.; Vignali, D.A. Cutting edge: Molecular analysis of the negative regulatory function of lymphocyte activation gene-3. J. Immunol. 2002, 169, 5392–5395. [Google Scholar] [CrossRef]
- He, W.; Tan, G.S.; Mullarkey, C.E.; Lee, A.J.; Lam, M.M.; Krammer, F.; Henry, C.; Wilson, P.C.; Ashkar, A.A.; Palese, P.; et al. Epitope specificity plays a critical role in regulating antibody-dependent cell-mediated cytotoxicity against influenza A virus. Proc. Natl. Acad. Sci. USA 2016, 113, 11931–11936. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primers’ Name | Sequence (5′-3′) | Target Fragment/bp |
|---|---|---|
| pLAG3-109-peF | CGGGATCCATGGGGGCTCCTGC (BamH I) a | 1200 |
| pLAG3-1305-peR | CCCAAGCTTGAGGTGGCCTGTTTTCTGG (Hind III) | |
| pLAG3-eeF | CCCAAGCTTATGAGGGAGGCTCACTTCC (Hind III) | 1521 |
| pLAG3-eeR | CGGGATCCGAGCTGCTCTGGCTG (BamH I) | |
| pLAG3-109-peF | CGGGATCCATGGGGGCTCCTGC (BamH I) | 417 (pLAG3-1) |
| pLAG3-525-peR | CCCAAGCTTGTTCAAAATGACCCAATGC (Hind III) | |
| pLAG3-502-peF | CGGGATCCATGACCTCGCATTGGGTCATT (BamH I) | 477 (pLAG3-2) |
| pLAG3-978-peR | CCCAAGCTTCTGCTGCCCCTGAAGATG (Hind III) | |
| pLAG3-643-peF | CGGGATCCATGCAAATCAGCCCCTTGGAC (BamH I) | 663 (pLAG3-3) |
| pLAG3-1305-peR | CCCAAGCTTGAGGTGGCCTGTTTTCTGG (Hind III) | |
| pLAG3-643-eeF | CCCAAGCTTATGCAAATCAGCCCCTTGGAC (Hind III) | 663 (pLAG3-3) |
| pLAG3-1305-eeR | CGGGATCCGAGGTGGCCTGTTTTCTG (BamH I) | |
| pLAG3-qF | ACCTGGCAACCCGAAGAAA | 217 |
| pLAG3-qR | GCGAGACAGCTCCGTGAAGTA | |
| PRRSV-ORF7-qF | TAAGATCATCGCCCAACAAAAC | 145 |
| PRRSV-ORF7-qR | ACACAATTGCCGCTCACTAGG | |
| pGAPDH-qF | GGTGAAGGTCGGAGTGAACG | 153 |
| pGAPDH-qR | CCGTGGGTGGAATCATACTG |
| siRNA Name | Sequence (5′-3′) | Target | |
|---|---|---|---|
| siRNA684 | Sense | CCUACAGAGAUGGCUUCAATT | pLAG3 |
| Anti-sense | UUGAAGCCAUCUCUGUAGGTT | ||
| siRNA987 | Sense | CUGUCACUUUGGCAGUCAUTT | pLAG3 |
| Anti-sense | AUGACUGCCAAAGUGACAGTT | ||
| siRNA1347 | Sense | CUGGAGCUAUUAGCUUUCATT | pLAG3 |
| Anti-sense | UGAAAGCUAAUAGCUCCAGTT | ||
| NC | Sense | UUCUCCGAACGUGUCACGUTT | None |
| Anti-sense | ACGUGACACGUUCGGAGAATT | ||
| Antibody | OD450 Values | AI | Possible Match |
|---|---|---|---|
| 1C2 (1:1000) | 1.148 ± 0.040 | - | - |
| 3E11 (1:1000) | 1.202 ± 0.059 | - | - |
| 1C2 (1:1000) + 3E11 (1:1000) | 1.216 ± 0.007 | 3.50% | <50% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.; Zheng, X.; Zheng, D.; Wang, X.; Zhao, Z.; Zhao, M.; Guo, Q.; Mu, Y. Monoclonal Antibody against Porcine LAG3 Inhibits Porcine Reproductive and Respiratory Syndrome Virus Infection. Vet. Sci. 2024, 11, 483. https://doi.org/10.3390/vetsci11100483
Wang H, Zheng X, Zheng D, Wang X, Zhao Z, Zhao M, Guo Q, Mu Y. Monoclonal Antibody against Porcine LAG3 Inhibits Porcine Reproductive and Respiratory Syndrome Virus Infection. Veterinary Sciences. 2024; 11(10):483. https://doi.org/10.3390/vetsci11100483
Chicago/Turabian StyleWang, Hui, Xu Zheng, Danyang Zheng, Xiaoqian Wang, Zhiqian Zhao, Mi Zhao, Qiang Guo, and Yang Mu. 2024. "Monoclonal Antibody against Porcine LAG3 Inhibits Porcine Reproductive and Respiratory Syndrome Virus Infection" Veterinary Sciences 11, no. 10: 483. https://doi.org/10.3390/vetsci11100483
APA StyleWang, H., Zheng, X., Zheng, D., Wang, X., Zhao, Z., Zhao, M., Guo, Q., & Mu, Y. (2024). Monoclonal Antibody against Porcine LAG3 Inhibits Porcine Reproductive and Respiratory Syndrome Virus Infection. Veterinary Sciences, 11(10), 483. https://doi.org/10.3390/vetsci11100483